Molecular tectonics: ribbon type coordination networks based on porphyrins bearing two pyridine or two pyridine N-oxide units†
Emmanuel
Deiters
,
Véronique
Bulach
* and
Mir
Wais Hosseini
*
Laboratoire de Chimie de Coordination Organique (UMR 7140), Université Louis Pasteur, Institut Le Bel, 4 rue Blaise Pascal, 67000, Strasbourg, France. E-mail: hosseini@chimie.u-strasbg.fr; Fax: 33 390241325; Tel: 33 390241323
First published on 6th September 2007
Abstract
Upon combining porphyrin based tectons bearing at the 5,10 meso positions either two pyridine or two pyridine N-oxide groups, with Cd2+ or Cu2+, four ribbon-type 1-D networks were obtained and structurally characterised in the crystalline phase by X-ray diffraction methods. Although, based on tecton’s structural features, the formation of ribbon-type architectures could be anticipated, their deformation, resulting from the geometry of the connecting metal centres as well as the packing forces in the solid state, could not be predicted.
Introduction
The rational design of coordination polymers in the solid state with predicted connectivity and structure is a subject of current investigations.1–9 One of the reasons for this increasing interest is related to the possibility of generating an infinite number of periodic molecular architectures upon combining a large variety of organic and metallic units. The approach called molecular tectonics10–13 is particularly well suited for the design and formation of coordination networks. Indeed, in principle, the shape, dimension and the topology of the final network may be coded within the structure of tectons or construction units by the number, nature and geometry of the coordinating sites on the organic tecton and by the number and disposition of coordination sites on the metal ions serving as connectors. However, the predictability of the connectivity and consequently the nature of the final architecture generated by self-assembly processes still remains an unresolved challenge. The diversity of architectures that may be formed strongly depends on the structural flexibility of tectons. In order to restrict the number of possible structures, on must use conformationally well defined units. With respect to that issue, owing to their rigidity, functionalised meso-arylporphyrin derivatives bearing coordination sites at their periphery are particularly interesting tectons. This type of molecules have been largely used for the formation of either discrete architectures14–21 or infinite coordination networks.22–32Furthermore, another interesting feature associated with the use of porphyrins as tectons is related to the fact that upon combining either free porphyrin derivatives or their metal complexes, one may generate a large variety of homo- or hetero-metallic architectures.
Pursuing our studies on the formation of discrete assemblies or infinite networks based on porphyrin derivatives,33–37 in particular porphyrins bearing pyridyl groups and to explore the role played by the pyridine N-oxide (PyNO) groups in the formation of coordination networks, we have investigated the formation of homo- as well hetero-bimetallic ribbon type networks by combining porphyrins bearing either two pyridine (Py) 1 or two pyridine N-oxide groups 2 located at two adjacent meso positions (Scheme 1) and external connector presenting four free coordination sites.
 | ||
| Scheme 1 | ||
In this contribution, we report on the formation of both homo- and hetero-metallic ribbon type architecture based on the combination of compounds 1 and 2 with Cd2+ and Cu2+ cations.
Results and discussion
Considering V-shape construction units such as tectons 1 and 2 (Fig. 1(a)) or their metal complexes (Fig. 1(b)) offering two coordinating sites in cis disposition, depending on the connecting metal centres one may envisage different types of architecture (Fig. 1). In principle, the combination with metal cations offering two free coordination sites in cis disposition, should either lead to the formation of discrete metallamacrocycles15–18,37 (Fig. 1(c)–(e)) or zigzag-type (Fig. 1(f), (g)) infinite 1-D networks. Combining tectons 1 and 2 or their metal complexes with metal centres or complexes offering four free coordination sites should afford ribbon type homometallic (Fig. 1(h) and (i)) or heterometallic architectures (Fig. 1(j)). The reverse situation, i.e. combination of a tetradentate tecton such as meso-tetrapyridineporphyrin or its Zn complex with V-shape connector such as HgX2 (X = Br, I) complexes has been shown to generate ribbon type 1-D networks.27–31 | ||
| Fig. 1 Schematic representation of the formation of discrete or infinite architectures resulting from the combination of V-shape tectons offering two coordinating sites with metal centres possessing either two or four coordination sites. | ||
In order to explore the possibility of forming ribbon-type coordination networks, tecton 1, bearing two pyridine units as monodentate coordinating site, was combined with CdBr2. The latter was chosen as connector because on one hand, it is a neutral complex and thus its combination with the neutral tecton 1 would lead to neutral coordination networks and on the other, Cd(II) often adopts the octahedral coordination geometry with the two halide anions occupying the two apical positions. Upon slow diffusion of a MeOH solution of CdBr2 into a solution of 1 in CHCl3 crystalline material was obtained after several days (see Experimental section). Structural investigation by X-ray diffraction on single crystal revealed that the crystal (triclinic, P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) ) is composed of 1, CdBr2 and CHCl3 solvent molecules. The Cd(II) ion, on an inversion centre, is hexacoordinated and adopts a distorted D4h geometry with four pyridines in the equatorial plane and two bromides in the axial positions. The independent Cd–N distances are equal to 2.388(2) and 2.446(2) Å while the Cd–Br bond length is 2.7557(3) Å (Fig. 2 top). The porphyrin backbone is slightly saddle-shaped with an average deviation of the Cβ atoms relative to the porphyrin mean plane (24 atoms) close to 0.4 Å. Due to the mutual bridging between tecton 1 and CdBr2, the overall architecture is a 1-D ribbon-type neutral network. The ribbon is almost flat, indeed, the four connected porphyrin backbones 1 by Cd2+ cation are parallel but not coplanar with 1.0 Å separation between porphyrin units along the ribbon axis and 1.7 Å between the two adjacent units perpendicular to the ribbon axis. The distance between the centroids of two porphyrin rings is 14.2 Å for the adjacent units perpendicular to the axis of the ribbon and 14.5 Å for consecutive porphyrins along the ribbon axis. Within the ribbon, the Cd⋯Cd distance is 14.5 Å. Dealing with packing of 1-D networks, the adjacent ribbons are stacked with the shortest distance between the centroid of two porphyrins of two adjacent ribbons of 5.04 Å (Fig. 2 bottom). CHCl3 molecules are located in the interspaces between consecutive ribbons with no specific interactions with the porphyrin or CdB2 units.
) is composed of 1, CdBr2 and CHCl3 solvent molecules. The Cd(II) ion, on an inversion centre, is hexacoordinated and adopts a distorted D4h geometry with four pyridines in the equatorial plane and two bromides in the axial positions. The independent Cd–N distances are equal to 2.388(2) and 2.446(2) Å while the Cd–Br bond length is 2.7557(3) Å (Fig. 2 top). The porphyrin backbone is slightly saddle-shaped with an average deviation of the Cβ atoms relative to the porphyrin mean plane (24 atoms) close to 0.4 Å. Due to the mutual bridging between tecton 1 and CdBr2, the overall architecture is a 1-D ribbon-type neutral network. The ribbon is almost flat, indeed, the four connected porphyrin backbones 1 by Cd2+ cation are parallel but not coplanar with 1.0 Å separation between porphyrin units along the ribbon axis and 1.7 Å between the two adjacent units perpendicular to the ribbon axis. The distance between the centroids of two porphyrin rings is 14.2 Å for the adjacent units perpendicular to the axis of the ribbon and 14.5 Å for consecutive porphyrins along the ribbon axis. Within the ribbon, the Cd⋯Cd distance is 14.5 Å. Dealing with packing of 1-D networks, the adjacent ribbons are stacked with the shortest distance between the centroid of two porphyrins of two adjacent ribbons of 5.04 Å (Fig. 2 bottom). CHCl3 molecules are located in the interspaces between consecutive ribbons with no specific interactions with the porphyrin or CdB2 units.
 | ||
| Fig. 2 A portion of the structure of the almost flat ribbon obtained upon combining the 5,10-dipyridylporphyrin 1 with CdBr2 showing the interconnection of consecutive porphyrins 1 by Cd(II) cations (top) and the packing of consecutive ribbons (bottom). C atoms of consecutive ribbons are differentiated by different shading for sake of clarity. H atoms are not presented for clarity. For bond distances and angles see text. | ||
In order to generate heterobimetallic ribbon type coordination networks, the tecton 1 was metallated with Cu2+. The combination of 1-Cu2+ with Cd(NO3)2, afforded crystalline material which was analysed using X-ray diffraction on single crystals. The crystal was composed of 1-Cu2+, Cd(NO3)2, MeOH and CHCl3 solvent molecules. As mentioned above, again a ribbon type architecture is observed. The difference here is related to the presence of Cu2+ within the core of the porphyrin 1 and thus, the ribbon formed is of heterobimetallic type (Fig. 3). The ribbon is generated by interconnection of metallaporphyrin tecton 1-Cu2+ by Cd(II) centres which lie on a mirror plane. The Cd(II) cation, due to the disorder of one of the two nitrate anions (occupancy 0.5), is either penta- or hexa-coordinated and is coordination sphere is composed of four N atoms of pyridines belonging to four consecutive 1-Cu2+ (Cd–N distance of 2.340(8) and 2.373(8) Å) and either one or two O atom belonging to nitrate anions (dCd–O of 2.307(12) and 2.38(5) Å (occupancy of 0.5)). Four pyridines belonging to four tectons 1-Cu2+ are located in the equatorial plane while nitrate anions are located in the apical positions.
 | ||
| Fig. 3 A portion of the structure of the ribbon obtained upon combining the 1-Cu2+ with Cd(NO3)2 showing the interconnection of consecutive porphyrins 1-Cu2+ by Cd(II) cations. H atoms are not presented for clarity. For bond distances and angles see text. | ||
The copper is located within the tetraaza core of the porphyrin with Cu–N distance in the range of 1.971(9) and 1.985(9) Å. The porphyrin backbone is saddle-shaped with an average deviation for the Cb relative to the porphyrin mean plane close to 0.5 Å. Within a ribbon, two consecutive Cd(II) centres are separated by 14.1 Å while the distances between two coppers complexed by adjacent porphyrins are 13.6 Å for two units located perpendicular to the ribbon axis (along c) and 14.6 Å for two porphyrins along the ribbon axis (along b).
In marked contrast with the almost flat ribbon obtained by combining 1 with CdBr2 mentioned above, in the present case, the consecutive metallated porphyrins are not parallel but tilted and consequently, the ribbon is severely puckered (Fig. 4). The dihedral angles between the mean planes of two facing and adjacent porphyrins are 39.1 and 56.4°, respectively. The puckered arrangement adopted by the ribbon may be related to the packing of the consecutive ribbons. Indeed, while along the a axis adjacent ribbons are stacked, along the c axis adjacent ribbons are interlaced (Fig. 4). Disordered solvent molecules (CHCl3 and MeOH) are located between ribbons along the a axis. The shortest Cd⋯Cd distance along this direction is 8.7 Å.
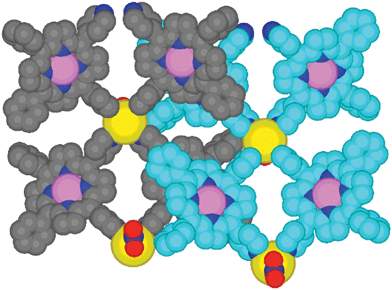 | ||
| Fig. 4 A portion of the structure showing the packing of consecutive heterobimetallic ribbons. C atoms of consecutive ribbons are differentiated by different shading for sake of clarity. H atoms are not presented for clarity. For bond distances and angles see text. | ||
Although pyridine is one of the most often used coordinating groups for the formation of coordination networks, pyridine N-oxide (PyNO) has been demonstrated to be another interesting neutral unit for the generation of infinite frameworks.38–44 In particular, Schröder and co-workers showed that the combination of 4,4′-bipyridine N-oxide with lanthanide cations leads to the formation of a variety of coordination networks.39
Dealing with porphyrin derivatives, we have shown37 that the oxidation of the meso-pyridyl groups to pyridine N-oxide leads to a less sterically demanding and more flexible coordinating site. Indeed, for the PyNO group, the binding takes place through the oxygen atom thus increasing the size of the tecton by ca. 1.3 Å for each PyNO group with respect to the non-oxidized precursor. Furthermore, when compared to pyridine for which the binding of the metal is achieved in a linear fashion because of the disposition of the lone pair of nitrogen, in the case of PyNO, owing to the orientation of the lone pairs centred on the oxygen atom, the binding of the metal centre occurs in a bent fashion with a M–O–N bond angle of ca. 120°. Moreover, due to the presence of two lone pairs on the oxygen atom, the PyNO group may also act as a bridging ligand. Because the N–O–M angle is different from zero, for tectons such as 2 bearing two PyNO groups, depending on the relative orientation of the M–O bonds, three different conformations (in–in, out–out and in–out) may be envisaged (Fig. 5).
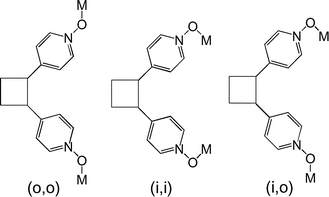 | ||
| Fig. 5 Schematic representation of the three possible conformations in the solid state for binding of metal centres by tectons bearing two PyNO groups: out-out (o, o), in–in (i, i) and in–out (i, o). | ||
The propensity of the tecton 2 was explored by combining the latter with Cu2+ salts. Upon slow diffusion of a solution containing 2 in CHCl3 into a MeOH solution of CuBr2 a crystalline solid was obtained. The structural study by X-ray diffraction on single crystal revealed that the crystal (triclinic, P, Z = 1) is composed of 2, Cu2+ cation, (CuBr4)2– complex and H2O solvent molecules. The porphyrin 2 has been metallated by CuBr2 during the crystallisation process. The copper is located in the centre of the cavity with a Cu–N distance of ca. 2.00 Å. The porphyrin backbone is slightly saddle-shaped (maximum deviation for Cβ equal to 0.49 Å). The consecutive 2-Cu2+ units are interconnected by Cu2+ cations through four Cu–O bonds. The connecting Cu2+ cation, located on a centre of inversion, adopts a square planar geometry with its coordination sphere composed of four O atoms belonging to four different 2-Cu2+ units (average Cu–O bond distance of ca. 1.96 Å). Consequently, the overall architecture is a cationic homometallic ribbon-type network (Fig. 6).
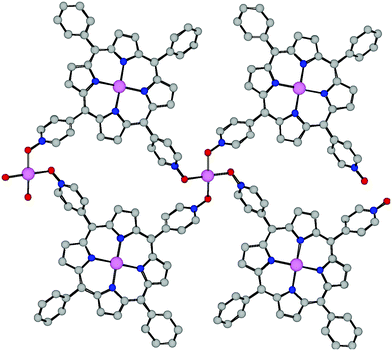 | ||
| Fig. 6 A portion of the structure of the ribbon obtained upon combining the 2 with CuBr2 showing the interconnection of consecutive porphyrins 2-Cu2+ by Cu(II) cations. H atoms are not presented for clarity. For bond distances and angles see text. | ||
Considering the orientation of the oxygen lone pairs, the in–out disposition (Fig. 5) is observed with independent Cu–O–N bond angles of 117.4(3) and 120.0(3)°. Within a ribbon, the Cu2+ atoms complexed by the porphyrin core are separated by 14.0 and 14.5 Å for the two facing units and two adjacent porphyrins, respectively. The distance between bridging coppers is 14.0 Å. The homobimetallic ribbons are almost flat with parallel arrangement of facing porphyrin backbones. The consecutive porphyrins are not coplanar and distant by ca. 0.8 Å. Considering the packing, the ribbons are parallel to each other with the shortest Cu⋯Cu distance of 8.19 Å.
As stated above, the interconnection of 2-Cu2+ by Cu2+ cations leads to a positively charged network. The charge neutrality in the crystalline phase is ensured by disordered (CuBr4)2– anion located between the ribbons. The anion may be described as (CuBr3, Br)2– species with the copper centre disordered over two positions (occupancy of 0.5). Indeed, the observation of three Cu–Br bonds distance of ca. 2.4 Å and one Cu–Br distance of 5.04 Å indicating the absence of interaction between the anion and copper cation, is in favour of the hypothesis. The shortest distance between (CuBr3, Br)2– anions and adjacent ribbons is equal to 3.08 Å and involves bromides located in the apical position of the connecting copper present in the ribbons.
Three disordered water molecules (occupancy of 0.5) are also present between the ribbons with no specific interactions with the 1D network.
Starting from 2 and Cu(BF4)2 we have obtained a similar homobimetallic ribbon. Again, during the crystallisation process, the porphyrin 2 has been metallated. The porphyrin units 2-Cu2+ are connected through external copper bridges. Contrary to the case mentioned above, here the connecting Cu(II) cation is five-coordinated and adopts a distorted pyramidal geometry with one PyNO in the apical position and the equatorial plane composed by three PyNO and one chloride arising from the decomposition of CHCl3 (Fig. 7). Again, the Cu-porphyrin adopts a in–out conformation with Cu–O–N angles ranging from 114.9(3) to 123.8(3)°. Interestingly, due to the presence of a PyNO in the apical position, the ribbons are severely twisted. The dihedral angle between the mean planes of two adjacent porphyrins is closed to 39°, while facing porphyrins are parallel but not coplanar with a separation of 3.6 Å between them.
 | ||
| Fig. 7 A portion of the structure of the ribbon obtained upon combining the 2 with Cu(BF4)2 showing the interconnection of consecutive porphyrins 2-Cu2+ by Cu(II) cations and the presence of Cl– anions on the bridging metal. H atoms are not presented for clarity. For bond distances and angles see text. | ||
The homometallic ribbon crystallizes in the P space group with two copper porphyrins and one CuCl+ moiety in the asymmetric unit. The neutrality is achieved by the presence of BF4– anions located between the ribbons.
The saddle-shape deformation of the two independent porphyrin backbones is similar to that observed in the previous cases with an average deviation for the Cβ relative to the porphyrin mean plane equal to 0.4 Å. In both porphyrins, the copper is located in the centre of the cavity with Cu–N distances equal to 2.00(1) Å. Considering the bridging copper ion, the three equatorial Cu–O bond distances are close to 1.96 Å while the axial Cu–O bond length is equal to 2.339(4) Å. The Cu–Cl bond distance is equal to 2.264(2) Å.
Within the ribbon, the connecting copper centres are separated by 14.3 Å and the Cu(por)⋯Cu(por) distances are close to 13.9 Å between facing porphyrins while the distance between adjacent porphyrins is equal to 18.0 Å. This long distance compared to previous cases is induced by the peculiar coordination of the four PyNO around the five-coordinated connecting copper cation. The ribbons are arranged in a parallel fashion and along the a axis, the Cu⋯Cu distance is ca. 9.7 Å. Disordered solvent molecules (one CHCl3 and 3/2MeOH in the asymmetric unit) are located in the empty space between ribbons.
Conclusions
We have shown that porphyrin based ligands bearing either two pyridyl (compound 1) or pyridyl N-oxide (compound 2) groups at two adjacent meso positions are suitable construction units for the generation of 1-D coordination networks. The combination of bismonodentate tectons 1 and 2 with metal centres offering four free coordination sites leads under self-assembly conditions to ribbon-type homo- or hetero-bimetallic coordination networks in the crystalline phase. Interestingly, all three ribbons generated display similar characteristics. Although, based on binding features of tecton 1 and metal cations used, it was possible to anticipate the formation of the observed ribbons, for the tecton 2 bearing two PyNO groups, owing to its propensity to generate different isomers (different orientation of the two coordinated metal centres), the formation of the ribbon-type architecture could not be predicted. Furthermore, concerning the shape of the ribbon, two out of the four ribbons obtained are almost flat whereas the other two are severely twisted. This deformation could not be anticipated. Based on tectons 1, 2 and analogues, the formation of heterobimetallic coordination networks bearing transition metal cations and lanthanide ions is currently under investigation.Experimental
Porphyrin 1 was synthesized according to literature procedure26 and the porphyrin 2 was obtained upon oxidation of 1 by m-chloroperbenzoic acid.45 The copper complex 1-Cu2+ was obtained following a reported procedure.36Microanalyses were performed by the Service de Microanalyses de la Fédération de Recherche de Chimie, Université Louis Pasteur, Strasbourg or by Service de Microanalyse-Vernaison, CNRS, Lyon, France.
Synthesis of ribbon (1,![[thin space (1/6-em)]](https://www.rsc.org/images/entities/h3_char_2009.gif) CdBr2)n
CdBr2)n
Crystals suitable for X-ray analysis were obtained within 5–7 days at room temperature by slow diffusion of a MeOH solution of CdBr2 (5 eq./2mL) into a solution of the porphyrin 1 in CHCl3 (0.5 mg/0.5 mL). C84H56N12CdBr2·3CHCl3 (1863.73): calc.: C 56.06, H 3.19, N 9.02. Found: C 55.93, H 3.14, N 8.90%.
Synthesis of ribbon (1-Cu2+,Cd(NO3)2)n
Crystals suitable for X-ray analysis were obtained within 5–7 days at room temperature by slow diffusion of a MeOH solution of Cd(NO3)2 (5 eq./2 mL), into a solution of the complex 1-Cu2+ in CHCl3 (0.5 mg/0.5 mL). (C84H52N12Cu2)·Cd(NO3)2·0.5 CHCl3·0.5CH3OH (1668.64): calc.: C 60.82, H 3.30, N 11.75. Found: C 61.01, H 3.36, N 11.55%.
Synthesis of ribbon (2-Cu2+,Cu2+,CuBr4)n
Crystals suitable for X-ray analysis were obtained within 10–12 days at room temperature by slow diffusion of a MeOH solution of CuBr2 (5 eq./2 mL), into a solution of the porphyrin 2 in CHCl3–CH3OH (1–1) (0.5 mg/0.5 mL). C84H52Br4Cu4N12O3·3H2O (1921.22): calc.: C 52.51, H 3.05, N 8.75. Found: C 52.67, H 3.17, N 8.60%.
Synthesis of ribbon (2-Cu2+,CuCl(BF4))n
Crystals suitable for X-ray analysis were obtained within 12–15 days, at room temperature by slow diffusion of a MeOH solution of Cu(BF4)·6H2O (5 eq./2 ml) into a solution of the porphyrin 2 in CHCl3–CH3OH (1 : 1) (0.5 mg/0.5 mL). (C84H52N12O4Cu2)Cu·BF4Cl·0.75CHCl3·1.5CH3OH (1743.90): calc.: C 59.40, H 3.40, N 9.64. Found: C 59.29, H 3.36, N 9.82%.
Crystallography
Data were collected at 173(2) K on a Bruker APEX8 CCD Diffractometer equipped with an Oxford Cryosystem liquid N2 device, using graphite-monochromated Mo-Kα (λ = 0.71073) radiation. For all structures, diffraction data were corrected for absorption and structural determination was achieved using the APEX (1.022) package. All hydrogen atoms have been calculated except those connected to disordered atoms.![[thin space (1/6-em)]](https://www.rsc.org/images/entities/b_char_2009.gif) CdBr2)n.
2(C42H28N6)·CdBr2·4CHCl3; C88H60Br2CdCl12N12, M = 1983.10, triclinic, space groupP, a = 9.4956(4), b = 14.4774(6), c = 16.5058(6) Å, α = 82.058(2), β = 79.919(2), γ = 74.550(2)°, U = 2143.27(15) Å3, Z = 1, µ = 1.614 mm–1, refl. measured: 38
CdBr2)n.
2(C42H28N6)·CdBr2·4CHCl3; C88H60Br2CdCl12N12, M = 1983.10, triclinic, space groupP, a = 9.4956(4), b = 14.4774(6), c = 16.5058(6) Å, α = 82.058(2), β = 79.919(2), γ = 74.550(2)°, U = 2143.27(15) Å3, Z = 1, µ = 1.614 mm–1, refl. measured: 38![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 653, independent refl.: 12431, Rint = 0.0317. Final R indices [I > 2σ(I)]: R1 = 0.0500, wR2 = 0.1565.
Cd(NO3)2)n.
2(CuC42H26N6)·Cd(NO3)2·CHCl3·1/2CH3OH; C85.5H55CdCl3Cu2N14O6.5, M = 1728.26, orthorhombic, space groupPbcm, a = 8.7394(4), b = 27.7882(16), c = 31.3773(16) Å, U = 7620.0(7) Å3, Z = 4, µ = 1.001 mm–1, refl. measured: 55803, independent refl.: 8890, Rint = 0.1622. Final R indices [I > 2σ(I)]: R1 = 0.1123, wR2 = 0.2858.
Cu2+,CuBr4)n.
Cu3C84H52N12O4·CuBr4·3H2O; C84H58Br4Cu4N12O7, M = 1921.22, triclinic, space groupP, a = 8.1872(3), b = 14.4491(5), c = 16.8506(5) Å, α = 80.7490(10), β = 78.4670(10), γ = 75.4150(10)°, U = 1877.34(11) Å3, Z = 1, µ = 3.312 mm–1, refl. measured: 50820, independent refl.: 11013, Rint = 0.0419. Final R indices [I > 2σ(I)]: R1 = 0.0795, wR2 = 0.2533.
CuCl(BF4))n.
4(CuC42H26N6O2)·2(CuCl(BF4)·2CHCl3·3CH3OH; C173H118B2Cl8Cu6F8N24O11, M = 3547.37, triclinic, space groupP, a = 9.7001(3), b = 17.1346(5), c = 24.9317(8) Å, α = 86.6580(10), β = 79.721(2), γ = 73.9430(10)°, U = 3918.1(2) Å3, Z = 1, µ = 1.016 mm–1, refl. measured: 90211, independent refl.: 17993, Rint = 0.0480. Final R indices [I > 2σ(I)]: R1 = 0.0781, wR2 = 0.2195.
653, independent refl.: 12431, Rint = 0.0317. Final R indices [I > 2σ(I)]: R1 = 0.0500, wR2 = 0.1565.
Cd(NO3)2)n.
2(CuC42H26N6)·Cd(NO3)2·CHCl3·1/2CH3OH; C85.5H55CdCl3Cu2N14O6.5, M = 1728.26, orthorhombic, space groupPbcm, a = 8.7394(4), b = 27.7882(16), c = 31.3773(16) Å, U = 7620.0(7) Å3, Z = 4, µ = 1.001 mm–1, refl. measured: 55803, independent refl.: 8890, Rint = 0.1622. Final R indices [I > 2σ(I)]: R1 = 0.1123, wR2 = 0.2858.
Cu2+,CuBr4)n.
Cu3C84H52N12O4·CuBr4·3H2O; C84H58Br4Cu4N12O7, M = 1921.22, triclinic, space groupP, a = 8.1872(3), b = 14.4491(5), c = 16.8506(5) Å, α = 80.7490(10), β = 78.4670(10), γ = 75.4150(10)°, U = 1877.34(11) Å3, Z = 1, µ = 3.312 mm–1, refl. measured: 50820, independent refl.: 11013, Rint = 0.0419. Final R indices [I > 2σ(I)]: R1 = 0.0795, wR2 = 0.2533.
CuCl(BF4))n.
4(CuC42H26N6O2)·2(CuCl(BF4)·2CHCl3·3CH3OH; C173H118B2Cl8Cu6F8N24O11, M = 3547.37, triclinic, space groupP, a = 9.7001(3), b = 17.1346(5), c = 24.9317(8) Å, α = 86.6580(10), β = 79.721(2), γ = 73.9430(10)°, U = 3918.1(2) Å3, Z = 1, µ = 1.016 mm–1, refl. measured: 90211, independent refl.: 17993, Rint = 0.0480. Final R indices [I > 2σ(I)]: R1 = 0.0781, wR2 = 0.2195.
CCDC reference numbers 656906 ((2-Cu2+,CuCl(BF4))n), 656907 ((1-Cu2+,Cd(NO3)2)n), 659598 ((1,CdBr2)n) and 659599 ((2-Cu2+,Cu2+,CuBr4)n).
For crystallographic data in CIF or other electronic format see DOI: 10.1039/b708352a
Acknowledgements
Université Louis Pasteur, Institut Universitaire de France, the CNRS and the Ministry of Education and Research are acknowledged for financial support and for a scholarship to E. D.References
- S. R. Batten and R. Robson, Angew. Chem., Int. Ed., 1998, 37, 1460 CrossRef.
- A. J. Blake, N. R. Champness, P. Hubberstey, W.-S. Li, M. A. Withersby and M. Schröder, Coord. Chem. Rev., 1999, 193, 117 CrossRef.
- M. W. Hosseini, in NATO ASI Series, ed. D. Braga, F. Grepiono and G. Orpen, Serie c, Kluwer, Dordrecht, Netherlands, 1999, vol. 538, p. 181 Search PubMed.
- B. Moulton and M. J. Zaworotko, Chem. Rev., 2001, 101, 1629 CrossRef CAS.
- M. Eddaoui, D. B. Moler, H. Li, B. Chen, T. M. Reineke, M. O’Keeffe and O. M. Yaghi, Acc. Chem. Res., 2001, 34, 319 CrossRef CAS.
- G. F. Swiergers and T. J. Malefetse, Chem. Rev., 2000, 100, 3483 CrossRef CAS.
- C. Janiak, Dalton Trans., 2003, 2781 RSC.
- S. Kitagawa, Angew. Chem., Int. Ed., 2004, 43, 2434.
- (a) G. Férey, Actual. Chim., 2007, 304, 3 Search PubMed; (b) G. Férey, C. Mellot-Draznieks, C. Serre and F. Millange, Acc. Chem. Res., 2005, 38, 218.
- M. Simard, D. Su and J. D. Wuest, J. Am. Chem. Soc., 1991, 113, 4696 CrossRef.
- S. Mann, Nature, 1993, 365, 499 CrossRef CAS.
- M. W. Hosseini, Acc. Chem. Res., 2005, 38, 313 CrossRef CAS.
- M. W. Hosseini, CrystEngComm, 2004, 6, 318 RSC.
- C. M. Drain and J.-M. Lehn, J. Chem. Soc., Chem. Commun., 1994, 2313 RSC.
- (a) E. Iengo, F. Scandola and E. Alessio, Struct. Bonding, 2006, 121, 105 CAS; (b) F. Scandola, C. Chiorboli, A. Prodi, E. Iengo and E. Alessio, Coord. Chem. Rev., 2006, 250, 1471 CrossRef CAS.
- (a) E. Iengo, B. Milani, E. Zangrando, S. Geremia and E. Alessio, Angew. Chem., Int. Ed., 2000, 39, 2344; (b) E. Iengo, E. Zangrando and E. Alessio, Eur. J. Inorg. Chem., 2003, 2371 CrossRef CAS; (c) E. Iengo, E. Zangrando, R. Minatel and E. Alessio, J. Am. Chem. Soc., 2002, 124, 1003 CrossRef CAS; (d) E. Iengo, E. Zangrando, M. Bellini, E. Alessio, A. Prodi, C. Chiorboli and F. Scandola, Inorg. Chem., 2005, 44, 9752 CrossRef CAS.
- (a) S. J. Lee and J. T. Hupp, Coord. Chem. Rev., 2006, 250, 1710 CrossRef CAS; (b) R. V. Slone and J. T. Hupp, Inorg. Chem., 1997, 36, 5422 CrossRef CAS.
- (a) P. J. Stang, J. Fan and B. Olenyuk, Chem. Commun., 1997, 1453 RSC; (b) J. Fan, J. A. Whiteford, B. Olenyuk, M. D. Levin, P. J. Stang and E. B. Fleischer, J. Am. Chem. Soc., 1999, 121, 2741 CrossRef CAS; (c) M. Schmitz, S. Leiniger, J. Fan, A. M. Arif and P. J. Stang, Organometallics, 1999, 18, 4817 CrossRef CAS.
- (a) H. L. Anderson, C. A. Hunter, M. N. Meah and J. K. M. Sanders, J. Am. Chem. Soc., 1990, 112, 5780 CrossRef CAS; (b) S. Anderson, H. L. Anderson and J. K. M. Sanders, J. Chem. Soc., Perkin Trans. 1, 1995, 2255 RSC; (c) J. K. M. Sanders, in Comprehensive Supramolecular Chemistry, ed. J. L. Atwood, J. E. D. Davies, D. D. MacNicol, F. Vögtle, J. P. Sauvage and M. W. Hosseini, Elsevier, 1996, vol. 9, pp. 131–164 Search PubMed; (d) A. Vidal-Ferran, Z. Clyde-Watson, N. Bampos and J. K. M. Sanders, J. Org. Chem., 1997, 62, 240 CrossRef CAS.
- (a) K. Ogawa and Y. Kobuke, Angew. Chem., Int. Ed., 2000, 39, 4070 CrossRef CAS; (b) N. Fujita, K. Biradha, M. Fujita, S. Sakamoto and K. Yamagushi, Angew. Chem., Int. Ed., 2001, 40, 1718 CrossRef CAS.
- A. Ikeda, M. Ayabe, S. Shinkai, S. Sakamoto and K. Yamaguchi, Org. Lett., 2000, 2, 3707 CrossRef CAS.
- (a) K. S. Suslick, P. Bhyrappa, J.-H. Chou, M. E. Kosal, S. Nakagaki, D. W. Smithenry and S. R. Wilson, Acc. Chem. Res., 2005, 38, 283 CrossRef CAS; (b) J. H. Chou, M. E. Kosal, H. S. Nalwa, N. A. Rakow and K. S. Suslick, in The Porphyrin Handbook, ed. K. M. Kadish, K. M. Smith and R. Guilard, Academic Press, New York, 2000, vol. 6, pp. 43–132 Search PubMed.
- (a) B. F. Abrahams, B. F. Hoskins and R. Robson, J. Am. Chem. Soc., 1991, 113, 3606 CrossRef CAS; (b) B. F. Hoskins, D. M. Michail and R. Robson, Nature, 1994, 369, 727 CrossRef CAS.
- U. Michelsen and C. A. Hunter, Angew. Chem., Int. Ed., 2000, 39, 764 CrossRef CAS.
- L. Carlucci, G. Ciani, D. M. Proserpio and F. Porta, CrystEngComm, 2005, 7, 78 RSC.
- (a) E. B. Fleischer and A. M. Shachter, Inorg. Chem., 1991, 30, 3763 CrossRef CAS; (b) S. Maji, A. Kumar, K. Pal and S. Sarkar, Inorg. Chem., 2005, 44, 7277 CrossRef CAS.
- C. V. K. Sharma, G. A. Broker, J. G. Huddleston, J. W. Baldwin, R. M. Metzger and R. D. Rogers, J. Am. Chem. Soc., 1999, 121, 1137 CrossRef CAS.
- L. Pan, B. C. Noll and X. Wang, Chem. Commun., 1999, 157 RSC.
- (a) L. Pan, S. Kelly, X. Huang and J. Li, Chem. Commun., 2002, 2334 RSC; (b) L. Pan, X. Huang, H.-L. N. Phan, T. J. Emge, J. Li and X. Wang, Inorg. Chem., 2004, 43, 6878 CrossRef CAS.
- C. M. Drain, F. Nifiatis, A. Vasnko and J. D. Batteas, Angew. Chem., Int. Ed., 1998, 37, 2344 CrossRef CAS.
- D. J. Ring, M. C. Aragoni, N. R. Champness and C. Wilson, CrystEngComm, 2005, 7, 621 RSC.
- (a) I. Goldberg, Chem. Commun., 2005, 1243 RSC; (b) I. Goldberg, Chem.–Eur. J., 2000, 21, 3863 CrossRef CAS; (c) H. Krupitsky, Z. Stein, I. Goldberg and C. E. Strouse, J. Inclusion Phenom. Mol. Recognit. Chem., 1994, 18, 177 Search PubMed.
- B. Zimmer, V. Bulach, M. W. Hosseini, A. De Cian and N. Kyritsakas, Eur. J. Inorg. Chem., 2002, 3079 CrossRef CAS.
- B. Zimmer, M. Hutin, V. Bulach, M. W. Hosseini, A. De Cian and N. Kyritsakas, New J. Chem., 2002, 26, 1532 RSC.
- E. Deiters, V. Bulach and M. W. Hosseini, Chem. Commun., 2005, 3906 RSC.
- E. Deiters, V. Bulach and M. W. Hosseini, New J. Chem., 2006, 30, 1289 RSC.
- E. Deiters, V. Bulach, N. Kyritsakas and M. W. Hosseini, New J. Chem., 2005, 29, 1508 RSC.
- B. Q. Ma, S. Gao, H.-L. Sun and G.-X. Xu, J. Chem. Soc., Dalton Trans., 2001, 130 RSC.
- (a) D.-L. Long, A.-J. Black, N. R. Champness, C. Wilson and M. Schröder, J. Am. Chem. Soc., 2001, 123, 3401 CrossRef CAS; (b) D.-L. Long, A. J. Black, N. R. Champness, C. Wilson and M. Schröder, Chem.–Eur. J., 2002, 8, 2026 CrossRef CAS; (c) R. J. Hill, D.-L. Long, M. S. Turvey, A. J. Black, N. R. Champness, P. Hubberstey, C. Wilson and M. Schröder, Chem. Commun., 2004, 1, 1792 RSC; (d) R. J. Hill, D.-L. Long, N. R. Champness, P. Hubberstey and M. Schröder, Acc. Chem. Res., 2005, 38, 337.
- S. Tanase, M. Andruh, A. Muller, M. Schmidtmann, C. Mathoiere and G. Rombaut, Chem. Commun., 2001, 1084 RSC.
- S.-L. Ma, W.-X. Zhu, G.-H. Huang, D.-Q. Yuan and X. Yan, J. Mol. Struct., 2003, 646, 89 CrossRef CAS.
- A. Nedelcu, Z. Zak, A. M. Madalan, J. Pinkas and M. Andruh, Polyhedron, 2003, 22, 789 CrossRef CAS.
- Y. Xu, D. Yuan, L. Han, E. Ma, M. Wu, Z. Lin and M. Hong, Eur. J. Inorg. Chem., 2005, 2054 CrossRef CAS.
- D. Gonzalez Mantero, A. Neels and H. Stoeckli-Evans, Inorg. Chem., 2006, 45, 3287 CrossRef CAS.
- J. J. Posakony, R. C. Pratt, S. J. Rettig, B. R. James and K. A. Skov, Can. J. Chem., 1999, 77, 182 CrossRef CAS.
Footnote |
| † The HTML version of this article has been enhanced with colour images. |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2008 |