A general system for evaluating the efficiency of chromophore-assisted light inactivation (CALI) of proteins reveals Ru(II) tris-bipyridyl as an unusually efficient “warhead”
Jiyong
Lee
,
Peng
Yu
,
Xiangshu
Xiao
and
Thomas
Kodadek
*
Division of Translational Research, Departments of Internal Medicine and Molecular Biology, University of Texas Southwestern Medical Center, 5323 Harry Hines Blvd, Dallas, TX 75390-9185. E-mail: Thomas.Kodadek@utsouthwestern.edu
First published on 9th October 2007
Abstract
Chromophore-assisted light inactivation (CALI) of proteins is a potentially powerful tool in biological research for the triggered disruption of protein function. It involves the creation of chimeric molecules that can bind specifically to the protein target and can also sensitize the photo-generation of singlet oxygen, which inactivates the target protein . There remains a need for more efficient chromophores for singlet oxygen generation. Here we report a general and convenient system with which to evaluate the efficiency of chromophores in CALI both in crude extracts and in living cells. We employ this system to show that a readily available derivative of ruthenium(II) tris-bipyridyl dication is an unusually efficient “warhead” for CALI, exhibiting a performance markedly superior to the commonly used organic fluorophore, fluorescein.
Introduction
There is great interest in the development of techniques for the selective inactivation of protein function as a tool for the study of biological systems. By far the most general and powerful method to do so in mammalian cells is RNA interference (RNAi) technology, which results in the destruction of the message that encodes the protein of interest.1,2 Another general and powerful approach is the construction of transgenic animals lacking the gene that encodes the protein of interest. However, all such genetic methods have significant limitations, one of which is that they do not allow for the possibility of carrying out time-resolved studies of the effects of protein knockouts.3 Thus, there has been a great deal of interest in chemical approaches to protein inactivation.Of the many strategies that have been explored, chromophore-assisted light inactivation (CALI) holds the promise of being a particularly useful tool.4–7 In this technique, a chromophore capable of generating singlet oxygen when irradiated is somehow delivered to the target protein . Singlet oxygen is an extraordinarily reactive and short-lived molecule and thus will rapidly inactivate only proteins in the immediate vicinity of its point of origin. Thus, this technique provides high spatial and temporal control over protein inactivation.
Unfortunately, the practical utility of this technology is currently limited. There are two major barriers. The first is the paucity of high-affinity, high-specificity ligands for the protein of interest. In most cases, chromophore-conjugated antibodies are employed, but these molecules are cell-impermeable and the use of a macromolecular targeting agent may place the chromophore some distance away from the target protein when bound. Cell permeable small molecules hold great promise as targeting agents and will probably dominate this area in the future. However, while the selection of cell permeable protein -binding small molecules from combinatorial libraries is advancing rapidly, it remains challenging to obtain ligands with affinities in the low nM region, which would be ideal for this application. For this reason, various strategies have been reported by which engineered proteins can be inactivated by CALI.8–10 One of the most effective of these is to tag the target protein with the sequence CCXXCC (where X can be several amino acids), which is a high affinity and relatively specific receptor for biarsenical fluorescein derivatives.8,9 Of course, this approach is most useful in cases where the native analogue of the tagged protein has also somehow been eliminated.
The second limitation of this technology is the efficiency of singlet oxygen generation exhibited by the chromophores used routinely in CALI. The most common “warheads” employed in CALI experiments are organic fluorophores with relatively long-lived triplet states.4–7 While some cases of efficient protein inactivation using organic fluorophore-containing constructs have been reported, there remains a need for better singlet oxygen generators since these molecules are sensitive to self-bleaching.
Recently, we have become interested in developing a comprehensive approach for CALI of native proteins . In previous reports, we have demonstrated that protein -binding peptoids (oligo-N-alkylglycines) can be consistently and rapidly isolated from combinatorial libraries.11–14 The screening conditions that we have developed routinely provide peptoids that bind their target with good specificity and equilibrium dissociation constants in the low µM range. Furthermore, these peptoids are generally cell permeable15,16 and thus peptoid libraries provide a promising source of specific binding agents required for CALI of native proteins (again however, the challenge of maturing these ligands to nM binders remains). At the same time, we have begun to turn our attention to the development of more efficient “warheads” for use in CALI reagents that overcome the current limitations of organic fluorophores. In this paper, we report our initial efforts towards this goal. First, we designed a convenient system that allows the efficiency of protein inactivation of any chromophore to be compared to any other (Fig. 1). As is discussed below, this system allows CALI warheads to be evaluated both in crude extracts as well as in living cells. Using this system, we compared the efficacy of a Ru(II) tris(bipyridyl) dication (Ru(bpy)32+) derivative with fluorescein, one of the most efficient and commonly used organic chromophores. A wealth of literature is available showing that Ru(bpy)32+ is an efficient photocatalyst for singlet oxygen generation and thus might be an excellent CALI warhead.17–22 We demonstrate that the ruthenium complex is indeed a more efficient CALI warhead than fluorescein and is capable of supporting essentially quantitative inactivation of the target protein . Ru(bpy)32+ derivatives can be attached easily to synthetic protein -binding molecules and thus are likely to be of considerable utility in the next generation of CALI reagents.
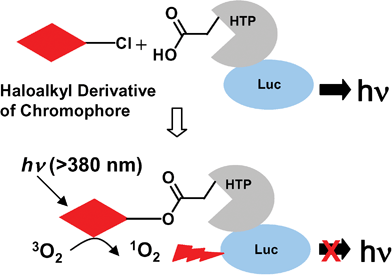 | ||
| Fig. 1 A system to evaluate CALI efficiencies of different chromophores. A haloalkyl derivative of the chromophore of interest is used to label a target protein , 3×Flag-Luc-HTP-Myc. The photoexcited chromophore converts triplet oxygen to singlet oxygen in the vicinity of the target protein , leading to inactivation. The efficiency of target protein inactivation is determined by measuring the remaining luciferase activity. | ||
Results
Covalent attachment of small molecules to the target protein in complex biological mixtures
In order to facilitate the comparison of different chromophores for their efficiency in singlet oxygen generation and subsequent protein inactivation, we sought to develop a system that would eliminate the need for complex biological antibody-based or synthetic targeting agents. To do so, we made use of the commercially available, mutant haloalkane dehalogenase, the HaloTag protein (HTP).23 Wild-type haloalkane dehalogenases from Xanthobacter autotrophicus GJ10 (XaDHL)24 and Rhodococus rhodochrous (RrDHL)25 hydrolyze halogen-containing alkane substrates to the corresponding alcohols via a mechanism that involves formation of a substrate-enzyme ester intermediate. This occurs through attack of an active site carboxylate (Asp106) anion on the primary alkyl chloride (Fig. 1).26,27 In the wild-type protein , this ester intermediate is subsequently hydrolyzed. However, mutation of a residue in the active site (His272 to Phe) provides a protein that cannot hydrolyze the covalent intermediate. This allows HTP fusion proteins to be labeled covalently with a variety of haloalkane-containing compounds (generically called HaloTags). The labeled product is known to be very stable. While a few other methods exist for the delivery of chimeric small molecules to protein tags, the ease of synthesis of chloroalkane derivatives makes this technique particularly convenient. Since HTP does not exhibit additional, easily assayed activities, we constructed an expression vector for an epitope-tagged Luciferase-HTP (3×Flag-Luc-HTP-Myc) fusion protein . Luciferase activity is easily monitored in cells and in crude extracts and is a convenient target for CALI experiments. Thus, this system should allow the efficient covalent delivery of a primary haloalkane derivative of the chromophore to a luciferase fusion protein , whose activity is monitored before and after irradiation.First, to test if the target protein 3×Flag-Luc-HTP-Myc can be efficiently labeled by HaloTag ligands, a lysate prepared from HeLa cells transfected with the 3×Flag-Luc-HTP-Myc expression vector was prepared and incubated with increasing concentrations of a primary haloalkane derivative of biotin (B-HT; Fig. 2). After a brief incubation, streptavidin-agarose beads were employed to pull out biotinylated proteins . As shown in Fig. 3A, the use of increasing amounts of B-HT followed by streptavidin-agarose pull-out resulted in the loss of luciferase activity in a dose-dependent fashion up to a concentration of 100 nM under the conditions employed. However, addition of higher concentrations of B-HT did not result in increased pull-out of luciferase activity. About 50% of the input was still observed in supernatant, indicating that the labeling reaction was incomplete. Pull-downs with anti-Myc agarose or anti-Flag agarose removed almost all luciferase activity from the cell lysate (Fig. 3B), arguing that the inability of 50% of the protein to be biotinylated could not be ascribed to proteolysis of half of the fusion protein , a conclusion confirmed by Western blot (Fig. 5B). Instead, these data suggest that while most of the fusion protein is intact, only about half of the molecules that display luciferase activity also have active HTP capable of being alkylated by a HaloTag ligand. This phenomenon of bifunctional fusion proteins expressed with only one of the activities intact is not uncommon.
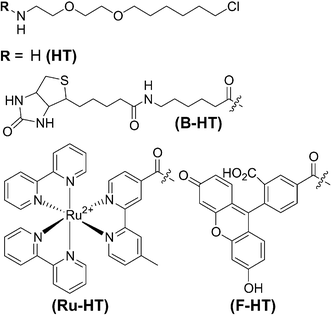 | ||
| Fig. 2 Chemical structures of the HaloTag reagents used in this study. | ||
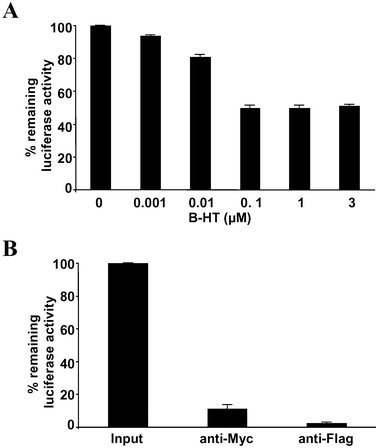 | ||
| Fig. 3 Labeling of target protein with the HaloTag reagent. (A) Extract from HeLa cells expressing the 3×Flag-Luc-HTP-Myc fusion protein was incubated with increasing concentrations of B-HT. Biotinylated protein was then precipitated with streptavidin-agarose. The relative level of luciferase activity remaining in the supernatant was then measured. (B) The same extract was incubated with immobilized anti-Myc or anti-Flag antibodies and the luciferase activity remaining in the supernatant was measured. The data represent the mean of triplicate experiments; error bars indicate standard deviations. | ||
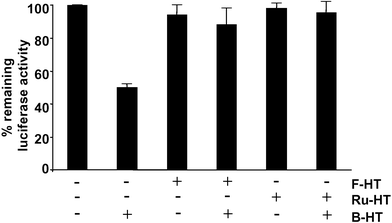 | ||
| Fig. 4 Comparison of the 3×Flag-Luc-HTP-Myc labeling efficiencies of Ru–HT and F–HT. Extract from HeLa cells expressing the 3×Flag-Luc-HTP-Myc fusion protein was incubated with either F-HT or Ru-HT. Subsequently, B-HT was added and the biotinylated material was precipitated with streptavidin-agarose. The amount of luciferase activity remaining in the supernatant, representing fusion protein that had reacted with F-HT or B-HT or contains an inactive HTP domain, was measured. The data represent the mean of triplicate experiments; error bars indicate standard deviations. | ||
Delivery of fluorescein and Ru(II) tris(bipyridyl) dication derivatives to the Luc-HTP fusion protein and comparison of their efficacy in CALI
We next tested whether primary haloalkyl derivatives of Ru(bpy)32+ (Ru-HT; Fig. 2) and fluorescein (F-HT; Fig. 2) can label the target protein . Since we are comparing different chromophores for their CALI efficiencies, it is important for the ligands to have same efficiency in the labeling reaction and so this was tested first. Cell lysate prepared from HeLa cells expressing 3×Flag-Luc-HTP-Myc was first incubated with 100 nM of Ru-HT or F-HT and then treated with an excess of B-HT (1 µM). If any active protein fails to link to either Ru-HT or F-HT, it will be labeled by B-HT and will be removed from the solution by subsequent streptavidin-agarose pull-down. By measuring remaining luciferase activity of the solution, we can indirectly evaluate the labeling efficiencies of Ru-HT and F-HT. As shown in Fig. 4, for both Ru-HT and F-HT, no significant decreases of luciferase activities were observed in the B-HT treated-solutions. This means that all of the active target proteins were already labeled during pretreatment with Ru-HT or F-HT. More importantly, Ru-HT and F-HT showed the same efficiencies of protein labeling within experimental error.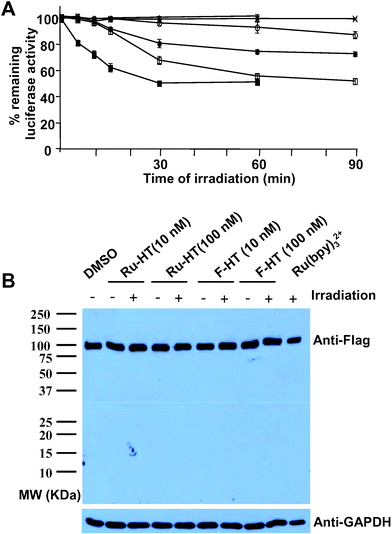 | ||
| Fig. 5 Comparison of Ru–HT and F–HT as CALI “warheads”. (A) Extract from HeLa cells expressing the 3×Flag-Luc-HTP-Myc fusion protein was incubated with DMSO (×), F-HT (10 nM: ○, 100 nM: ●), Ru-HT (10 nM: □, 100 nM: ■), or Ru(bpy)32+ (100 nM: ▲). After irradiation for the indicated times at room temperature, the remaining luciferase activity was measured. Data represent mean of triplicate experiments; error bars indicate standard deviations. (B) Western blot showing no proteolysis during experimental procedures. 2 µL of HeLa cell extract in PBS (total volume: 50 µL, pH 7.4) was incubated with compounds as indicated for 30 min at room temperature and irradiated for 30 min at room temperature. Samples were run on SDS-PAGE gel, transferred to nitrocellulose membrane, and probed with anti-Flag antibody or anti-GAPDH (loading control). | ||
To evaluate the CALI efficiencies of the inorganic and organic chromophores, a HeLa cell lysate containing 3×Flag-Luc-HTP-Myc and Renilla luciferase was again incubated with Ru-HT or F-HT to allow alkylation of the active fusion protein . The sample was then irradiated with visible light using a Xenon arc lamp and a filter that blocks wavelengths below 380 nm. As shown in Fig. 5A, the rate of protein inactivation was dependent on the concentration of the Ru-HT compound. In the case 100 nM of Ru-HT was used, maximal inactivation was achieved after 30 min irradiation. However, slower inactivation of the target protein could be achieved with as little as 10 nM Ru-HT. Recall that since 50% of the protein is incapable of being alkylated, this represents essentially quantitative inactivation of the accessible target protein . Note that no proteolysis of the target protein was observed during labeling and irradiation (Fig. 5B), excluding the possibility that residual luciferase activity can be from cleaved protein . In the absence of irradiation, but in the present of Ru-HT, there was no loss of luciferase activity. Furthermore, Ru(bpy)32+, i.e., the chromophore not incorporated into a HaloTag, also had no effect (Fig. 5A). This shows that the ruthenium complex must be delivered to the fusion in order to be effective and that luciferase inactivation is triggered only after irradiation. Interestingly, under identical conditions, the sample treated with F-HT exhibited little loss of luciferase activity. Note the decrease of luciferase activity by 100 nM of F-HT is even smaller than that of 10 nM of Ru-HT. Since the labeling efficiencies of Ru-HT and F-HT are the same, these data indicate that Ru-HT is a more efficient CALI reagent than one of the best organic fluorescent dyes, fluorescein.
Evidence that singlet oxygen is the protein -damaging agent in Ru-HT-mediated CALI
The reactive oxygen species (ROS) involved in protein inactivation in CALI is mainly either singlet oxygen or hydroxyl radical. Since the half-maximal radius of inactivation of hydroxyl radical (15 Å) is shorter than that of singlet oxygen (40 Å),6 CALI mediated by hydroxyl radical is generally considered to give higher specificity.7 Laser excitation of Malachite Green, a commonly used CALI dye, was shown to generate hydroxyl radical,4 while fluorescein-mediated protein inactivation occurs via singlet oxygen generation.6 We assumed that this is also the case for the ruthenium complex, since photo-excited Ru(bpy)32+ (triplet state) is known to convert triplet oxygen to singlet oxygen by energy transfer.18 To confirm that singlet oxygen is indeed the major vector for protein inactivation in Ru-HT mediated CALI, we tested inhibition of protein inactivation by ROS quenching molecules. As shown in Fig. 6, sodium azide (NaN3), a potent singlet oxygen quencher 28,29 inhibited Ru-HT mediated inactivation of target protein . On the other hand, D-mannitol, a hydroxyl radical or superoxide anion quencher ,30 had no effect. Imidazole, a less potent singlet oxygen quencher than sodium azide,31 also showed some extent of inhibition. We conclude that, as expected, CALI mediated by Ru-HT indeed proceeds via a singlet oxygen intermediate.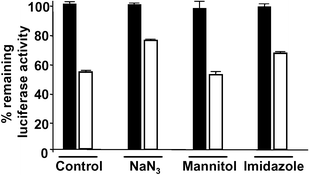 | ||
| Fig. 6 Evidence that Ru-HT-mediated protein inactivation involves singlet oxygen. Extract from HeLa cells expressing the 3×Flag-Luc-HTP-Myc fusion protein was incubated with Ru-HT (100 nM) for 30 min at room temperature. NaN3 (30 mM), D-mannitol (65 mM), or imidazole (30 mM) were added immediately before irradiation. Open bars: irradiated (30 min) sample, filled bars: non-irradiated sample. Data represent mean of triplicate experiments; error bars indicate standard deviations. | ||
Ru-HT inactivates target protein inside living cells
All of the experiments described above were carried out in cell extracts. However, CALI is most useful if it can be performed in living cells. Since Ru(bpy)32+ is known to be cell permeable,32 we speculated that Ru-HT should be able to pass through the cell membrane and label HTP-containing target proteins . To test this, HeLa cells expressing 3×FLAG-Luc-HTP-Myc as well as Renilla luciferase were treated with Ru-HT and irradiated with visible light. As shown in Fig. 7, cells treated with 1 µM Ru-HT and irradiated for 30 minutes exhibited a 40% inactivation of luciferase activity relative to cells treated with DMSO only that were also irradiated. When the extracellular concentration of Ru-HT was increased to 10 µM, 63% of the target luciferase activity was lost. Note that the specific activity of the protein may well be higher in living cells relative to that observed in the crude extract, explaining the ability to knock out more than 50% of the activity. The Renilla luciferase activity was unaffected by Ru-HT and irradiation. Furthermore, incubation of the cells with 10 µM Ru(bpy)32+ had no effect on the activity of the 3×FLAG-Luc-HTP-Myc protein . Taken together, these data argue that the Ru-HT can indeed permeate cells and that its ability to inactivate the target protein requires formation of the covalent intermediate and irradiation, as expected. Under the same conditions, irradiation of cells treated with F-HT also resulted in inactivation of the target protein , but less efficiently than was observed using the Ru-HT reagent. This suggests that, as was observed in vitro, the Ru(bpy)32+ is a more efficient CALI warhead than fluorescein, although we cannot absolutely rule out the possibility that the fluorescein-containing reagent is somewhat less permeable. Finally, it is important to note that under all of the conditions employed in this experiment, neither the ruthenium complex nor irradiation affected cell viability (data not shown).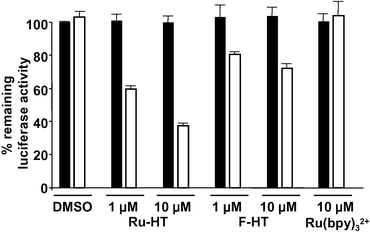 | ||
| Fig. 7 Ru-HT-mediated CALI in living cells. HeLa cells expressing 3×Flag-Luc-HTP-Myc were incubated with DMSO (control), Ru-HT, F-HT, or Ru(bpy)32+ and irradiated. After lysis, the luciferase activities of lysates were measured and presented as described before. Open bars: irradiated sample, filled bars: non-irradiated sample. Data represent mean of triplicate experiments; error bars indicate standard deviations. | ||
Discussion
We have developed a general system to compare the efficiencies of different chromophores as reagents for CALI. This is based on the delivery of a primary haloalkyl derivative of the chromophore to a fusion protein containing the target linked to HTP, which reacts irreversibly with the primary alkyl chloride. Since such derivatives are synthesized easily, this approach should be convenient for testing the efficiency, in a standardized fashion, of new potential CALI agents as they arise. Using this system, we found that a Ru(bpy)32+ has much higher efficiencies in singlet oxygen generation and subsequent target protein inactivation than conventional organic fluorophore, fluorescein. Indeed, Ru(bpy)32+ can mediate quantitative protein inactivation in vitro. In addition, the Ru(bpy)32+-based CALI reagent employed in this study was found to be cell permeable and thus capable of triggering target protein inactivation inside living cells.There is no evidence that the Ru-HT molecule exhibits significant off-target effects at lower concentrations of the ruthenium complex. The experiments reported here all contained two luciferase reporter activities, one fused to HTP (firefly) and one that is not (Renilla). There was no indication that Ru-HT and photolysis affected the activity of the Renilla luciferase. However, for in vitro CALI, the use of high concentrations of untargeted Ru(bpy)32+ at 1 µM or extended photolysis periods (≥1 h) did show inactivation of both target protein and Renilla luciferase. Thus, these reagents are best employed at concentrations of <1 µM and irradiation times should be held to 30 min or less, at least using the light source that we employed.
The greater efficiency of protein inactivation observed using the Ru(bpy)32+ warhead as compared to fluorescein is consistent with the photophysical and photochemical properties of the two molecules. Singlet oxygen is generated from the reaction of ground state triplet oxygen with the triplet state of the excited fluorophore. The quantum yield for triplet formation of fluorescein is relatively low (0.1 in MeOH)21 in comparison to Ru(bpy)32+ (0.86 in MeOH) due to the latter's access to a metal to ligand charge transfer state (MLCT).33
Photochemical stability of fluorophores to singlet oxygen can be another factor affecting the efficiency of CALI reagent. In other words, the sensitivity of the CALI warhead to photobleaching limits the number of singlet oxygen generation events that can occur. While organic fluorophores, commonly used in CALI, are known to be very sensitive to photobleaching, Ru(bpy)32+ is stable to singlet oxygen as shown in previous studies of photo-oxidation of 1, 3-cyclopentadiene by Ru(bpy)32+.34,35 Thus, it is likely that the Ru(bpy)32+ warhead has a far greater turnover number for singlet oxygen generation in a CALI experiment than does fluorescein or most other organic fluorophores. Finally, it is worth mentioning that the Ru-HT/HTP-based system reported here may also be of practical utility for studies of protein function. If the HTP fusion protein is somehow made the only form of the target protein present in a cell (for example by gene replacement in yeast or using RNAi in mammalian cells), then most or all of its activity could be knocked out using the experiments reported here, though currently, the efficiency of this approach would appear to be limited by the specific activity of the HTP fusion protein .
In conclusion, we have reported a general and convenient system for the comparison of different chromophores for CALI efficiency. This technique has been used to identify the Ru(bpy)32+ as an unusually efficient warhead for this purpose. We anticipate that appropriate small molecule–Ru(bpy)32+ chimeras may well be useful tools for the rapid and specific inactivation of target protein activities. Note that although this study employed a relatively low powered light source (since this was the best source available to us) and thus required irradiation times of many minutes, the use of high-powered lasers should allow protein inactivation in a matters of second and also allow for focused inactivation in different sub-cellular compartments. Efforts to develop such reagents are underway.
Experimental section
Materials and instruments
All chemicals, unless stated, were purchased from Aldrich Chemical Co., and were used without further purification. 1H NMR spectra were recorded on a Varian Inova–400 spectrometer at operating frequency of 400 MHz. Chemical shift (δ) are given in ppm; downfield shifts are reported as positive values from tetramethylsilane (TMS) standard at 0.00 ppm. Mass spectra (MS) were measured with Voyager–DE™ PRO (Applied Biosystems) for MALDI-TOF. Irradiation was done using 150 W Xenon arc lamp (Oriel, Stamford, CT). Light was filtered first through 10 cm of distilled water and then through a 380- to 2 500-nm cut-on filter (Oriel 49470). Samples were positioned at 25 cm distance from the light source and light intensity at the distance was 50 mW cm–2.Syntheses of HaloTag reagents
To a stirred solution of HT36(2.8 mg, 0.01 mmol) in DMF (200 µL) was added bis-(2,2′-bipyridin)-4′-methyl-4-carboxy-bipyridin-ruthenium-N-succinimidylester-bis-(hexafluorophosphate) (5.0 mg, 0.005 mmol) and DIPEA (5 µL). After 4 h at room temperature, the reaction mixture was diluted with dH2O (0.1% TFA, 3 mL) and directly purified by HPLC to afford Ru-HT as a red solid (75% yield). B-HT and previously reported F-HT37 were prepared in same way using Sulfo-NHS-LC-Biotin (Pierce) and 5-carboxyfluorescein succinimide ester (Molecular Probes), respectively. Ru-HT 1H NMR (DMSO-d6): δ 9.14-9.11 (m, 2H), 8.84–8.82 (m, 4H), 8.77 (bs, 1H), 8.18–8.14 (m, 4H), 7.86 (d, 1H, J = 6.0 Hz), 7.80–7.79 (m, 1H), 7.76 (d, 1H, J = 5.6 Hz), 7.70–7.69 (m, 3H), 7.56 (d, 1H, J = 5.6 Hz), 7.53–7.49 (m, 4H), 7.39 (d, 1H, J = 6.0 Hz), 2.52 (S, 3H), 1.65–1.61 (m, 2H), 1.44–1.40 (m, 2H), 1.30–1.22 (m, 4H); MALDI/TOF: [M]+ calculated 833.24, found 833.23. B–HT 1H NMR (DMSO-d6): 7.86 (t, 1H, J = 5.2 Hz), 7.72 (t, 1H, J =5.2 Hz), 4.30–4.27 (m, 1H), 4.12–4.09 (m, 1H), 3.62–3.59 (m, 2H), 3.46–3.45 (m, 4H), 3.38–3.33 (m, 4H), 3.17–3.13 (m, 2H), 3.08–3.06 (m, 1H), 2.97 (dd, 2H, J = 12.8, 4.8 Hz), 2.80 (dd, 1H, J = 12.4, 4.8 Hz), 2.57–2.52 (m, 2H), 2.04–2.00 (m, 4H), 1.70–1.67 (m, 2H), 1.60–1.54 (m, 1H), 1.50–1.43 (m, 8H), 1.38–1.26 (m, 10H), 1.22–1.18 (m, 2H); MALDI/TOF: [M+H]+ calculated 563.30, found 563.47.Plasmid
The vector backbone of the host plasmid that is used to construct the 3×Flag-Luc-HTP-Myc plasmid was derived from the mammalian expression vector pEGFP-N3 with multiple convenient restriction sites after the CMV promoter. The DNA sequences encoding different parts of the fusion construct were ligated into the host vector sequentially. The sequence encoding the full length luciferase (Luc) was amplified through PCR using pGL3 basic (Promega) as template, along with forward primer (5′-GGACGCTAGCGAAGACGCCAAAAACATAAAG-3′) and reverse primer (5′-GAAAGATCTCACGGCGATCTTTCCGCCCTTC-3′). The PCR product was digested with NheI/BglII and inserted into the digested host plasmid. The sequence encoding the full length haloalkane dehalogenase (HTP) was amplified through PCR using pHT2 (Promega) as template, along with forward primer (5′-GACCAGAGATCTGCCACCATGGGATCCGAAATC-3′) and reverse primer (5′-GAAAACAAGCTTGCCGGCCAGCCCGGGGAG-3′). The PCR product was digested with BglII/HindIII and inserted into the digested host plasmid. The sequence encoding the start codon followed by the 3×Flag tag was inserted into the BamHI/NheI sites, and the sequence encoding the Myc tag followed by the stop codon was inserted into the HindIII/NotI sites of the host plasmid.Cell culture, transfection, and preparation of cell lysate
HeLa cells (American Type Culture Collection, CCL-2) were grown in 10 cm plates at 37 °C under 5% CO2 in Dulbeco's modified Eagle medium (Invitrogen) supplemented with 10% (v/v) fetal calf serum (Invitrogen). Cells were allowed to reach 70% confluence before transfection. Cells were transfected by the Lipofectamine™ Plus method (invitrogen) with 8 µg of plasmid for 3×Flag-Luc-HTP-Myc and 80 ng of Renilla reniformis luciferaseplasmid (pRL-SV40). After transfection, cell were maintained at 37 °C under 5% CO2. After 30 h, cells were lysed with 1× passive lysis buffer (Promega) including protease inhibitor cocktail. Total protein concentration of cell extract was measured (1.59 mg mL–1) using the BCA protein assay kit (Pierce).Measurement of luciferase activities
Luciferase activities were measured using dual-luciferaseassay kit (Promega) following manufacture's protocol. The firefly luciferase activity in sample was measured and normalized with the Renilla luciferase activity and then presented as the percentage of the activity normalized of input (control).Labeling of target protein with HaloTag reagents.
Immunoprecipitation
2 µL of HeLa cell lysate in PBS (total volume: 50 µL, pH 7.4) including BSA (4 mg ml–1) was incubated with PBS (40 µL, pH 7.4), anti-Myc agarose affinity gel (Sigma) in PBS (40 µL, 1 : 1 v/v, pH 7.4), or anti-Flag M2 agarose affinity gel (Sigma) in PBS (40 µL, 1 : 1 v/v, pH 7.4) for 30 min at room temperature. After centrifugation, the remaining luciferase activities of the solutions were measured as described above.In vitro CALI
1 µl of HeLa cell lysate in PBS (total volume: 20 µL, pH 7.4) including BSA (4 mg ml–1) in transparent 96 well plate was incubated with DMSO or compounds for 30 min at room temperature in the dark. Samples were irradiated (without lid) for different times at room temperature. Luciferase activities of the solution were measured as described above.In vivo CALI
HeLa cell were seeded in 96-well plate (2.5 × 104 cells per well) and grown in DMEM (10% FBS) for 24 h before transfection. Cells in each well were transfected as above with 100 ng of 3×Flag-HTP-Luc-Myc plasmid and 1 ng of Renilla reniformis luciferaseplasmid (pRL-SV40). After transfection, cells were maintained at 37 °C under 5% CO2 for 15 h. Medium was replaced with fresh DMEM (w/o phenol red, 10% FBS) and cells were treated with compounds. After 3 h at 37 °C, media was replaced with fresh DMEM (w/o phenol red, 10% FBS) to remove excess compound in media. Cells were irradiated at room temperature for 30 min as described above. After dark incubation for 30 min at 37 °C, cells were lysed with 1× passive lysis buffer (20 µL per well, Promega). Luciferase activities of the lysates were measured as described above.Acknowledgements
This work was supported by a contract from the National Heart, Lung, and Blood Institute (NO1-HV-28185) and a grant from the Welch Foundation (I-1299).References
- S. M. Elbashir, J. Harborth, W. Lendeckel, A. Yalcin, K. Weber and T. Tuschl, Nature, 2001, 411, 494–498 CrossRef CAS.
- J. Harborth, S. M. Elbashir, K. Bechert, T. Tuschl and K. Weber, J. Cell Sci., 2001, 114, 4557–4565 CAS.
- J. A. Wilson and C. D. Richardson, Curr. Opin. Mol. Ther., 2003, 5, 389–396 Search PubMed.
- J. C. Liao, J. Roider and D. G. Jay, Proc. Natl. Acad. Sci. U. S. A., 1994, 91, 2659–2663 CrossRef CAS.
- D. G. Jay and T. Sakurai, Biochim. Biophys. Acta, 1999, 1424, M39–48 CAS.
- S. Beck, T. Sakurai, B. K. Eustace, G. Beste, R. Schier, F. Rudert and D. G. Jay, Proteomics, 2002, 2, 247–255 CrossRef CAS.
- B. K. Eustace, A. Buchstaller and D. G. Jay, Brief Funct. Genomic Proteomic, 2002, 1, 257–265 Search PubMed.
- K. W. Marek and G. W. Davis, Neuron, 2002, 36, 805–813 CrossRef CAS.
- O. Tour, R. M. Meijer, D. A. Zacharias, S. R. Adams and R. Y. Tsien, Nat. Biotechnol., 2003, 21, 1505–1508 CrossRef CAS.
- K. M. Marks, P. D. Braun and G. P. Nolan, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 9982–9987 CrossRef CAS.
- P. G. Alluri, M. M. Reddy, K. Bachhawat-Sikder, H. J. Olivos and T. Kodadek, J. Am. Chem. Soc., 2003, 125, 13995–14004 CrossRef CAS.
- P. Alluri, B. Liu, P. Yu, X. Xiao and T. Kodadek, Mol. BioSyst., 2006, 2, 568–579 RSC.
- X. Xiao, P. Yu, H. S. Lim, D. Sikder and T. Kodadek, Angew. Chem., Int. Ed., 2007, 46, 2865–2868 CrossRef CAS.
- H.-S. Lim, C. T. Archer and T. Kodadek, J. Am. Chem. Soc., 2007, 129, 7750–7751 CrossRef CAS.
- P. Yu, B. Liu and T. Kodadek, Nat. Biotechnol., 2005, 23, 746–751 CrossRef CAS.
- Y. U. Kwon and T. Kodadek, J. Am. Chem. Soc., 2007, 129, 1508–1509 CrossRef CAS.
- J. S. Winterle, D. S. Kliger and G. S. Hammond, J. Am. Chem. Soc., 1976, 98, 3719–3721 CrossRef CAS.
- J. N. Demas, E. W. Harris and R. P. McBride, J. Am. Chem. Soc., 1977, 99, 3547–3551 CrossRef CAS.
- S. S. Miller, K. Zahir and A. Haim, Inorg. Chem., 1985, 24, 3978–3980 CrossRef CAS.
- Q. G. Mulazzani, H. Sun, M. Z. Hoffmann, W. E. Ford and M. A. J. Rodgers, J. Phys. Chem., 1994, 98, 1145–1150 CrossRef CAS.
- M. C. DeRosa and R. J. Crutchley, Coord. Chem. Rev., 2002, 233–234, 351–371 CrossRef CAS.
- A. Abdel-shafi, D. R. Worrall and A. Y. Ershov, Dalton Trans., 2004, 30–36 RSC.
- G. V. Los and K. Wood, Methods Mol. Biol., 2007, 356, 195–208 Search PubMed.
- D. B. Janssen, A. Scheper, L. Dijkhuizen and B. Witholt, Appl. Environ. Microbiol., 1985, 49, 673–677 CAS.
- A. N. Kulakova, T. M. Stafford, M. J. Larkin and L. A. Kulakov, Plasmid, 1995, 33, 208–217 CrossRef CAS.
- F. Pries, J. Kingma, M. Pentenga, G. van Pouderoyen, C. M. Jeronimus-Stratingh, A. P. Bruins and D. B. Janssen, Biochemistry, 1994, 33, 1242–1247 CrossRef CAS.
- F. Pries, J. Kingma, G. H. Krooshof, C. M. Jeronimus-Stratingh, A. P. Bruins and D. B. Janssen, J. Biol. Chem., 1995, 270, 10405–10411 CrossRef CAS.
- C. S. Foote, T. T. Fujimoto and Y. C. Chang, Tetrahedron Lett., 1972, 1, 45–48 CrossRef.
- N. Hasty, P. B. Merkel, P. Radlick and D. R. Kearns, Tetrahedron Lett., 1972, 1, 49–52 CrossRef.
- G. Ambrosio and J. T. Flaherty, Cardiovasc. Drugs Ther., 1992, 6, 623–632 CrossRef CAS.
- A. Grimmett, Adv. Heterocycl. Chem., 1970, 12, 103–183 CAS.
- J. R. Newcomb, B. Rivnay, C. M. Bastos, T. D. Ocain, K. Gordon, P. Gregory, S. M. Turci, K. A. Sterne, M. Jesson, J. Krieger, J. C. Jenson and B. Jones, Inflammation Res., 2003, 52, 263 CAS.
- A. A. Abdel-Shafi, P. D. Beer, R. J. Mortimer and F. Wilkinson, Helv. Chim. Acta, 2001, 84, 2784–2795 CrossRef CAS.
- M. Suzuki, O. Bartels, R. Gerdes, G. Schneider, D. Wöhrle, G. Schulz-Ekloff, M. Kimura, K. Hanabusa and H. Shirai, Chem. Lett., 1999, 579–580 CrossRef CAS.
- M. Suzuki, O. Bartels, R. Gerdes, G. Schneider, D. Wöhrle, G. Schulz-Ekloff, M. Kimura, K. Hanabusa and H. Shirai, Phys. Chem. Chem. Phys., 2000, 2, 109–114 RSC.
- Y. Zhang, M. K. So, A. M. Loening, H. Yao, S. S. Gambhir and J. Rao, Angew. Chem., Int. Ed., 2006, 45, 4936–4940 CrossRef CAS.
- K. V. Wood, G. V. Los, R. F. Bulleit, D. Klaubert, M. Mcdougall and C. Zimprich, PCT Int. Appl., 2004, WO 2004072232 Search PubMed.
| This journal is © The Royal Society of Chemistry 2008 |