Is chemical crosslinking necessary for the photoinduced bending of polymer films?†
Jun-ichi
Mamiya
a,
Akira
Yoshitake
a,
Mizuho
Kondo
a,
Yanlei
Yu
b and
Tomiki
Ikeda
*a
aChemical Resources Laboratory, Tokyo Institute of Technology, R1-11, 4259 Nagatsuta, Midori-ku, Yokohama 226-8503, Japan. E-mail: tikeda@res.titech.ac.jp; Web: http://www.res.titech.ac.jp./polymer/ Fax: +81-45-924-5275; Tel: +81-45-924-5240
bDepartment of Materials Science, Fudan University, 220 Handan Road, Shanghai, China 200433
First published on 1st November 2007
Abstract
Freestanding crosslinked liquid-crystalline polymer films obtained by self-assembly through intermolecular hydrogen bonding showed photoinduced bending and unbending. The structural change at the microscopic level, caused by trans–cisphotoisomerization of the azobenzene moieties at the hydrogen-bonded crosslinks, is successfully converted into a macroscopic deformation in the liquid-crystalline polymer films.
Soft materials have attracted much attention because of their dynamic properties.1 For effective muscle-like actuation, soft materials with stratified structures and high molecular orders are necessary. Crosslinked liquid-crystalline polymers (CLCPs) are superior soft materials that possess both the order of LCs and the elasticity of polymer networks.2,3 CLCPs exhibit a spontaneous contraction along the director axis when heated above nematic–isotropic (I) phase transition temperatures.4 When azobenzene chromophores are incorporated into CLCPs, they can undergo the contraction isothermally because of the change in LC alignment caused by light.5 Furthermore, a three-dimensional deformation, bending, of CLCP films containing azobenzenes has been observed upon exposure to light.6 Due to the limitation of absorption of photons, a surface contraction caused by the photoinduced change in alignment of LCs contributes to the bending. By using photodeformation of these materials, one can convert light energy into mechanical work directly (photomechanical effects).3,5–7
Hydrogen bonds have been used for the preparation of a wide variety of self-assembled soft materials, among which LC phases with well-defined structures have been formed by exploiting intermolecular hydrogen bonding.8 Various synthetic approaches for the construction of dynamically functional materials using hydrogen bonds have been demonstrated in such self-assembly systems as supramolecular LCs and polymers.8,9 However, there are few examples of supramolecular crosslinking of polymers by low-molecular-weight (LMW) crosslinkers.10 Crosslinked polymers by supramolecular interactions are expected to function as recyclable materials responsive to external stimuli, because LMW crosslinkers can be connected and disconnected reversibly.
Since the hydrogen bonds crosslink polymer chains to form three-dimensional polymer networks, if an azobenzene moiety as a photoresponsive group is introduced into the crosslinker, trans–cisisomerization of the azobenzenes may influence the crosslinking of the three-dimensional polymer networks. In this communication, we investigated the photoresponsive behavior of azobenzene CLCP films formed by self-assembly. We succeeded in fabricating recyclable CLCP films that could crosslink/de-crosslink reversibly using the hydrogen bonding at the crosslinks, and we showed photoinduced bending and unbending as well.
The structure of the LC copolymer (1) used in this study is shown in Fig. 1, which has both a carboxyl group and an azobenzene moiety. The mesomorphic properties of the copolymer 1 were studied by differential scanning calorimetry (DSC). The copolymer 1 exhibited a glass transition (Tg) at 38 °C, and an LC–I phase transition at 106 °C upon heating (Fig. 2). Crosslinkers 2 and 3 that are capable of recognizing hydrogen-bond donor molecules at the pyridyl ends were selected as hydrogen-bond acceptors (Fig. 1). A complex of the copolymer and the crosslinker was obtained from a THF solution containing equimolar amounts of the carboxyl group and the pyridine moiety. In the infrared (IR) spectra of the complexes of 1 + 2 and 1 + 3, the absorption bands corresponding to the hydrogen-bonded O–H stretch of the carboxyl groups were observed at 1930 and 2500 cm−1.11 On the other hand, in the single copolymer 1, no absorption due to the hydrogen bonding appeared. It was also found that when each crosslinker was mixed with the copolymer 1, the temperature range of the LC phase in the complexes became broader than that in the copolymer 1 (Fig. 2). It is observed clearly that the mesophases of the complexes are stabilized by the formation of the hydrogen bonding between 1 and the crosslinkers
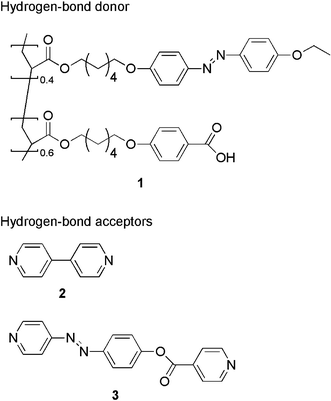 | ||
| Fig. 1 Chemical structures of the hydrogen-bond donor (1) and acceptors (2 and 3) used in this study. | ||
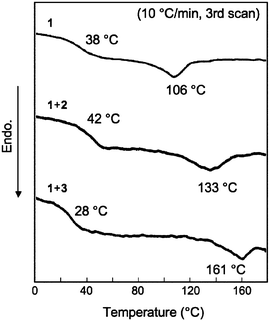 | ||
| Fig. 2 DSC thermograms of the copolymer and the complexes on heating (scan rate = 10 °C min−1). | ||
Hydrogen-bonded azobenzene CLCPs were sandwiched between two sodium chloride (NaCl) plates with rubbing treatment. The cell thickness was controlled with silica spacers with a diameter of 20 μm. Freestanding films were obtained after the NaCl substrates were dissolved in water. The optical anisotropy of the CLCP films was evaluated by polarizing optical microscopy. The regular maximum and minimum values with 90° separations showed that the azobenzene mesogens are preferentially aligned along the rubbing direction.
We observed the photoresponsive behavior of the hydrogen-bonded CLCP films above their Tgs of 1 + 2 and 1 + 3. In 1 + 2, no bending was observed upon exposure to UV light at 366 nm and the film showed no deformation upon further visible-light irradiation at 60 °C (Fig. 3a). On the other hand, when the CLCP film of 1+3 was exposed to 366 nm light at 50 °C, the film bent toward the actinic light source along the alignment direction of the mesogens (Fig. 3b). The bent films reverted to the initial flat states when irradiated with visible light at >540 nm. Here, crosslinker 2 has no photoresponsive property, while crosslinker 3 shows photoisomerization behavior. The photoinduced bending and unbending of the hydrogen-bonded films of 1 + 3 is similar to that of the chemically-bonded films reported previously.6 These results indicate that a structural change caused by photoisomerization of the azobenzene moieties at the crosslinks plays an important role in the photoinduced bending of the hydrogen-bonded CLCP films. In other words, it means that the crosslinks formed by non-covalent bonds can convert the motion of the mesogens into a macroscopic change of the CLCP films.
 | ||
| Fig. 3 Photoresponsive behavior of the hydrogen-bonded CLCP films of 1 + 2 (a) and 1 + 3 (b). Size of the films: 2 mm × 3 mm × 20 μm. UV light intensity, 18 mW cm−2; visible light intensity, 21 mW cm−2. | ||
Irradiation with UV light gives rise to trans–cisisomerization of the azobenzene mesogens in CLCPs. However, the CLCP films of 1 + 2 showed no bending upon UV-light irradiation. This is presumably because the photoisomerization of the azobenzene mesogens in the copolymer 1 induces no decrease in alignment order of the hydrogen-bonded LC structure. On the other hand, trans-azobenzenes in both the copolymer and the crosslinker isomerize to cis-azobenzenes in the CLCP films of 1 + 3. By photoisomerization of the azobenzene moieties at the crosslinks, hydrogen-bonds are partly destroyed, leading to a decrease in alignment order of the hydrogen-bonded LC structure only in the film surface (Fig. 4). As the azobenzene mesogens are aligned parallel to the rubbing direction, an anisotropic contraction is generated along the alignment direction of the azobenzene moieties upon irradiation with UV light.
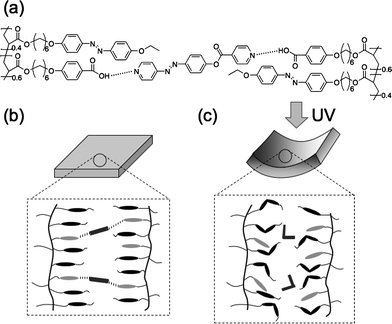 | ||
| Fig. 4 Plausible mechanism of bending in the hydrogen-bonded CLCP films of 1 + 3. (a) Network structures of the hydrogen-bonded CLCP films consisting of the copolymers and the crosslinkers. Schematic illustration of molecular alignment in the hydrogen-bonded CLCP film before (b) and after (c) irradiation with UV light. | ||
Next, the recyclability of the hydrogen-bonded CLCP films was investigated (ESI†). The chemically-bonded CLCP films were undissolved in various solvents. However, the hydrogen-bonded CLCP films were dissolved in THF. Reprecipitation from the THF solution in diethyl ether led to dissociation of the hydrogen bonding. We could reconstruct the CLCP films by self-assembly through hydrogen bonding and repeatedly induce the photoinduced bending of the reformed CLCP films.
In summary, freestanding hydrogen-bonded CLCP films containing azobenzenes have been obtained. The supramolecular crosslinking of the linear copolymers with LMW crosslinkers was found to be a useful strategy for constructing the CLCP films. The structural change at the microscopic level is successfully converted to a macroscopic deformation of the hydrogen-bonded CLCP films by trans–cisphotoisomerization of the azobenzene moieties at the crosslinks. This is the first example of a supramolecular photomechanical system based on hydrogen-bonded CLCP films, which converts light energy directly into mechanical work.
Notes and references
- (a) M. V. Gandhi and B. S. Tomson, Smart Materials and Structures, Chapman and Hall, London, 1992 Search PubMed; (b) Polymer Sensors and Actuators, ed. Y. Osada and D. E. DeRossi, Springer, Berlin, 2000 Search PubMed; (c) Polymer Gels and Networks, ed. Y. Osada and A. R. Khokhlov, Marcel Dekker Inc., New York, 2002 Search PubMed.
- (a) M. Warner and E. M. Terentjev, Liquid Crystal Elastomers, Oxford University Press, Oxford, UK, 2003 Search PubMed; (b) P.-G. de Gennes, M. Hébert and R. Kant, Macromol. Symp., 1997, 113, 39–49 CAS; (c) P. Xie and R. Zhang, J. Mater. Chem., 2005, 15, 2529–2550 RSC.
- T. Ikeda, J. Mamiya and Y. Yu, Angew. Chem., Int. Ed., 2007, 46, 506–528 CrossRef CAS.
- (a) H. Wermter and H. Finkelmann, e-polymers, 2001 Search PubMed , no. 13; (b) J. Naciri, A. Srinivasan, H. Jeon, N. Nikolov, P. Keller and B. R. Ratna, Macromolecules, 2003, 36, 8499–8505 CrossRef CAS.
- (a) H. Finkelmann, E. Nishikawa, G. G. Pereira and M. Warner, Phys. Rev. Lett., 2001, 87, 015501 CrossRef CAS; (b) M.-H. Li, P. Keller, B. Li, X. Wang and M. Brunet, Adv. Mater., 2003, 15, 569–572 CrossRef CAS.
- (a) T. Ikeda, M. Nakano, Y. Yu, O. Tsutsumi and A. Kanazawa, Adv. Mater., 2003, 15, 201–205 CrossRef CAS; (b) Y. Yu, M. Nakano and T. Ikeda, Nature, 2003, 425, 145 CrossRef CAS; (c) Y. Yu, M. Nakano and T. Ikeda, Pure Appl. Chem., 2004, 76, 1435–1445; (d) Y. Yu, M. Nakano, A. Shishido, T. Shiono and T. Ikeda, Chem. Mater., 2004, 16, 1637–1643 CrossRef CAS; (e) M. Kondo, Y. Yu and T. Ikeda, Angew. Chem., Int. Ed., 2006, 45, 1378–1382 CrossRef CAS; (f) Y. Yu, T. Maeda, J. Mamiya and T. Ikeda, Angew. Chem., Int. Ed., 2007, 46, 881–883 CrossRef CAS.
- (a) M. Camacho-Lopez, H. Finkelmann, P. Palffy-Muhoray and M. Shelley, Nat. Mater., 2004, 3, 307–310 CrossRef CAS; (b) K. D. Harris, R. Cuypers, P. Scheibe, C. L. van Oosten, C. W. M. Bastiaansen, J. Lub and D. J. Broer, J. Mater. Chem., 2005, 15, 5043–5048 RSC; (c) N. Tabiryan, S. Serak and X. M. Dai, Opt. Express, 2005, 13, 7442–7448 CrossRef CAS.
- (a) L. Brunsveld, B. J. B. Folmer, E. W. Meijer and R. P. Sijbesma, Chem. Rev., 2001, 101, 4071–4097 CrossRef CAS; (b) T. Kato, N. Mizoshita and K. Kishimoto, Angew. Chem., Int. Ed., 2006, 45, 38–68 CrossRef CAS.
- (a) T. Kato and J. M. J. Fréchet, J. Am. Chem. Soc., 1989, 111, 8533–8534 CrossRef CAS; (b) M.-J. Brienne, J. Gabard, J.-M. Lehn and I. Stibor, J. Chem. Soc., Chem. Commun., 1989, 1868–1870 RSC; (c) M. Suárez, J.-M. Lehn, S. C. Zimmerman, A. Skoulios and B. Heinrich, J. Am. Chem. Soc., 1998, 120, 9526–9532 CrossRef CAS; (d) T. Kato, H. Kihara, T. Uryu, A. Fujishima and J. M. J. Fréchet, Macromolecules, 1992, 25, 6836–6841 CrossRef CAS; (e) T. Kato, J. M. J. Fréchet, P. G. Wilson, T. Saito, T. Uryu, A. Fujishima, C. Jin and F. Kaneuchi, Chem. Mater., 1993, 5, 1094–1100 CrossRef CAS.
- (a) T. Kato, H. Kihara, U. Kumar, T. Uryu and J. M. J. Fréchet, Angew. Chem., Int. Ed. Engl., 1994, 33, 1644–1645 CrossRef; (b) R. J. Thibault, P. J. Hotchkiss, M. Gray and V. M. Rotello, J. Am. Chem. Soc., 2003, 125, 11249–11252 CrossRef CAS; (c) A. V. Medvedev, E. B. Barmatov, A. S. Medvedev, V. P. Shibaev, S. A. Ivanov, M. Kozlovsky and J. Stumpe, Macromolecules, 2005, 38, 2223–2229 CrossRef CAS; (d) T. Suzuki, S. Shinkai and K. Sada, Adv. Mater., 2006, 18, 1043–1046 CrossRef CAS.
- (a) S. L. Johnson and K. A. Rumon, J. Phys. Chem., 1965, 69, 74–86 CrossRef CAS; (b) J. Y. Lee, P. C. Painter and M. M. Coleman, Macromolecules, 1988, 21, 954–960 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details. See DOI: 10.1039/b715855f |
| This journal is © The Royal Society of Chemistry 2008 |