Mendable polymers
Sheba D. Bergman and Fred Wudl*
Department of Chemistry, University of California, Santa Barbara, Santa Barbara 90316, USA. E-mail: Wudl@chem.ucsb.edu; Fax: +1-805-893-4120; Tel: +1-805-893-5817
First published on 27th November 2007
Abstract
Recent advances in polymer and materials chemistry have led to the development of materials that exhibit the ability to undergo repair. Depending on the structure and chemical composition of the material, the repair process can occur either as a result of external stimuli or autonomously and can be a one-time event or an indefinitely repeatable process. Several systems have been demonstrated recently in the literature; however, a larger number of polymeric systems have the potential to exhibit similar mending capability. These polymeric systems all share one property in common—reversibility, either in the polymerization process or in the cross-linking process. This Feature Article will outline the various known systems that encompass the ability to mend, either by external stimuli or autonomously. In addition, several non-reversible composite systems will be presented that exhibit one-time autonomous healing.
 Sheba D. Bergman | Dr Sheba D. Bergman graduated from Tel Aviv University in 2005, completing her Ph.D. thesis under the supervision of Prof. Moshe Kol. She was the 2005 recipient of the prestigious Rotschild fellowship, and was appointed as a post-doctoral fellow in MIT in the group of Prof. Stephen L. Buchwald. She is currently a post-doctoral associate in the group of Prof. Fred Wudl at the University of California, Santa Barbara, and is an active member of the Center for Polymers and Organic Solids. Her principal research interests are the development of novel polymeric materials that exhibit both re-mendability and autonomous healing. |
 Fred Wudl | Fred Wudl is the Dean M. Willard Professor of Chemistry and Materials and Director of the Exotic Materials Institute at the University of California, Los Angeles. He received his B.S. (1964) and PhD (1967) degrees in chemistry from UCLA. After postdoctoral research with R. B. Woodward at Harvard, he joined the faculty of the State University of New York at Buffalo. He then moved, first in 1972 to AT&T Bell Laboratories, and subsequently to the University of California, Santa Barbara in 1982, then onto UCLA in 1997. Currently he is interested in the optical and electrooptical properties of processable conjugated polymers as well as in the organic chemistry of fullerenes and the design and synthesis of self-mending polymers. |
1. Introduction
Modern polymer science has reached a very high level of sophistication. It is now possible to prepare synthetic polymers with well-defined molecular weight as well as relatively easily controlled design of properties. In the recent past, it has become necessary to develop “smart” materials, including polymers. One family of smart polymers is represented by materials that will repair completely, ideally without external input. One of the approaches to polymer mending is to use reversible polymerization. Reversible polymers have been known for some time but the concept has been exploited in our research group over the last decade because the reversibility can be applied to repair at the molecular level.Polymers have a finite lifetime, their inherent properties degrade with age through accumulated stresses and strains (wear-and-tear). The cracking or breaking of a material starts at the microscopic level, and usually goes unnoticed until the material fails and requires mending. Mending at the macroscopic level can be done with adhesives, but does not refurbish the original properties of the polymer. Mending at the microscopic level, as enabled by a reversible polymerization process, fully restores the original properties of the material, and this process can be repeated many times. Historically, such reversible polymerizations would constitute the formation and cleavage of covalent bonds. The development of supramolecular chemistry has brought about a new type of these polymerizations, that are based on the formation and severing of non-covalent interactions. Recent advances in composite material manufacture have brought about the development of “smart” materials, where one-time healing is achieved without the necessity of a reversible polymerization process.
This Feature Article will outline the various known systems that encompass the ability to mend, either by external stimuli or autonomously. Hence, reversible polymerization, reversible cross-linking, and composite systems that have been pre-designed to undergo self-repair will be outlined, with particular focus on the systems that have been demonstrated to be mendable. Due to space limitations, we will only briefly mention biologically inspired systems, as they are outside the scope of this Feature Article.
2. Reversible systems
Reversible systems are broadly defined as polymeric systems that can revert to either their monomeric, oligomeric, or non-cross-linked state. For the polymer to be stable under normal working conditions, the reverting process would normally require an external stimulus for it to occur. For mending purposes, ideally the material would revert to its constituents after cracking but could be repaired by applying the conditions that were used to polymerize it. These systems can be categorized by the nature of the reversible bonds—covalent and non-covalent.2.1. Covalently bound systems
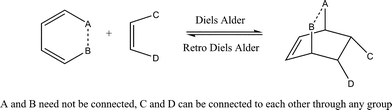 | ||
| Scheme 1 General mechanism of Diels–Alder cycloaddition reaction. | ||
2.1.1.1. Furan-maleimide based polymers. Furan-maleimide based polymers have recently been reviewed.1 Probably the earliest report of incorporation of furan-maleimide moieties into polymers for the purpose of achieving thermal reversibility was reported in 1969 by Craven.2 Since then several patents and journal articles have been published, all concerning the fabrication of a thermally reversible polymer network bearing DA-reactive furan and maleimide units, either as pendant groups (for reversible cross-linking), or as part of the polymer backbone (for reversible polymerization).
The pioneering work of Stevens and Jenkins tackled the possibility of fabricating a thermally reversible polymer network bearing DA-reactive furan and maleimide pendant groups.3 However, this idea was not exploited until a decade later, when Saegusa and coworkers described the reversible cross-linking of modified poly(N-acetylethyleneimine)s bearing either maleimide or furancarbonyl pendant moieties (Scheme 2).4 The authors claimed that mixing the two modified complementary polymers resulted in a highly cross-linked material. Subsequent heating to 80 °C for two hours in a polar solvent regenerated the two starting linear precursors in quantitative yield. The validity of this work, as well as of later studies of the same system,5 is interesting, partly because furancarbonyl moieties are not expected to be reactive dienes for the DA reaction.
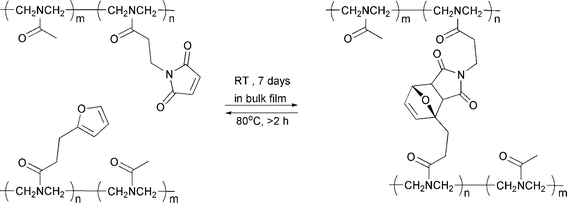 | ||
| Scheme 2 Reversible polymer cross-linking via Diels–Alder cycloaddition reaction between pendaent furan and maleimide moieties.4 | ||
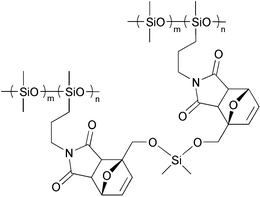
A later work by Canary and Stevens described the thermally reversible cross-linking of polystyrene bearing maleimide pendant groups with di-2-furfuryl adipate (Scheme 3).6 The RDA reaction of this system was achieved at 150 °C, but at this temperature, thermal instability of the furfuryl moiety ensued, and posed a serious limitation to the viability of their system. This was also observed by Gandini and coworkers while studying the DA/RDA reaction of N,N′-methylenediphenylbismaleimide (MDPBM) with random copolymers of methyl methacrylate with 2-furfuryl acrylate.7 The polymers could easily be cross-linked in solution, but the resultant gels did not revert entirely by the RDA reaction in bulk (TGA) and the residual proportion of network was greater with increasing furan content in the initial copolymer. The same group later conducted a systematic study of styrene copolymers bearing pendant 2-furfuryloxy moieties in conjuction with MDPBM.8 In this work it was demonstrated that at a temperature of ca. 130 °C, in the presence of a DA “trap” (in this case, 2-methylfuran, that reacted with the released MDPBM), the DA/RDA cycle was optimized and side reactions related to the furfuryl moieties were avoided. A similar study concerning copolymers with elastomeric properties has been performed as well.9 A furan acrylic copolymer and an oligoether-bridged bis-maleimide were cross-linked to yield a high flexibility system.9 Alternatively, the reverse approach could be applied as well, where an elastomeric polydimethylsiloxane (PDMS) copolymer bearing pendant maleimide moieties was cross-linked with a flexible difuran derivative (Fig. 1).9 Upon heating these materials to ca. 90 °C, the RDA reaction ensues. Huglin and coworkers have also studied a copolymer-based DA/RDA system, using MDPBM as the cross-linker.10–13 The swelling and mechanical properties of the networks as well as the kinetics of the DA cross-linking and the RDA cross-cleavage were studied. UV spectroscopy measurements were used to determine the rate of maleimide consumption and regeneration during the DA and RDA reactions, respectively.
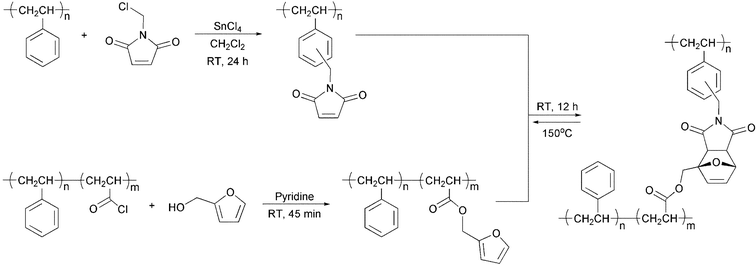 | ||
| Scheme 3 Reversible cross-linking of polystyrene via Diels–Alder cycloaddition reaction between pendent furan and maleimide moieties.6 | ||
Several early works concerning polymers formed by the DA reaction of bis-furan and bis-maleimide monomers demonstrated the feasibility of such polymerization. However, detailed structural analyses of these polymers were absent, and in most cases the degree of polymerization (DP) was quite low (10–20).14–19 Noteworthy is the observation by Kuramoto et al. of partial reversibility at 90 °C when the polymer was prepared from di-2-furfuryl adipate and bismaleimidodiphenylmethane (Fig. 2).20 Also, Goussé and Gandini have synthesized and studied a number of low DP polymer systems composed of difuran and bis-maleimide monomers.21 The same group also prepared 2-furfurylmaleimide (FM), and its subsequent DA oligomers.
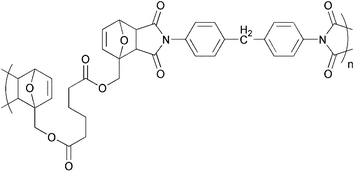 | ||
| Fig. 2 Structure of the polymer prepared from di-2-furfuryl adipate and bismaleimidodiphenylmethane.20 | ||
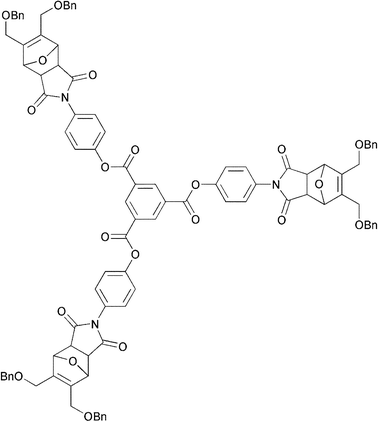
The first work to deliberately study the thermal reversibility of such materials was reported by McElhanon and Wheeler.22 In this work, the studied DA dendrimer (Fig. 3) exhibited roughly 40% dissociation of the DA links after 1 h at 110 °C, and full restoration of the original structure upon cooling to 65 °C over a couple of days. This work was the first example of a thermally labile-reassembling dendrimer exploiting the DA/RDA approach. The same group has successfully exploited furan-maleimide reversible DA chemistry for the design of debondable epoxy resins.23–26 The built-in reversibility of the material enabled de-bonding at 90 °C.
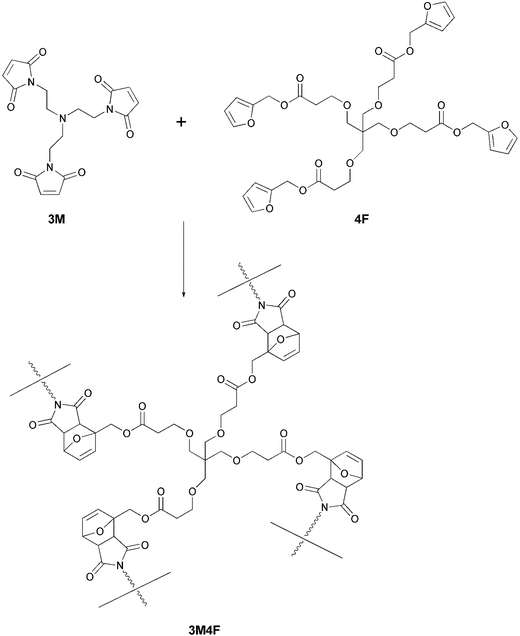
Despite these and other earlier studies,27,28 our laboratory was the one to employ the DA/RDA strategy to prepare thermally re-mendable polymers.29–31 Multifunctional furan- and maleimide-based monomers were used to form highly cross-linked polymeric networks. The first system that was developed is shown in Scheme 4, where a tris-maleimide (3M) and a tetra-furan (4F) were allowed to react to afford a clear solid DA-step-growth polymer (3M4F).29 These polymers were submitted to heating/cooling cycles and their structural changes followed by solid-state 13C NMR spectroscopy studies. These measurements clearly demonstrated the occurrence of the RDA reaction at ca. 120 °C. Samples of this polymer were stressed to complete failure and subsequently healed by heating to ca. 90–120 °C, followed by cooling to room temperature (Fig. 4). The healed polymer exhibited ca. 57% of the original polymer strength. Subsequently, an improved system was designed which employed 4F in conjunction with either 2ME or 2MEP (Fig. 5).30 The polymer 2MEP4F exhibited crack-healing with as much as 83% recovery of the polymer's original strength (Fig. 6). Thus, it was shown that the DA/RDA principle provides a simple and efficient way to prepare re-mendable polymers, which can go through repeated cycles of cracking and re-mending at the same site. The limitations of this system are the working temperature of the materials (<120 °C), too low for many applications, and the costly synthesis of the monomers that is problematic for large-scale production.
 | ||
| Fig. 4 Thermal mending of the polymer. (A) Mending efficiency obtained by fracture toughness testing of compact tension test specimens. Values for the original and healed fracture toughness were determined by the propagation of the starter crack along the middle plane of the specimen at the critical load. (B) Image of a broken specimen before thermal treatment. (C) Image of the specimen after thermal treatment. (D) SEM image of the surface of a healed sample: the left side is the as-healed surface and the right side is the scraped surface. (E) Enlarged image of the boxed area in (D). Reproduced with permission from ref. 29. | ||
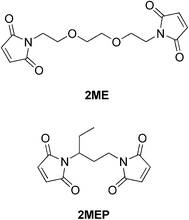 | ||
| Fig. 5 Structures of 2ME and 2MEP.30 | ||
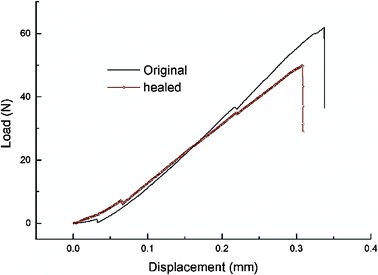 | ||
| Fig. 6 Load vs. displacement diagram of fracture toughness testing of specimens of original and healed polymer 2MEP4F. Reproduced with permission from ref. 30. | ||
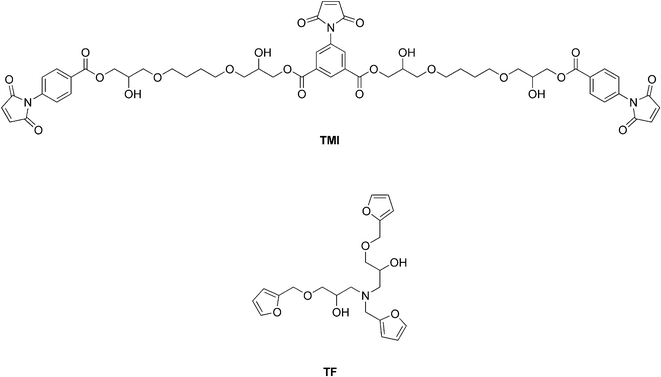
Since the publication of this work, a number of related polymer systems have been reported in the literature. Liu and Hsieh reported a similar cross-linked polymer constructed from the tris-maleimide and tris-furan monomers TMI and TF, respectively (Fig. 7).32 The polymer exhibited thermal re-mendability and removability through DA and RDA reactions (Fig. 8). These materials were shown to be applicable as advanced encapsulants and structural materials.
 | ||
| Fig. 8 SEM micrographs of (a) pristine crosslinked polymer, (b) knife-cut sample, (c) thermally mended sample (50 °C; 12 h), and (d) thermally mended sample (50 °C; 24 h). Reproduced with permission from ref. 32. | ||
Watanabe and Yoshie reported the synthesis and recycling of polymers made up of bis-furan terminated poly(ethylene adipate) (PEA2F) and multi-maleimide linkers (3M and 2M) (Fig. 9).33 They did not study the healing of the polymer.
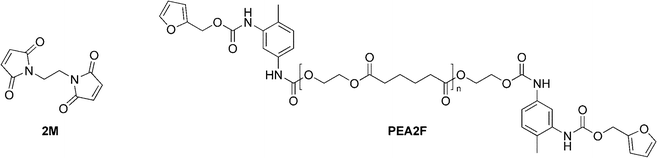 | ||
| Fig. 9 Structures of PEA2F and 2M.33 | ||
Liu and Chen designed polyamides containing various amounts of maleimide and furan pendant groups that exhibited thermally reversible cross-linking behavior via DA and RDA reactions.34 Films of these polymers were cut and then subjected to thermal mending. For these polymers, only partial healing was observed (Fig. 10).
 | ||
| Fig. 10 Test of healing ability: (a) knife cut cross-linked film surface; (b) thermally self-repaired cross-linked film surface: 120 °C, 3 h and 50 °C, 5 d. Reproduced with permission from ref. 34. | ||
An exciting application of these polymer systems has been reported by Gotsmann et al.35 A novel furan-maleimide polymer was designed that allows switching between two different states: a rigid, highly crosslinked, low-temperature state, and a deformable, fragmented, high-temperature state. The switching between these two states was exploited in this work for data-storage and lithographic applications. At high temperature, the polymer exists as dissociated fragments forming an easily deformable medium. At low temperature, the fragments associate to give a cross-linked, macromolecular network that persists as a stable solid, freezing in written structures and resisting mechanical wear. The Diels–Alder cross-linking functions well at the nanometer scale and microsecond timescale, which are necessary for functional switches.
Very recently, McElhanon and coworkers reported the synthesis of thermally labile dendrimers based on the reversible furan-maleimide DA reaction.36 They were able to prepare first through fourth generation benzyl aryl ether-based dendrons that contained furan moieties at their focal point that upon reaction with a bis-maleimide central linker, afforded the corresponding dendrimers. These dendrimers were found to undergo thermal degradation at 95 °C and thermal reassembly at 60 °C.
2.1.1.2. Dicyclopentadiene based polymers. Polymers of cyclopentadiene have been known since the early twentieth century, when Staudinger and Bruson discovered that cyclopentadiene (CP) undergoes a self-DA reaction to produce dicyclopentadiene (DCP), which is an even better DA substrate, and therefore continues to react with itself to produce a polymer.37
CP and its derivatives have been employed for some time as DA-active units in polymers, the major advantage being that they function as both diene and dienenophile, self-reacting to produce DCP and DCP-derived materials. Thus, only a single component monomer is required to achieve polymerization and cross-linking.
In 1961, Stille and Plummer reported the utilization of 1,n-alkane-bis(cyclopentadienes) as monomers for the production of high molecular weight DA-based polymers.38 They prepared homo-polymers from 1,6-bis(cyclopentadienyl)hexane, bis(cyclopentadienyl)nonane, and α,α′-bis(cyclopentadienyl)-p-xylene (Fig. 11), as well as copolymers of these monomers with the bis-dienophiles p-benzoquinone and N,N′-hexamethylenebismaleimide. However, the reversibility of these polymerizations was not studied.
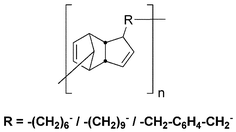 | ||
| Fig. 11 DCP-based polymers prepared from 1,6-bis(cyclopentadienyl)hexane, bis(cyclopentadienyl)nonane, and α,α′-bis(cyclopentadienyl)-p-xylene.38 | ||
CP derivatives have been employed as cross-linkers for a number of polymers, particularly chlorine-containing polymers, and several patents have been issued. Poly(vinyl chloride) (PVC), polychloroprene, polyepichlorohydrin (PECH), and polychloromethylstyrene (PCMS) have been cross-linked by reacting them with cyclopentadienyl sodium or lithium salts39–41 or dicyclopentadiene dicarboxylic acid (Thiele's acid);42–44 isobutylene-isoprene and ethylene-propylene rubbers have been cross-linked by reacting them with dimethylcyclopentadienylaluminium;45,46 and poly(dichlorophosphazene) has been cross-linked by reaction with sodium cyclopentadienylethoxide or its dimer.47 DCP derivatives have also been employed as cross-linking monomers in the polymerization of vinyl monomers40 and epoxide monomers48 to produce thermally reversible cross-linked polymers. The DCP-based cross-links exhibited thermal reversibility via RDA reaction. Upon heating, thermal dissociation of the DCP-based cross-links was evident from the rapid decrease in specific viscosity of polymer solutions.
The work of Kennedy and Castner is particularly noteworthy, as it was the first to actually demonstrate the thermal reversibility of such cross-linked systems.46 They were able to demonstrated the reversibility of the DA cross-linking by thermally reversing the reaction at 215 °C in the absence and presence of maleic anhydride. In addition, samples of the polymers were cut into small pieces and subjected to “curing” at 170 °C (high enough temperature to effect cracking of DCP); this process was repeated three times. After the first and second curing cycles, the cyclopentadienylated polymer behaved as a cross-linked elastomer. The cyclopentadienylated polymer assumed the shape of the mold cavity and was continuous and smooth. A stretching experiment revealed that on releasing the tension the polymer snapped back to its original shape. In the third curing cycle the derivatized polymer did not flow in the mold, and the pieces did not coalesce. No explanation was offered for this behavior, though one can speculate that the reversal temperatures were sufficiently high to cause undesirable decomposition reactions.
Despite these reports, DCP-based systems have not been reported in the context of re-mendable polymers. Only recently preliminary results of a DCP-based monomer that upon heating yields the DA-polycondensation polymer that can be reversibly mended were orally reported.49
2.1.1.3. Anthracene-based polymers. Few reports have appeared in the literature concerning thermally reversible polymers based on anthracene DA chemistry. In 1979, Grigoras and coworkers reported the synthesis of anthracene-methacrylic acid adducts, and their subsequent DA-polymerization, either thermal or Lewis acid (TiCl4) induced (Scheme 5).50 The [4 + 2] cycloaddition polymer structure was supported by spectroscopic data. A subsequent study performed by the same group revealed that the molecular weights were extremely low (around 2500).51
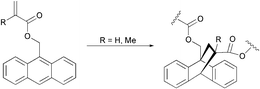 | ||
| Scheme 5 DA-polymerization of anthracene-methacrylic acid adducts.50 | ||
Stevens reported the Diels–Alder polymerization of N-(2-anthryl)maleimide (Mw = 14![[hair space]](https://www.rsc.org/images/entities/char_200a.gif) 000);52 later work performed by Simionescu and Grigoras reported the polymerization of N-(1-anthryl)maleimide to yield very low molecular weight oligomers (Mw = 5800) (Fig. 12).53 Grigoras and Colotin recently reported the formation of DA-type oligomers by allowing the reaction between a bis-anthracene with various bis-maleimides.54 In all anthracene-maleimide cases, the low molecular weights obtained were rationalized by steric effects and the reversibility of the reaction. The healing capabilities of these systems have not been studied.
000);52 later work performed by Simionescu and Grigoras reported the polymerization of N-(1-anthryl)maleimide to yield very low molecular weight oligomers (Mw = 5800) (Fig. 12).53 Grigoras and Colotin recently reported the formation of DA-type oligomers by allowing the reaction between a bis-anthracene with various bis-maleimides.54 In all anthracene-maleimide cases, the low molecular weights obtained were rationalized by steric effects and the reversibility of the reaction. The healing capabilities of these systems have not been studied.
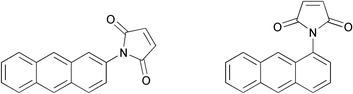 | ||
| Fig. 12 Left: structure of N-(2-anthryl)maleimide. Right: structure of N-(1-anthryl)maleimide. | ||
Jones et al. reported the reversible cross-linking of polymers bearing pendent anthracene moieties with maleimides (Fig. 13).55 In this report, severing of the cross-links occurred only partially upon heating to 250 °C. Although not stated in the original work, it is reasonable to assume that at this temperature competing decomposition reactions prevent full reversibility of the process.
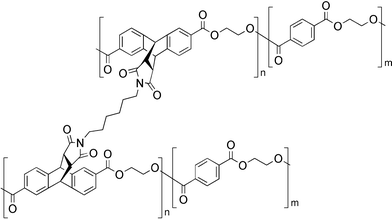 | ||
| Fig. 13 Structure of the reversibly cross-linked anthracene-maleimide based polymer.55 | ||
2.1.1.4. Other DA-based polymers. An interesting report by Brand and Klapper reported the thermally reversible DA polymerization of α,ω-bis(3-furylmethyl)pentaethylene glycol and α,ω-bis(trans-4,4,4-trifluorocrotonylethyl)polyethylene glycol (Fig. 14).56 Under inert conditions, the authors were able to cycle the polymerization/de-polymerization multiple times. This system was studied in the context of variable viscosity, and did not demonstrated re-mendability in the solid state.
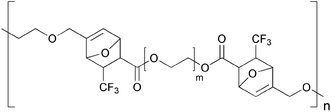
While several thiol containing polymers have been prepared previously,57 the first intentional use of thiol groups for reversible cross-linking was reported in 1993 by Chujo et al. (Scheme 6).58 In this report, a redox-reversible hydrogel system based on thiol-modified poly(N-acetylethyleneimine) was designed, exploiting the reversible interconversion between disulfide (SS) groups and thiols (SH). The same year, Tesoro and Sastri issued a patent concerning the preparation of a polyimide with reversible disulfide cross-links.59
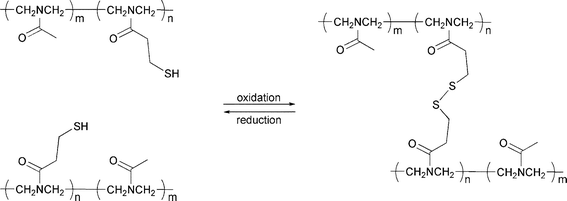 | ||
| Scheme 6 Reversible polymer cross-linking by formation and cleavage of disulfide bridges under oxidizing and reducing conditions, respectively.58 | ||
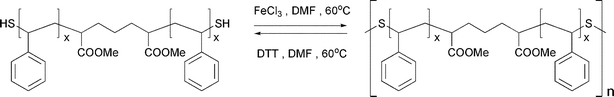
Tsarevsky and Matyjaszewsky employed atom transfer radical polymerization (ATRP) for the preparation of well defined polystyrene “blocks”, bridged by disulfide bonds (Scheme 7).60 The internal disulfide bond could be cleaved by reduction with dithiothreitol to yield the corresponding thiol-terminated polystyrene. The thiol end groups could be efficiently coupled back to the starting disulfide by oxidation with FeCl3, yielding a high molecular weight product.
Bowman and coworkers exploited the photoinduced reversible cleavage of allyl sulfide linkages to induce plasticity, actuation, and equilibrium shape changes without residual stress (Scheme 8).61 The networks studied in this work were produced from pentaerythritol tetra(3-mercaptopropionate) (PETMP) and triethyleneglycol divinyl ether (TEGDVE), which were additionally varied by adding different amounts of a comonomer, 2-methyl-7-methylene-1,5-dithiacyclooctane (MDTO). All the networks exhibited rubber-like behavior, i.e. application of stress caused the samples to strain; when the stress was released the specimens returned to their original length. When the process was repeated with irradiation, stress relaxation was observed due the irradiation, and the samples' dimensions were altered and remained altered when the stress was removed. Under those conditions, homolytic photolysis occurs and the radicals formed diffuse via addition–fragmentation chain transfer through the allyl sulfide functionalities.
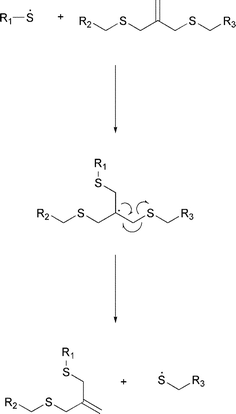
To date, there are no reports in the literature of using such systems for affecting healing of polymers.
Otsuka et al. have introduced dynamic covalent polymers incorporating thermally reversible alkoxyamine units in the main chain (Scheme 9).62 In this study, the thermal structural reorganization behavior of these polymers was systematically investigated, and it was found that the exchange reaction between the derivatives occurs above 60 °C. The reversibility of these systems has been exploited for the preparation of degradable graft co-polymers,63 and a thermodynamic cross-linked polymer,64 where a thermodynamic covalent cross-linking system utilizing alkoxyamine units as thermally reversible covalent bonds was designed. Poly(methacrylic ester)s containing alkoxyamine units on the side chain were thermally cross-linked as a result of radical exchange reaction of alkoxyamine moieties (Fig. 15).64 Cleaving of the cross-links was carried out by heating the cross-linked polymer in the presence of an excess amount of alkoxyamine. It was found that the cross-linked points thermally dissociate and that the reaction system is reversible under stoichiometric control.
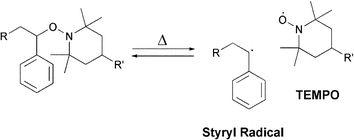 | ||
| Scheme 9 Radical exchange reaction of alkoxyamine derivatives.64 | ||

Chang and Aklonis have prepared polymers that undergo reversible photo-cross-linking as part of an ongoing study concerning the influence of aging on the creep behavior of cross-linked polymer networks.65 Two systems were prepared: (i) polyurethanes with disulfide linkages, where UV irradiation results in photochemical scission of the disulfide bonds and a net decrease in the cross-link density of the network; (ii) polyurethanes with anthracene derivatives of various types, where UV irradiation causes photochemical dimerization of the anthracene units and leads to a net increase in cross-link density. Both systems were shown to exhibit tunable cross-link density; i.e., the photo-processes could be reversed thermally. The authors did not synthesize a hybrid elastomer of the two systems that would combine the increase and decrease capabilities in cross-link density, but stated that such a hybrid could be made and would be of potential interest.
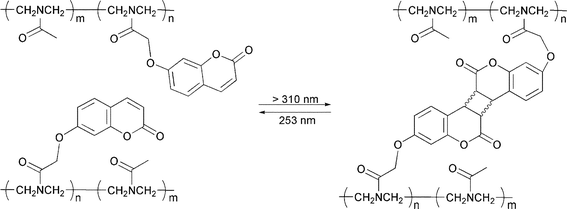
Saegusa and coworkers prepared hydrogels made of polyoxazoline-bearing pendant coumarin moieties (Scheme 10).66 Gelation was achieved by photo-induced (λ > 300 nm) [2 + 2] cycloaddition of the coumarin moieties, and the process could be reversed photochemically (λ = 253 nm) to get back a soluble polymer. In a later work, the same group achieved similar photogelating polyoxazolines bearing pendant anthracene moieties.67 Torii et al. also reported photo-cross-linkable polymers with various anthryl side chains.68 Zheng et al. prepared PEG-based hydrogels via the photodimerization of anthracene groups.69
Coursan et al. reported the preparation of polystyrene end-capped with an anthrylmethyl ether group (Scheme 11).70 Irradiation at 366 nm resulted in dimerization of the polystyrene chains via the anthryl end groups. Irradiation at lower wavelength (λ = 280 nm), resulted in restoration of the original ω-anthrylpolystyrene chains. The authors showed that this process could be cycled more than ten times without any detectable degradation of the photosensitive polymer.
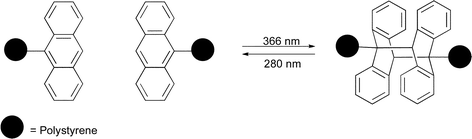 | ||
| Scheme 11 Reversible photo-dimerization of anthrylmethyl ether end-capped polystyrene.70 | ||
Chen and Geh prepared acrylate-based copolymers bearing pendant 4-methylcoumarin chromophores that undergo reversible photo-induced cross-linking.71 Upon irradiation of thin films of these copolymers with 300 nm or 350 nm light, the pendant 4-methylcoumarins dimerize, forming a cross-linked polymeric network. This process was reversed by irradiation with 254 nm light, which caused photo-cleavage.
Chung et al. presented a 1,1,1-tris(cinnamoyloxymethyl)ethane (TCE) based polymer (Fig. 16) that undergoes photodimerizaion to produce a highly cross-linked matrix.72 The authors claimed that these networks, once cracked, can undergo photo-induced healing by irradiating with light of λ > 280 nm. Flexural strength measurements indicated a drastic decrease in strength of the healed vs. the original polymer specimens, raising doubts regarding the claimed re-mendability of this polymer.
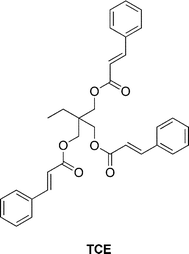 | ||
| Fig. 16 Structure of 1,1,1-tris(cinnamoyloxymethyl)ethane (TCE).72 | ||
Matsui et al. have recently reported a novel molecularly imprinted polymer (MIP) prepared by photo-cross-linking poly(methacrylic acid) (PMAA) with anthryl moieties as photo-cross-linking sites in the presence of the antimalarial drug cinchonidine as a template molecule.73 Subsequent exposure of this MIP to shortwave UV light resulted in cleavage of the anthracene dimer cross-links, as observed by the reduced cinchonidine binding capacity.

2.2. Non-covalently bound systems
Non-covalently bound systems are systems where the polymerization and/or the cross-linking occur by intermolecular interactions of the monomer units and/or the side chains. The reversibility of non-covalent interactions renders these supramolecular polymers with dynamic features such as the ability to change in length and/or cross-linking extent, constitution, and structure. As a result, one of their inherent traits is the ability to self-repair and self-heal. While this concept has been known for some time, useful materials have been made only recently. Since supramolecular polymers have been extensively reviewed, the interested reader is referred to the recent literature on the subject.76–82 Here we will present some of the latest interesting and potentially useful systems that have been developed.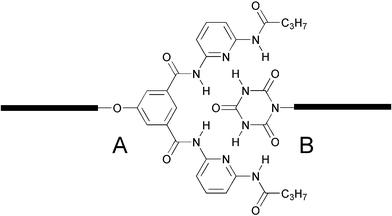 | ||
| Fig. 18 Structure of a cyanuric wedge (ADA–ADA array) and a corresponding diaminopyridine-substituted isophthalamide receptor (DAD-DAD array) assembled into a well-defined six hydrogen-bonded system.83 | ||
Meijer and coworkers have introduced supramolecular polymers that utilize the cooperative effect and directionality of quadruple hydrogen bonds.84–101 These polymers were fabricated by employing 2-ureido-4-pyrimidone (UPy) end groups that form dimers held together by self-complementary DDAA (donor–donor⋯acceptor–acceptor) hydrogen bonds. Monomers containing two and three binding sites led to the formation of linear and cross-linked polymers, respectively. The high dimerization constant (>106 M−1 in CHCl3)85,86 leads to a high degree of polymerization. The polymeric networks generated by this method, above ca. 90 °C, dissociate and melt, and behave much like thermoplastic elastomers (Fig. 19). As a consequence, these polymeric systems can undergo thermal mending.
 | ||
| Fig. 19 Illustration of the phase changes in a rubbery material based on Kraton®, poly(ethylene-co-butylene), modified with UPy, exemplifying the diversity in phase behavior because of the supramolecular interactions. Reproduced with permission from ref. 97. | ||
The ureido-pyrimidone based materials represent the first example of a truly reversible supramolecular polymer network that can be easily synthesized from commercially available starting materials, where the ureido-pyrimidone dimerization is strong enough to construct supramolecular materials possessing acceptable mechanical properties. Meijer et al. have recently reviewed their progress in the field.97
Interest from industry in this reversible polymeric system led to the inauguration of SupraPolix, whose purpose is the manufacture of materials based on supramolecular polymers. It is claimed that incorporation of even a small amount of UPy in existing plastics dramatically simplifies the processing of the material. It is further claimed that production is cheaper and faster, though there should be additional costs incurred in the modification of conventional polymers. The materials should be easily recycled and be consumer-friendly. For further information, see www.suprapolix.com.
Zimmerman and co-workers have designed a UG and DAN system that forms a high stability, high fidelity complex that exceeds that of DNA base-pairing (Fig. 20).102–106 These units were incorporated as end-functional groups in poly(butyl methacrylate) (PBMA) and poly(ethylene glycol) (PEG) segments, enabling the assembly of multi-block supramolecular copolymers.102 The degree of polymerization was shown to depend on the concentration and ratio of the blocks in the mixture.
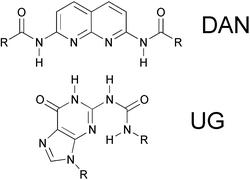 | ||
| Fig. 20 Structures of UG and DAN.102 | ||
Yagai et al. reported supramolecular polymers based on bis(melamine) that can non-covalently polymerize upon binding with cyanurates or barbiturates through complementary triple hydrogen-bonding interaction (Fig. 21).107 The same group has also designed binary hydrogen-bonded supramolecular polymers composed of a bis(melamine) and barbiturate-type merocyanine dyes.108 The supramolecular polymers obtained exhibit well-defined one-dimensional fibrous or two-dimensional sheet-like macroscopic structures, which are dependent on the blended merocyanine dyes, due to the formation of supramolecular polymers.
 | ||
| Fig. 21 Supramolecular polymerization in cyclohexane solution via hydrogen bonding. Reproduced with permission from ref. 107. | ||
Rotello and coworkers have employed bis-thymine units to non-covalently cross-link a complementary diamidopyridine-functionalized copolymer via hydrogen bonding.109–113 In non-competitive solvents, discrete micro-spherical aggregates were formed due to specific three-point polymer cross-linking, the size of which could be controlled by the spacer structure. This cross-linking process was shown to be completely thermally reversible, with complete dissolution observed at 50 °C and reformation of the aggregates upon return to ambient temperature. This process could be repeated multiple times, with the lower particle dispersity observed arising from the annealing process.
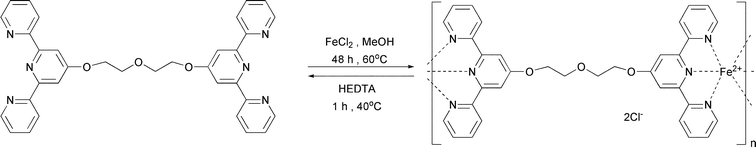
Rowan and coworkers prepared a series of metallo-supramolecular polymers by appending the terdentate ligand 2,6-bis(benzimidazolyl)-4-oxypyridine to the ends of various (macro)monomer units, such as penta(ethylene glycol) and poly(tetrahydrofuran) (Mn = 2000 or 4800 g mol−1), followed by complexation with a metal ion (e.g., Fe(II), Co(II), Zn(II), or Cd(II)), which can bind to the ligand in 1:2 ratio (Fig. 22).118 Self assembled linear supramolecular polymers were obtained, some of which form thermoplastic elastomeric films in which the ionic blocks and soft poly(tetrahydrofuran) segments are phase separated. Additional work from the same group demonstrated the same metallo-supramolecular polymer formation in the presence of Zn(II) with a similar bridged ligand.119
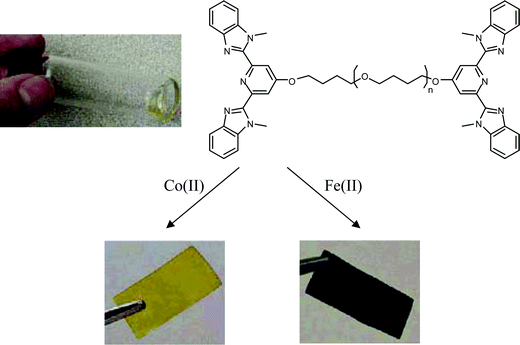 | ||
| Fig. 22 Formation of metallo-supramolecular polymer films by complexation of the 2,6-bis(1′-methylbenzimidazolyl)pyridine-terminated oligomers with Fe(II) and Co(II). Reproduced with permission from ref. 118. | ||
Recently, Lehn and coworkers have prepared neutral metallo-supramolecular polymers by self-assembly through multiple condensation–coordination–deprotonation reactions between a bishydrazide, an aldehyde, and either a Zn(II) or Ni(II) salt (Fig. 23).120 The polymers obtained all had around 20 repeating units, and exhibited gum-like consistency and readily formed thin films. These polymers exhibited solution dynamic behavior as a result of the reversibility of the coordination bonds; ligand exchange and recombination was shown to occur by 1H NMR studies of polymer blends. Thus far, the mechanical properties of these polymers have not been studied.

![Structure of a supramolecular [2]rotaxane polymer based on host–guest complexation of cyclodextrins. Reproduced with permission from ref. 121.](/image/article/2008/JM/b713953p/b713953p-f24.gif) | ||
| Fig. 24 Structure of a supramolecular [2]rotaxane polymer based on host–guest complexation of cyclodextrins. Reproduced with permission from ref. 121. | ||
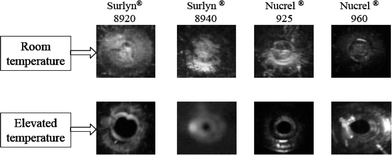
Several ionomers exhibit instantaneous and autonomous self-healing in response to projectile puncturing. Of the ionomers manufactured by DuPont, only Surlyn 8920, Surlyn 8940, Nucrel 960 and Nucrel 925 (Scheme 13) have been examined in depth for their self-healing properties. Surlyn 8940 is licensed by Reactive Target Systems and sold as React-A-Seal; it is currently being used at some shooting ranges as target backings to stop or slow stray rounds.127–129
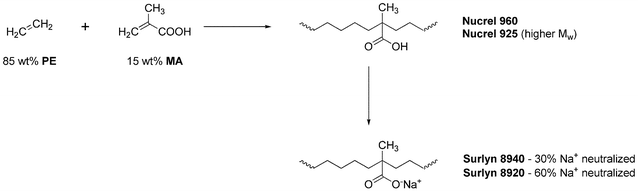 | ||
| Scheme 13 Preparation and structure of ionomers investigated for self-healing properties. | ||
Initially, the self-healing observed in these ionomers was largely attributed to the thermally responsive ionic crosslinking present in the Surlyn polymers.130,131 However, later studies127 revealed that the healing mechanism was more sensitive to temperature and projectile shape than to ionic content. This, combined with the fact that the hydrogen bond cross-linked Nucrel polymers performed similarly to Surlyn, indicated that the healing action occurs via thermally controlled reversible hydrogen bonding (Scheme 14).
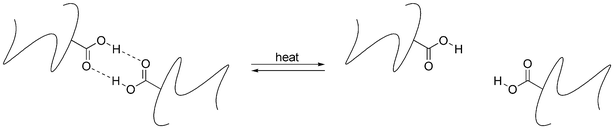 | ||
| Scheme 14 Thermally controlled hydrogen-bond cross-linking in Nucrel. | ||
Although the healing mechanism is activated by a sharp increase in temperature after projectile impact, elevated temperatures do not lead to healing of the damaged sample (Fig. 25). The healing response was found to be dependent on the shape of the projectile, both Surlyn and Nucrel faring better in puncture tests with pointed, rather than blunt, projectiles. Preliminary work has been performed on carbon nanotube reinforced ionomer composites with positive results.132 Of all the self-healing systems, self-healing ionomers are able to autonomously recover from the most devastating damage in a very short period of time, and are by far the least expensive to manufacture.
3. Hybrid covalent/non-covalent systems
Takata and coworkers have designed a recyclable polymer by utilizing a polyrotaxane network with reversible thiol-disulfide bridges (Scheme 15).136 Two or three coupled crown ether rings were assembled with a disulfide-bridged ammonium salt thread, resulting in a polymeric material with <Mn> = 5100, <Mw>/<Mn> = 7.1. The disulfide bridges were easily cleaved in the presence of an appropriate thiol at 60 °C, enabling the quantitative recovery of the original poly(crown ether) and ammonium salt thread.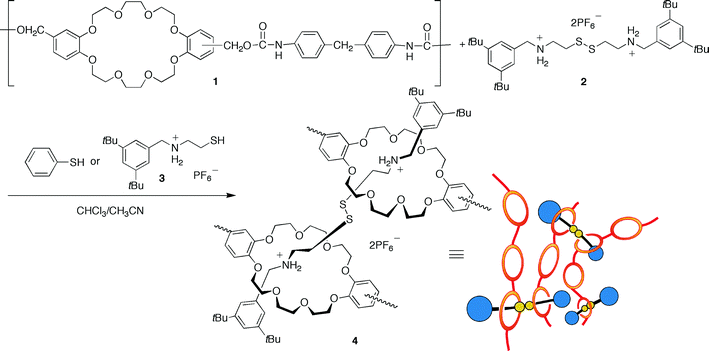 | ||
| Scheme 15 Synthesis of a polyrotaxane network. Reproduced with permission from ref. 136. | ||
Zhao and Moore have designed a reversible polymer based on metathesis of imines under acidic conditions (Scheme 16).137,138 While the monomeric units, m-phenylene ethylene oligomers, are covalently bound to each other, the polymerization process is driven by chain folding of the growing polymer, where solvophobic and aromatic stacking interactions shift the equilibrium in favor of longer chains. The equilibrium could be shifted back towards the starting materials simply by changing the solvent from acetonitrile to chloroform, where the folding propensity diminishes. The limitations of the system include very long synthesis times (required for the full equilibration), very broad polydispersity indices (PDIs), and limited solvent compatibility.
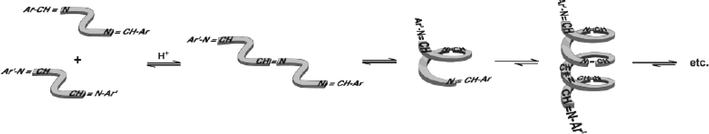 | ||
| Scheme 16 Schematic illustration of the metathesis polymerization driven by chain folding. Reproduced with permission from ref. 138. | ||
Rudkevich and coworkers have employed a combination of hydrogen bonding and chemical CO2 fixation in order to construct supramolecular polymers.139 CO2 combines rapidly with the primary amino groups of calix[4]arene tetraurea at ordinary temperatures and pressures to form carbamates, the latter are thermally unstable and release CO2 upon heating.
Recently, Kolomiets and Lehn described “double dynamers”—polymers that are dynamic on both molecular and supramolecular levels.140 These systems combine the high affinity sextuple hydrogen bond formation between a DAD–DAD receptor and a cyanuric acid wedge with reversible acylhydrazone bond formation by condensation of hydrazides with aldehydes.
4. Irreversible systems
Irreversible systems are polymeric systems that cannot be reverted back to their monomeric, oligomeric, or non cross-linked state. For such polymers, mending is achieved by a different approach—the use of “healing agents”, either embedded in the polymer matrix in a dormant form, or produced over time.4.1. Composite systems—“smart” materials
 | ||
| Fig. 26 Optical micrograph of hollow glass fibres (×200). Reproduced with permission from ref. 141. | ||
Zako and Takano introduced a method of impregnating small particles (50 μm) of thermoplastic adhesive in a glass/epoxy composite laminate.142 The cure temperature of the epoxy matrix was 110 °C. The embedded thermoplastic particles melted when damaged composites were subsequently heated to 120 °C for 10 min on a hot plate. In subsequent three point bend testing, the load–displacement curve indicated that stiffness was recovered in the repaired specimen (Fig. 27).
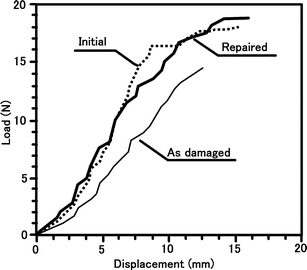 | ||
| Fig. 27 Load and displacement curves for virgin, damaged and repaired specimens under bending. Reproduced with permission from ref. 142. | ||
Trau et al. introduced a new method for achieving mending:143 electric-field induced colloidal aggregation was developed. This self-healing system consists of two concentric cylinders (the inner one has a thin layer of insulating ceramic coating), with an applied electrical field as shown in Fig. 28. Between the two cylinders is a colloidal dispersion of polystyrene or silica particles, which are used to repair defects that occur in the inner cylinder when high stress is applied.
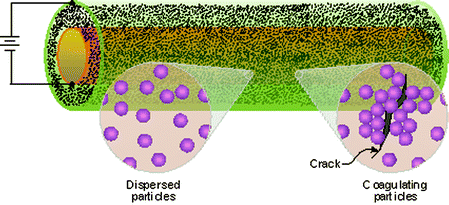 | ||
| Fig. 28 A schematic for a self-healing system that uses the electrohydrodynamic coagulation of particles to close a defect in a cylinder wall. Reproduced with permission from ref. 143. | ||
When a defect occurs in the insulating layer, underneath metal is exposed to create high current density at the damaged site, causing colloidal particles to coagulate around the defect.143 The defect is not completely repaired until the voids between the colloids are filled by metal electrodeposition within the void spaces by dissolving and electroplating of the anodic metal electrode (sacrificial anode) ultimately fills the void. This process produces a coating of a ceramic/metal composite at the site of the initial defect. One drawback to this self-healing technique may be the time scale at which the repair occurs, relative to other systems.
One of the most efficient ways to produce an autonomously self-healing material is to store healing agents inside composite materials that restore the original physical properties after damage. Healing agent storage methods have been developed based on the use of hollow tubes and fibers, particles and microcapsules; all of which can provide integral healing agent storage.
Dry and coworkers embedded methyl methacrylate (MMA) inside hollow porous polypropylene fibres within concrete (Fig. 29).144–147 Bleeding of MMA from these fibers was shown to reduce concrete permeability. In addition, it was shown that the release of crack-adhering adhesive from hollow glass pipettes into the concrete increased the ability to carry load under a subsequent flexural test by 20%. Similarly, Li et al. employed Superglue (ethyl cyanoacrylate) as a healing agent within 500 μm diameter hollow glass tubes embedded in cementitious composites.148
 | ||
| Fig. 29 Photo of samples in which the release of red-dyed chemical (dark areas) from the fiber into the white cement matrix can be seen. Reproduced with permission from ref. 147. | ||
Dry et al. have investigated damage-associated matrix micro-cracking.149–154 A single repair fibre was embedded in a polymer matrix and tests performed to visually verify the release of the healing agent. Motuku et al. further developed this concept by considering different critical parameters, such as method of storage (glass, copper and aluminium tubing), and healing agents (vinyl ester 411-C50 and EPON-862 epoxy).155 In their report, glass tubing was found to be an effective encapsulation medium for the healing agent, allowing its release into the matrix upon fiber breakage. In both works,154,155 dye release accompanied the healing agent; however, this combination resulted in an inability to cure and thus no improvement in mechanical properties was reported.
White and coworkers have achieved autonomous mending of cracks by embedding an appropriate catalyst and microcapsules of monomer healing agent throughout a polymer matrix (Fig. 30 and 31).156–162 Upon cracking (a), the microcapsules burst (Fig. 31, left) and release a healing agent, dicyclopentadiene (DCP) monomer. The healing agent bleeds out filling the crack via capillary action (b), and subsequently reacts with a first generation Grubb's catalyst embedded in the surrounding matrix and undergoes ring opening metathesis polymerisation (ROMP) (Fig. 31, right), thus healing the crack and restoring its mechanical properties (c). Early investigations of this system exhibited varying healing efficiencies, ranging from up to 67% recovery of virgin fracture toughness for manually injected monomer into the damage plane,156,157,159 down to 19% when the catalyst was directly embedded into the matrix. Recent work has demonstrated that the DCP healing agent worked very well at 80 °C giving a maximum healing efficiency of >70%.159,161
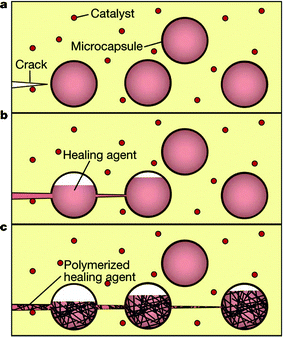 | ||
| Fig. 30 The self-healing concept where a microencapsulated healing agent is embedded in a structural composite matrix containing a catalyst capable of polymerizing the healing agent. (a) Cracks form in the matrix. (b) The crack ruptures the microcapsules, releasing the healing agent into the crack plane through capillary action. (c) The healing agent contacts the catalyst, triggering polymerization that bonds the crack faces closed. Reproduced with permission from ref. 156. | ||
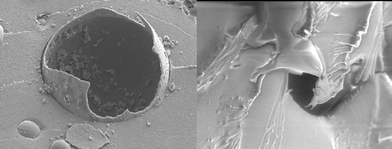 | ||
| Fig. 31 Left: a scanning electron micrograph shows the fracture plane of a self-healing epoxy with a ruptured urea-formaldehyde microcapsule in the center of the image. Right: a scanning electron micrograph shows the fracture plane of a self-healing epoxy and the polymerized healing agent coating the original fracture plane. A broken (emptied) microcapsule appears in the background. Reproduced with permission from ref. 156. | ||
Recent work by Liu et al. revealed that the healing process could be improved by using a blend of DCP with 5-ethylidene-2-norbornene (ENB) in a ratio of 1 : 3.163 A computational model of fatigue crack retardation in White's system,156 taking into account the reaction kinetics of the polymeric material, has been presented.164 While autonomous self-mending, this system has several limitations such as healing being limited to small cracks that enable capillary action, inability to heal multiple recurring cracks and incompatibility of different polymeric matrices.
Braun and coworkers improved on this concept by designing self-healing materials based on the tin-catalyzed polycondensation of phase-separated droplets containing hydroxyl end-functionalized polydimethylsiloxane (HOPDMS) and polydiethoxysiloxane (PDES) (Fig. 32).165 Polyurethane microcapsules of the catalyst, di-n-butyltin dilaurate (DBTL), are embedded in a vinyl ester matrix, and upon cracking, these capsules break and release the catalyst. The important advantages of this system over the previous one, mentioned above, include stability of the healing chemistry in ambient and workable (>100 °C) conditions, wide availability and relatively low cost of the components and facile fabrication due to the phase separation of the healing agent which is simply mixed into the polymer matrix. The disadvantages are the lower healing efficiency and the inherent properties of the polymer, which is not likely to find applications as a structural engineering material but rather as one where soft matter is required.
 | ||
| Fig. 32 Schematic of self-healing process: a) self-healing composite consisting of microencapsulated catalyst (yellow) and phase-separated healing-agent droplets (white) dispersed in a matrix (green); b) crack propagating into the matrix releasing catalyst and healing agent into the crack plane; c) a crack healed by polymerized PDMS (crack width exaggerated). Scanning electron microscopy (SEM) images of d) the fracture surface, showing an empty microcapsule and voids left by the phase-separated healing agent, and e) a representative microcapsule showing its smooth, uniform surface. Reproduced with permission from ref. 165. | ||
Pang and Bond have introduced a novel fiber reinforced plastic which also utilizes a bleeding mechanism to achieve self-repair and visualization of damage.166 The fibers are filled with a two part epoxy healing and fluorescent indicator system (Fig. 33), enabling the monitoring of bleeding by spectroscopic methods. This system exhibited up to 97% recovery of the original flexural strength. The release and infiltration of a UV fluorescent dye from fractured hollow fibers (Fig. 34) into damage sites was shown to be an effective method of quickly and easily highlighting damage. In a later study by the same group, a dilute epoxy healing agent and fluorescent dye-filled hollow-fiber reinforced epoxy matrix was manufactured (Fig. 35).167 This composite exhibited similar self-healing as well as convenient spectroscopic monitoring of the process and featured up to 93% restoration of flexural strength. Despite the gains in material longevity, this healing system ultimately decreases flexural stiffness of a structure when incorporated into laminate structures and it should also be quite difficult to fabricate on a large scale.
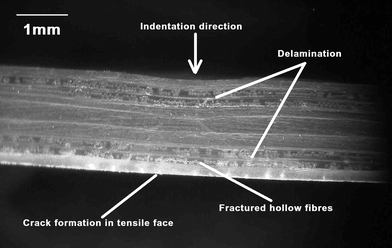 | ||
| Fig. 33 Impact damaged cross-section of composite laminate containing healing agent and UV dye filled filaments. Reproduced with permission from ref. 166. | ||
 | ||
| Fig. 34 Damaged hollow fibre composite material viewed under UV light shows ‘bleeding’ of the fluorescent dye mixed with the healing resin. Reproduced with permission from ref. 166. | ||
 | ||
| Fig. 35 Optical micrographs of the manufactured fibers and composites: (a) hollow glass fibers of 60 μm external diameter with a hollowness of 50% and (b) the same fibers within a Hexcel 913 epoxy matrix. Reproduced with permission from ref. 167. | ||
Stephenson and coworkers have designed self-healing coats for steel.168 Their system employs urea formaldehyde microcapsules (50–150 μm diameter) containing several types of film-forming compounds (healants) and corrosion inhibitors mixed into commercially available coating systems. These microcapsules release their corrosion inhibiting/self-healing constituents when mechanically ruptured, i.e. when the coating is damaged by impact or abrasion (Fig. 36). It was shown that the coating formulation significantly affects the self-healing capability of the microcapsules (Fig. 37); in general, layering of the coatings provided effective self-healing and reduced corrosion.
 | ||
| Fig. 36 (a) Release of red dye from a rupture of UF microcapsules caused by induced crack in coating. (b) Red dye release due to breaking UF microcapsules by scribing the coated layer. Reproduced with permission from ref. 168. | ||
 | ||
| Fig. 37 (a) Left: schematic layer-by-layer coating formulation. Right: optical micrograph of cross-section of layer-by-layer coating. Reproduced with permission from ref. 168. | ||
 | ||
| Fig. 38 Scanning electron micrographs of calcite precipitation induced by B. pasteurii immobilized in PU. a. Porous PU matrix without microbial cells showing open-cell structures. Bar, 1 mm. b. Distribution of microorganisms on the PU surface. Bar, 1 μm. c. Microorganisms densely packed in a pore of the PU matrix. Bar, 10 μm. d. Calcite crystals grown in the pore (shown in c) of the PU matrix. Bar, 10 μm. e. Calcite crystals grown extensively over the PU polymer. Bar, 500 μm. f. Magnified section pointed with an arrow in e shows crystals embedded with microorganisms. Bar, 20 μm. Reproduced with permission from ref. 173. | ||
5. Summary and outlook
Though the field is rather young, with the first publications with a view toward “smart” materials appearing only 6 years ago, substantial progress has already been made. Comparing reversible polymerization and autonomous healing as means of mending and re-mending cracked polymeric monoliths one can find advantages and disadvantages to both systems.Considering the reversible polymerization approach to stimuli responsive materials, the advantages are clear: i) repeated healing of multiple cracks has been demonstrated; ii) conceptual simplicity of reversible polymerization is certainly a benefit; and finally iii) the fact that the solids are single-component rather than composites has the advantage of facile and reproducible manufacturing. The disadvantages are equally clear: i) reversible polymerization requires an external agent in the form of heat or light for the healing step; ii) commercial monomers and polymers are currently not candidates; and iii) monomers are relatively expensive, particularly if they will be used in large-scale applications.
The autonomous healing approach clearly has the following advantages: i) it is truly autonomous, not requiring any outside intervention; ii) it can be applied to well-established commercial polymers such as polyepoxides; and iii) commercialization of the systems developed by the Bond166 group on a relatively large scale can be expected in the near future. The main disadvantage is that the healing is irreversible; i.e., once a crack has been healed and the material cracks again in the same place, it cannot be healed because the healing agent(s) is (are) depleted. This problem has very recently been addressed by the Illinois156–162 group through microvascularization,174 though the process of producing the monoliths is rather complex and currently applicable to only relatively small objects. Another disadvantage is that the catalyst used in the Illinois approach is moisture and oxygen sensitive, is expensive and, if a large part were to be produced, the cost of the catalyst would be prohibitive and the requisite quantities would not be available.
The future of these materials appears to be bright, particularly once they leave the comfortable womb of invention in academic research. Examination of the program of a recently concluded International Conference (http://www.selfhealingmaterials.nl/index_eng.htm) gives merit to the first sentence of this paragraph. The applications in the area of concrete mending by cyanoacrylate appear to be closest to large-scale implementation that, in the end, may very well be instrumental in preventing disastrous failures such as the bridge collapse in Minneapolis, MN, USA on August 1, 2007 (http://en.wikipedia.org/wiki/I-35W_Mississippi_River_Bridge).
References
- A. Gandini, Polím.: Ciênc. Tecnol., 2005, 15, 95 Search PubMed.
- J. M. Craven, US Pat., 3 435 003, 1969.
- M. Stevens and A. Jenkins, J. Polym. Sci., 1979, 17, 3675 Search PubMed.
- Y. Chujo, K. Sada and T. Saegusa, Macromolecules, 1990, 23, 2636 CrossRef CAS.
- Y. Imai, H. Itoh, K. Naka and Y. Chujo, Macromolecules, 2000, 33, 4343 CrossRef CAS.
- S. A. Canary and M. P. Stevens, J. Polym. Sci., Polym. Chem., 1992, 30, 1755 Search PubMed.
- H. Laita, S. Boufi and A. Gandini, Eur. Polym. J., 1997, 33, 1203 CrossRef.
- C. Goussé, A. Gandini and P. Hodge, Macromolecules, 1998, 31, 314 CrossRef CAS.
- R. Gheneim, C. Perez-Berumen and A. Gandini, Macromolecules, 2002, 35, 7246 CrossRef CAS.
- E. Goiti, M. B. Huglin and J. M. Rego, Polymer, 2001, 42, 10187 CrossRef CAS.
- E. Goiti, M. B. Huglin and J. M. Rego, Macromol. Rapid Commun., 2003, 24, 692 CrossRef CAS.
- E. Goiti, M. B. Huglin and J. M. Rego, Eur. Polym. J., 2004, 40, 219 CrossRef CAS.
- E. Goiti, M. B. Huglin and J. M. Rego, Eur. Polym. J., 2004, 40, 1451 CrossRef CAS.
- G. C. Tesoro and V. R. Sastri, Ind. Eng. Chem. Prod. Res. Dev., 1986, 25, 444 CrossRef CAS.
- X. He, V. R. Sastri and G. C. Tesoro, Makromol. Chem. Rapid Commun., 1989, 9, 191.
- R. O'Dell, Ph.D. Thesis, Lancaster University, UK, 1990.
- N. Kuramoto, K. Hayashi and K. Nagai, J. Polym. Sci., Polym. Chem., 1992, 30, 2501 Search PubMed.
- C. D. Diakoumakos and J. A. Mikroyannidis, J. Polym. Sci., Polym. Chem., 1992, 30, 2559 Search PubMed.
- C. D. Diakoumakos and J. A. Mikroyannidis, Eur. Polym. J., 1994, 30, 465 CrossRef CAS.
- N. Kuramoto, K. Hayashi and K. Nagai, J. Polym. Sci., Polym. Chem., 1994, 32, 2501 Search PubMed.
- C. Goussé and A. Gandini, Polym. Int., 1999, 48, 723 CrossRef CAS.
- J. R. McElhanon and D. R. Wheeler, Org. Lett., 2001, 3, 2681 CrossRef CAS.
- J. R. McElhanon, E. M. Russick, D. R. Wheeler, D. A. Loy and J. H. Aubert, J. Appl. Polym. Sci., 2002, 85, 1496 CrossRef CAS.
- D. A. Loy, D. R. Wheeler, J. R. McElhanon and M. L. Durbin-Voss, US Pat., 6 403 753, 2002.
- D. A. Loy, D. R. Wheeler, E. M. Russick, J. R. McElhanon and R. S. Sanders, US Pat., 6 337 384, 2002.
- J. H. Small, D. A. Loy, D. R. Wheeler, J. R. McElhanon and R. S. Saunders, US Pat., 6 271 335, 2001.
- K. Kamahori, S. Tada, K. Ito and S. Itsuno, Macromolecules, 1999, 32, 541 CrossRef CAS.
- C. Goussé and A. Gandini, Polym. Bull., 1998, 40, 389 CrossRef CAS.
- X. Chen, M. A. Dam, K. Ono, A. Mal, H. Shen, S. R. Nutt, K. Sheran and F. Wudl, Science, 2002, 295, 1698 CrossRef CAS.
- X. Chen, F. Wudl, A. K. Mal, H. Shen and S. R. Nutt, Macromolecules, 2003, 36, 1802 CrossRef CAS.
- F. Wudl and X. Chen, US Pat., 2004 014 933, 2004.
- Y. L. Liu and C. Y. Hsieh, J. Polym. Sci., Polym. Chem., 2006, 44, 905 Search PubMed.
- M. Watanabe and N. Yoshie, Polymer, 2006, 47, 4946 CrossRef CAS.
- Y. L. Liu and Y. W. Chen, Macromol. Chem. Phys., 2007, 208, 224 CrossRef CAS.
- B. Gotsmann, U. Duerig, J. Frommer and C. J. Hawker, Adv. Funct. Mater., 2006, 16, 1499 CrossRef CAS.
- M. L. Szalai, D. V. McGrath, D. R. Wheeler, T. Zifer and J. R. McElhanon, Macromolecules, 2007, 40, 818 CrossRef CAS.
- H. Staudinger and H. A. Bruson, Justus Liebigs Ann. Chem., 1926, 447, 97 CrossRef CAS; H. Staudinger and H. A. Bruson, Justus Liebigs Ann. Chem., 1926, 447, 110 CrossRef CAS.
- J. K. Stille and L. Plummer, J. Org. Chem., 1961, 26, 4026 CrossRef CAS.
- Y. Takeshita, M. Uoi, Y. Hirai and M. Uchiyama, Ger. Pat., 2 164 022, 1972.
- A. Harumi and K. Shyuzo, Jpn. Pat., 48 36293, 1973.
- M. Miura, F. Akutsu, T. Usui, Y. Ikebukuro and K. Nagakubo, Makromol. Chem., 1985, 186, 473 CrossRef CAS.
- X. N. Chen and E. J. Ruckenstein, J. Polym. Sci., Polym. Chem., 1999, 37, 4390 Search PubMed.
- E. J. Ruckenstein and X. N. Chen, J. Polym. Sci., Polym. Chem., 2000, 38, 818 Search PubMed.
- X. N. Chen and E. J. Ruckenstein, J. Polym. Sci., Polym. Chem., 2000, 38, 1662 Search PubMed.
- J. P. Kennedy and K. F. Castner, US Pat., 4 138 441, 1979.
- J. P. Kennedy and K. F. Castner, J. Polym. Sci.: Polym. Chem. Ed., 1979, 17, 2039 CrossRef CAS; J. P. Kennedy and K. F. Castner, J. Polym. Sci.: Polym. Chem. Ed., 1979, 17, 2055 CrossRef CAS.
- J. C. Salamone, Y. Chung, S. B. Clough and A. C. Watterson, J. Polym. Sci., Polym. Chem. Ed., 1988, 26, 2923 Search PubMed.
- X. N. Chen and S. K. Jiao, Acta Polym. Sin., 1999, 5, 564 Search PubMed.
- E. Murphy, E. Bolanos and F. Wudl, manuscript in preparation.
- S. Dumitrescu, M. Grigoras and A. Natansohn, J. Polym. Sci., Polym. Lett. Ed., 1979, 17, 553 CrossRef CAS.
- C. I. Simionescu, M. Grigoras and V. Bărboiu, J. Polym. Sci.: Polym. Chem. Ed., 1985, 23, 2089 CrossRef CAS.
- M. P. Stevens, J. Polym. Sci., Polym. Lett. Ed., 1984, 22, 467 CrossRef CAS.
- C. I. Simionescu and M. Grigoras, J. Polym. Sci., Polym. Lett., 1990, 28, 39 Search PubMed.
- M. Grigoras and G. Colotin, Polym. Int., 2001, 50, 1375 CrossRef CAS.
- J. R. Jones, C. L. Liotta, D. M. Collard and D. A. Schiraldi, Macromolecules, 1999, 32, 5786 CrossRef CAS.
- T. Brand and M. Klapper, Des. Monomers Polym., 1999, 2, 287 Search PubMed.
- C. S. Lee and W. H. Daly, Adv. Polym. Sci., 1974, 15, 61 CAS; G. Wulff and I. Schulze, Angew. Chem., Int. Ed. Engl., 1978, 17, 537 CrossRef.
- Y. Chujo, K. Sada, A. Naka, R. Nomura and T. Saegusa, Macromolecules, 1993, 26, 883 CrossRef CAS.
- G. C. Tesoro and V. R. Sastri, US Pat., 5 260 411, 1993.
- N. V. Tsarevsky and K. Matyjaszewsky, Macromolecules, 2002, 35, 9009 CrossRef CAS.
- T. F. Scott, A. D. Schneider, W. D. Cook and C. N. Bowman, Science, 2005, 308, 1615 CrossRef CAS.
- H. Otsuka, K. Aotani, Y. Higaki, Y. Amamoto and A. Takahara, Macromolecules, 2007, 40, 1429 CrossRef CAS.
- Y. Higaki, H. Otsuka and A. Takahara, Macromolecules, 2004, 37, 1696 CrossRef CAS.
- Y. Higaki, H. Otsuka and A. Takahara, Macromolecules, 2006, 39, 2121 CrossRef CAS.
- J. M. Chang and J. J. Aklonis, J. Polym. Sci., Polym. Lett. Ed., 1983, 21, 999 CrossRef CAS.
- Y. Chujo, K. Sada and T. Saegusa, Macromolecules, 1990, 23, 2693 CrossRef CAS.
- Y. Chujo, K. Sada, R. Nomura, A. Naka and T. Saegusa, Macromolecules, 1993, 26, 5611 CrossRef CAS.
- T. Torii, H. Ushiki and K. Horie, Polym. J., 1993, 25, 173 CAS.
- Y. Zheng, M. Micic, S. V. Mello, M. Mabrouki, F. M. Andreopoulos, V. Konka, S. M. Pham and R. M. Leblanc, Macromolecules, 2002, 35, 5228 CrossRef CAS.
- M. Coursan, J. P. Desvergne and A. Deffieux, Macromol. Chem. Phys., 1996, 197, 1599 CrossRef CAS.
- Y. Chen and J. L. Geh, Polymer, 1996, 37, 4481 CrossRef CAS.
- C. M. Chung, Y. S. Roh, S. Y. Cho and J. G. Kim, Chem. Mater., 2004, 16, 3982 CrossRef CAS.
- J. Matsui, Y. Ochi and K. Tamaki, Chem. Lett., 2006, 35, 80 CrossRef CAS.
- T. Ono, T. Nobori and J.-M. Lehn, Chem. Commun., 2005, 1522 RSC.
- T. Ono, S. Fujii, T. Nobori and J.-M. Lehn, Chem. Commun., 2007, 46 RSC.
- M. J. Serpe and S. L. Craig, Langmuir, 2007, 23, 1626 CrossRef CAS; A. Harada, A. Hashidzume and Y. Takashima, Adv. Polym. Sci., 2006, 201, 1.
- H. Hofmeier and U. S. Schubert, Chem. Commun., 2005, 2423 RSC.
- J. M. Lehn, Prog. Polym. Sci., 2005, 30, 814 CrossRef CAS.
- A. Ciferri, Supramolecular polymers, Taylor & Francis, Boca Raton, FL, 2nd edn, 2005 Search PubMed.
- G. Armstrong and M. Buggy, J. Mater. Sci., 2005, 40, 547 CrossRef CAS.
- L. Brunsveld, B. J. B. Folmer, E. W. Meijer and R. P. Sijbesma, Chem. Rev., 2001, 101, 4071 CrossRef CAS.
- J. S. Moore, Curr. Opin. Colloid Interface Sci., 1999, 4, 108 CrossRef CAS.
- V. Berl, M. Schmutz, M. J. Krische, R. G. Khoury and J.-M. Lehn, Chem.–Eur. J., 2002, 8, 1227 CrossRef CAS.
- R. P. Sijbesma, F. H. Beijer, L. Brunsveld, B. J. B. Folmer, J. H. K. Ky Hirschberg, R. F. M. Lange, J. K. L. Lowe and E. W. Meijer, Science, 1997, 278, 1601 CrossRef CAS.
- F. H. Beijer, R. P. Sijbesma, H. Kooijman, A. L. Spek and E. W. Meijer, J. Am. Chem. Soc., 1998, 120, 6761 CrossRef CAS.
- S. H. M. Söntjens, R. P. Sijbesma, M. H. P. van Genderen and E. W. Meijer, J. Am. Chem. Soc., 2000, 122, 7487 CrossRef.
- J. H. K. Ky Hirschberg, F. H. Beijer, H. A. van Aert, P. C. M. M. Magusin, R. P. Sijbesma and E. W. Meijer, Macromolecules, 1999, 32, 2696 CrossRef.
- R. F. M. Lange, M. van Gurp and E. W. Meijer, J. Polym. Sci., Polym. Chem., 1999, 37, 3657 Search PubMed.
- B. J. B. Folmer, R. P. Sijbesma, R. M. Versteegen, J. A. J. van der Rijt and E. W. Meijer, Adv. Mater., 2000, 12, 874 CrossRef CAS.
- A. El-ghayoury, E. Peeters, A. P. H. J. Schenning and E. W. Meijer, Chem. Commun., 2000, 1969 RSC.
- J. H. K. Ky Hirschberg, L. Brunsveld, A. Ramzi, J. A. J. M. Vekemans, R. P. Sijbesma and E. W. Meijer, Nature, 2000, 407, 167 CrossRef CAS.
- A. P. H. J. Schenning, P. Jonkheijm, E. Peeters and E. W. Meijer, J. Am. Chem. Soc., 2001, 123, 409 CrossRef CAS.
- A. El-Ghayoury, A. P. H. J. Schenning, P. A. van Hal, J. K. J. van Duren, R. A. J. Janssen and E. W. Meijer, Angew. Chem., Int. Ed., 2001, 40, 3660 CrossRef CAS.
- H. M. Keizer, R. P. Sijbesma, J. F. G. A. Jansen, G. Pasternack and E. W. Meijer, Macromolecules, 2003, 36, 5602 CrossRef CAS.
- H. M. Keizer, R. van Kessel, R. P. Sijbesma and E. W. Meijer, Polymer, 2003, 44, 5505 CrossRef CAS.
- A. W. Bosman, L. Brunsveld, B. J. B. Folmer, R. P. Sijbesma and E. W. Meijer, Macromol. Symp., 2003, 201, 143 CrossRef CAS.
- A. W. Bosman, R. P. Sijbesma and E. W. Meijer, Mater. Today, 2004, 7, 34 CrossRef CAS.
- G. B. W. L. Ligthart, H. Ohkawa, R. P. Sijbesma and E. W. Meijer, J. Am. Chem. Soc., 2005, 127, 810 CrossRef CAS.
- O. A. Scherman, G. B. W. L. Ligthart, R. P. Sijbesma and E. W. Meijer, Angew. Chem., Int. Ed., 2006, 45, 2072 CrossRef CAS.
- H. Kautz, D. J. M. van Beek, R. P. Sijbesma and E. W. Meijer, Macromolecules, 2006, 39, 4265 CrossRef CAS.
- H. Ohkawa, G. B. W. L. Ligthart, R. P. Sijbesma and E. W. Meijer, Macromolecules, 2007, 40, 1453 CrossRef CAS.
- T. Park and S. C. Zimmerman, J. Am. Chem. Soc., 2006, 128, 13986 CrossRef CAS.
- T. Park, E. M. Todd, S. Nakashima and S. C. Zimmerman, J. Am. Chem. Soc., 2005, 127, 18133 CrossRef CAS.
- T. Park, S. C. Zimmerman and S. Nakashima, J. Am. Chem. Soc., 2005, 127, 6520 CrossRef CAS.
- T. Park and S. C. Zimmerman, J. Am. Chem. Soc., 2006, 128, 11582 CrossRef CAS.
- E. M. Todd, J. R. Quinn, T. Park and S. C. Zimmerman, Isr. J. Chem., 2005, 45, 381 CrossRef CAS.
- S. Yagai, M. Higashi, T. Karatsu and A. Kitamura, Chem. Mater., 2004, 16, 3582 CrossRef CAS.
- S. Yagai, M. Higashi, T. Karatsu and A. Kitamura, Chem. Mater., 2005, 17, 4392 CrossRef CAS.
- F. Ilhan, M. Gray and V. M. Rotello, Macromolecules, 2001, 34, 2597 CrossRef CAS.
- F. Ilhan, T. H. Galow, M. Gray, G. Clavier and V. M. Rotello, J. Am. Chem. Soc., 2000, 122, 5895 CrossRef CAS.
- U. Drechsler, R. J. Thibault and V. M. Rotello, Macromolecules, 2002, 35, 9621 CrossRef CAS.
- R. J. Thibault, T. H. Galow, E. T. Turnberg, M. Gray, P. H. Hotchkiss and V. M. Rotello, J. Am. Chem. Soc., 2002, 124, 15249 CrossRef.
- R. J. Thibault, P. J. Hotchkiss, M. Gray and V. M. Rotello, J. Am. Chem. Soc., 2003, 125, 11249 CrossRef CAS.
- U. S. Schubert and C. Eschbaumer, Angew. Chem., Int. Ed., 2002, 41, 2892 CrossRef CAS.
- P. R. Andres and U. S. Schubert, Adv. Mater., 2004, 16, 1043 CrossRef CAS.
- R. Dobrawa and F. Würthner, J. Polym. Sci., Part A: Polym. Chem., 2005, 43, 4981 CrossRef CAS.
- S. Schmatloch, M. Fernández González and U. S. Schubert, Macromol. Rapid Commun., 2002, 23, 957 CrossRef CAS.
- J. B. Beck, J. M. Ineman and S. J. Rowan, Macromolecules, 2005, 38, 5060 CrossRef CAS.
- P. K. Iyer, J. B. Beck, C. Weder and S. J. Rowan, Chem. Commun., 2005, 319 RSC.
- C. F. Chow, S. Fujii and J.-M. Lehn, Angew. Chem., Int. Ed., 2007, 46, 5007 CrossRef CAS.
- A. Harada, J. Polym. Sci., Part A: Polym. Chem., 2006, 44, 5113 CrossRef CAS.
- L. Holliday, Ionic polymers, John Wiley & Sons, New York, 1975 Search PubMed.
- http://www2.dupont.com/Surlyn/en_US/; http://www2.dupont.com/Products/en_RU/Nucrel_en.html.
- A. Eisenberg and J. S. Kim, Introduction to ionomers, John Wiley & Sons, New York, 1998 Search PubMed.
- M. R. Tant, K. A. Mauritz and G. L. Wilkes, Ionomers: Synthesis, structure, properties, and applications, Chapman and Hall, New York, 1997 Search PubMed.
- A. D. Wilson and H. J. Prosser, Developments in Ionic Polymers, Applied Science Publishers, New York, 1983 Search PubMed.
- S. J. Kalista, Self-healing of thermoplastic poly(ethylene-co-methacrylic acid) copolymers following projectile puncture, M.Sc. Thesis, Virginia Tech, 2003, http://scholar.lib.vt.edu/theses/available/etd-12162003-103411/; S. J. Kalista, T. C. Ward and Z. Oyetunji, Mech. Adv. Mater. Struct., 2007, 14, 391 Search PubMed.
- S. J. Kalista, T. C. Ward and Z. Oyetunji, Proc. Ann. Meet. Adhes. Soc., 2003, 26, 176 Search PubMed.
- S. J. Kalista and T. C. Ward, Proc. Ann. Meet. Adhes. Soc., 2004, 27, 212 Search PubMed.
- R. Fall, Puncture reversal of ethylene ionomers-mechanistic studies, M.Sc. Thesis, Virginia Tech, 2001, http://scholar.lib.vt.edu/theses/available/etd-08312001-084412/.
- A. Huber and J. A. Hinkley, NASA Tech. Man., 2005, 213532 Search PubMed.
- S. J. Kalista and T. C. Ward, Proc. Ann. Meet. Adhes. Soc., 2006, 29, 244 Search PubMed.
- E. A. Fogleman, W. C. Yount, J. Xu and S. L. Craig, Angew. Chem., Int. Ed., 2002, 41, 4026 CrossRef CAS.
- J. Xu, E. A. Fogleman and S. L. Craig, Macromolecules, 2004, 37, 1863 CrossRef CAS.
- J. Kim, Y. Liu, S. Jung Ahn, S. Zauscher, J. M. Karty, Y. Yamanaka and S. L. Craig, Adv. Mater., 2005, 17, 1749 CrossRef CAS.
- T. Oku, Y. Furusho and T. Takata, Angew. Chem., Int. Ed., 2004, 43, 966 CrossRef CAS.
- D. Zhao and J. S. Moore, J. Am. Chem. Soc., 2002, 124, 9996 CrossRef CAS.
- D. Zhao and J. S. Moore, Macromolecules, 2003, 36, 2712 CrossRef CAS.
- H. Xu, E. M. Hampe and D. M. Rudkevich, Chem. Commun., 2003, 2828 RSC.
- E. Kolomiets and J.-M. Lehn, Chem. Commun., 2005, 1519 RSC.
- S. M. Bleay, C. B. Loader, V. J. Hawyes, L. Humberstone and P. T. Curtis, Composites, Part A, 2001, 32, 1767 Search PubMed.
- M. Zako and N. Takano, J. Int. Mater. Sys. Struct., 1999, 10, 836 Search PubMed.
- M. Trau, D. A. Saville and I. A. Aksay, Langmuir, 1997, 13, 6375 CrossRef CAS ; http://www.princeton.edu/%E2%88%BCcml/html/colloidalaggregation.html.
- C. Dry, Ceram. Trans., 1991, 16, 729 CAS.
- C. Dry, Smart Mater. Struct., 1994, 3, 118 CrossRef CAS.
- C. Dry and W. McMillan, Smart Mater. Struct., 1996, 5, 297 CrossRef CAS.
- C. Dry, Cem. Concr. Res., 2000, 30, 1969 CrossRef CAS.
- V. C. Li, Y. M. Lim and Y. W. Chan, Composites, Part B, 1998, 29B, 819 Search PubMed.
- C. Dry, M. Corsaw and E. Bayer, J. Adhes. Sci. Technol., 2003, 17, 79 CrossRef CAS.
- C. Dry and M. Corsaw, Cem. Concr. Res., 2003, 33, 1723 CrossRef CAS.
- C. Dry, Int. Pat., 2007 005 657, 2007.
- C. Dry, Compos. Struct., 1996, 35, 263 CrossRef.
- C. Dry and N. Sottos, Smart Mater. Struct., 1996, 5, 438.
- C. Dry and W. McMillan, Smart Mater. Struct., 1997, 6, 35 CrossRef.
- M. Motuku, U. K. Vaidya and G. M. Janowski, Smart Mater. Struct., 1999, 8, 623 CrossRef CAS.
- S. R. White, N. R. Sottos, P. H. Geubelle, J. S. Moore, M. R. Kessler, S. R. Sriram, E. N. Brown and S. Viswanathan, Nature, 2001, 409, 794 CrossRef CAS.
- M. R. Kessler and S. R. White, Composites, Part A, 2001, 32, 683 Search PubMed.
- M. R. Kessler, N. R. Sottos and S. R. White, Composites, Part A, 2003, 34, 743 Search PubMed.
- E. N. Brown, N. R. Sottos and S. R. White, Exp. Mech., 2002, 42, 372 Search PubMed.
- E. N. Brown, M. R. Kessler, N. R. Sottos and S. R. White, J. Microencapsulation, 2003, 20, 719 CrossRef CAS.
- E. N. Brown, S. R. White and N. R. Sottos, J. Mater. Sci., 2004, 39, 1703 CrossRef CAS.
- A. S. Jones, J. D. Rule, J. S. Moore, S. R. White and N. R. Sottos, Chem. Mater., 2006, 18, 1312 CrossRef CAS.
- X. Liu, J. K. Lee, S. H. Yoon and M. R. Kessler, J. Appl. Polym. Sci., 2006, 101, 1266 CrossRef CAS.
- S. Maiti, C. Shankar, P. H. Geubelle and J. Kieffer, J. Eng. Mater. Technol., 2006, 128, 595 CrossRef CAS.
- S. H. Cho, H. M. Andersson, S. R. White, N. R. Sottos and P. V. Braun, Adv. Mater., 2006, 18, 997 CrossRef CAS.
- J. W. C. Pang and I. P. Bond, Composites, Part A, 2005, 36, 183 Search PubMed.
- J. W. C. Pang, W. C. Jody and I. P. Bond, Compos. Sci. Technol., 2005, 65, 1791 CrossRef CAS.
- A. Kumar, L. D. Stephenson and J. N. Murray, Prog. Org. Coat., 2006, 55, 244 CrossRef CAS.
- U. K. Gollapudi, C. L. Knutson, S. S. Bang and M. R. Islam, Chemosphere, 1995, 30, 695 CrossRef CAS.
- S. Stocks-Fischer, J. K. Galinat and S. S. Bang, Soil Biol. Biochem., 1999, 31, 1563 CrossRef CAS.
- S. S. Bang, J. K. Galinat and V. Ramakrishnan, Enzyme Microb. Technol., 2001, 28, 404 CrossRef CAS.
- K. Bachmeier, A. E. Williams, J. Warmington and S. S. Bang, J. Biotechnol., 2002, 93, 171 CrossRef CAS.
- http://ssbang.sdsmt.edu/PDF%20Files/MECR.pdf.
- K. S. Toohey, N. R. Sottos, J. A. Lewis, J. S. Moore and S. R. White, Nat. Mater., 2007, 6, 581 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2008 |