[2.2]Paracyclophane: a pseudoconjugated spacer for long-lived electron transfer in phthalocyanine–C60 dyads
Axel
Kahnt
a,
Dirk M.
Guldi
*a,
Andrés
de la Escosura
b,
M. Victoria
Martínez-Díaz
b and
Tomás
Torres
*b
aDepartment of Chemistry and Pharmacy, University of Erlangen, Erlangen, 91081, Germany. E-mail: guldi@chemie.uni-erlangen.de; Fax: +4991318527340; Tel: +4991318527340
bDepartamento de Química Orgánica, Universidad Autónoma de Madrid, Cantoblanco, 28049, Madrid, Spain. E-mail: tomas.torres@uam.es; Fax: +34914973966; Tel: +34914974151
First published on 22nd October 2007
Abstract
Charge transfer features in two different ZnPc–C60 donor–acceptor conjugates were contrasted to highlight the benefits of integrating a [2.2]paracyclophane molecular building block instead of just a phenylene vinylene analogue.
Introduction
Most classical molecular designs for the synthesis and the optimization of artificial systems that can efficiently convert solar energy into useful chemical energy are based on the covalent linkage of a photoexcitable chromophore with an electron-acceptor or an electron-donor unit. The nature of the spacer in these systems is very important for the control of the distance, orientation and electronic coupling between the chromophores. Conjugated fragments are the most widely used bridges, since they allow the elongation of the lifetime of the radical ion pair state by spatial separation of the photoactive moieties. However, some drawbacks inherent to such spacers reduce the scope of these π-extended conjugated bridges: for example, for bridge lengths exceeding a certain critical value, they may switch from the superexchange mechanism to molecular wire behavior, which would limit their photovoltaic application.1 In contrast, non-conventional strategies such as π–π stacking through aromatic rings have received much less attention up to now.2 Arene–arene interactions could be advantageously involved in electron transfer processes between donor and acceptor photoactive units: full or partial elimination of the conjugated electronic path would allow the increase of the distance between the photoactive units with no adopting of molecular wire characteristics. A π–π stacked spacer may show, therefore, slow back electron transfer without diminishing electronic interaction between the donor and the acceptor moieties. The [2.2]paracyclophane3 (pCP) molecular building block can be used as a pseudoconjugated spacer in donor–acceptor systems for electron transfer, mediated and controlled by π–π stacking within the cyclophane moiety. Over the years, this paradigmatic molecule has played a significant role in structural/mechanistic studies and served as a valuable probe for understanding the interplay of aromaticity versus strain, and transannular π–π interactions. The use of chiral ligands with planar or central chirality based on paracyclophane systems represents another important research area that has been growing considerably in recent years.4 In contrast with classical conjugated connectors, this spacer can be viewed as a biased double potential well separated by a tunnelling barrier.2b,5 The linkage of a donor and an acceptor unit through a [2.2]paracyclophane moiety would provide the bias, promoting directional charge transfer through the barrier. Moreover, the sp3-hybridized lateral chains between the two benzene rings disrupt a possible through-bond pathway for electronic communication between the chromophores, ensuring the occurrence of electron transfer by a through-space path.Phthalocyanines6 (Pcs) are highly suited molecules for their integration in light energy conversion systems. They exhibit very high extinction coefficients around 700 nm for efficient photon harvesting, rich redox chemistry, and p-type semiconducting properties. In recent years, our group has been interested in the preparation of phthalocyanine–C60 (Pc–C60) dyads and related systems7 for studying photoinduced electron transfer and applications in photovoltaic devices. Although some promising results have already been reached, much effort is still needed, mainly to get longer-lived photoinduced charge-separated states.
On these bases, the covalent grafting of an electron-donor phthalocyanine to the upper cycle of the pCP and the electron-acceptor C60 unit to the lower cycle in the pseudo-para position could be an excellent strategy towards long-lived charge-separated states. In this paper, the synthesis and characterization of ZnPc–paracyclophane–C60 (1), consisting of a tri-(tert-butyl)phthalocyanine and a C60 unit directly attached to the pseudo-para positions of a central [2.2]paracyclophane moiety, is described (Chart 1). To demonstrate the through-space charge transfer effect between the benzene rings of the cyclophane core, a structurally related compound (2) bearing a phenylene vinylene spacer instead of [2.2]paracyclophane, has also been considered (Chart 1). Charge separation and recombination processes in both conjugates have been evaluated by photophysical studies.
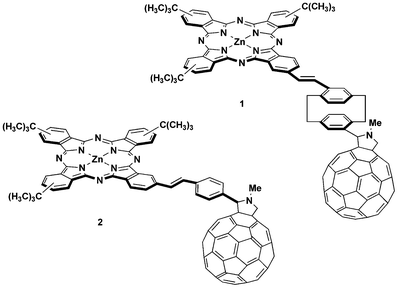 | ||
| Chart 1 tert-Butylphthalocyanine–fullerene donor–acceptor conjugates 1 and 2. | ||
Results and discussion
Synthesis
The synthetic strategy employed for the preparation of donor–acceptor conjugate 1 consisted of two subsequent reactions to attach the phthalocyanine and C60 units to the central pCP spacer: (i) a Heck reaction to attach the iodophthalocyanine 38 to the appropriate pCP derivative 4,2b,5 and (ii) a [3 + 2] cycloaddition of the azomethine ylide generated from the formyl derivative 5 and N-methylglycine to one of the double bonds of the fullerene (Scheme 1). Thus, [2.2]paracyclophane 4 was reacted with phthalocyanine3 in the presence of Pd(OAc)2, Et3N and Bu4N+Br– yielding 64% of compound 5. 1 was finally obtained in 35% yield by Prato reaction with C60 in the presence of sarcosine and ortho-dichlorobenzene (o-DCB) as solvent.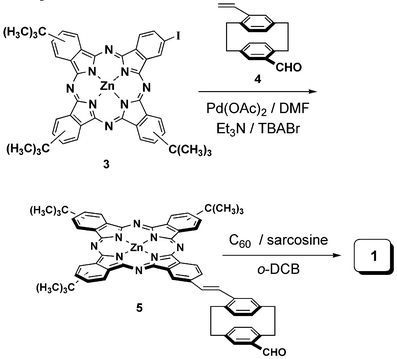 | ||
| Scheme 1 Synthesis of 1. | ||
All the compounds were purified by column chromatography on silica gel, and characterized by mass spectrometry, 1H-NMR, UV–vis and FT-IR spectroscopy. The 1H-NMR spectrum of the formyl derivative 5 shows the typical broad multiplets in the aromatic region, between 7.8 and 8.8 ppm, characteristic of tert-butyl substituted phthalocyanines. The presence of a complex mixture of regioisomers makes the assignment of the peaks in this region of the spectra difficult. Additionally, signals assigned to the pCP or phenylene vinylene groups are also present, together with the aldehyde resonance at 9.88 ppm. 1 also gives a well resolved 1H-NMR spectrum, in which it is possible to identify the pyrrolidine protons between 4.3 and 5.3 ppm, as well as the N–CH3 resonance around 3 ppm (Fig. 1). Assignment of the peaks in the spectrum of 1 was done by COSY NMR experiments. The singlet at 7.1 ppm corresponds to the pCP hydrogen in the ortho position with respect to the fulleropyrrolidine substituent. On the other hand, the proton para to the phthalocyanine unit gives a doublet with an ortho coupling constant at 6.5 ppm. The rest of the cyclophane protons appear as a multiplet around 6.8 ppm. Concerning the pyrrolidine signals, the COSY NMR spectrum shows a clear coupling between resonances at 4.4 and 5.0 ppm. Thus, the other pyrrolidine signal, a singlet at 5.3 ppm, can be assigned to the hydrogen of the asymmetric carbon atom. Interestingly, in 1, two sources of chirality are present. On one hand, the chirality inherent to the asymmetric functionalization of the [2.2]paracyclophane framework and, additionally, an asymmetric carbon atom at the pyrrolidine ring generated by the Prato reaction. However, the 1H-NMR spectrum of 1 did not show any signal splitting due to diastereoisomer mixtures. The MALDI-TOF mass spectrum of dyad 1 shows peaks corresponding to [M – C60]+ fragments, together with the molecular ions [M+] or [M + H]+ as major species.
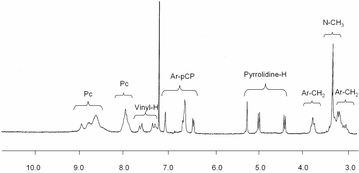 | ||
| Fig. 1 Partial 1H-NMR spectra in CDCl3 of 1. | ||
Photophysical studies
The electronic ground state absorption spectra of donor–acceptor conjugates 1 and 2 are essentially identical to that of ZnPc. In particular, a strong maximum is seen around 685 nm, which is flanked by minor maxima at 350 and 610 nm, which correspond to the Soret and Q-bands, respectively.9 An additional band is seen at 330 nm, which is a relatively good match for a fullerene centered transition.10The first insight into electron donor–acceptor interactions came from fluorescence assays. Typical measurements were conducted in the following way: solutions of ZnPc and either ZnPc–C601 or 2 were adjusted to exhibit identical absorbance of around 0.05 at the excitation wavelength of 610 nm. This ensures exclusive and likewise comparable excitation of the ZnPc chromophore, while avoiding inner filter effects that would result in intrinsic losses of the ZnPc fluorescence. ZnPc—as a reference system—emits strongly in the monitored range, that is, between 620 and 800 nm, from which we have determined an excited state energy of 1.8 eV and a lifetime of 3.1 ns.
Regardless of the particular linker in the ZnPc–C60 systems, that is, phenylene vinylene versus paracyclophane, we noted a strong quenching of the ZnPc centered fluorescence, see Fig. 2. The actual quantum yields are 0.3,11 0.0023, and 0.024 for ZnPc, ZnPc–phenylene vinylene–C60 (2), and ZnPc–paracyclophane–C60 (1) in THF, respectively. Importantly, the general shape of the fluorescence spectra is practically identical to that of ZnPc. Comparing the fluorescence lifetimes of the ZnPc unit allows for the estimation of the rate constant of fluorescence deactivation in the two donor–acceptor systems.
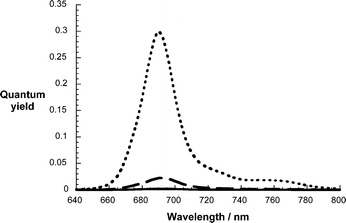 | ||
| Fig. 2 Fluorescence spectra of the reference (i.e., ZnPc, top spectrum) and the donor–acceptor conjugates (i.e., 2, bottom spectrum; 1, middle spectrum) in THF upon 610 nm photoexcitation. | ||
Transient absorption measurements helped to provide conclusive spectroscopic evidence in support of the proposed charge transfer mechanism. To guarantee comparable conditions, especially with the fluorescence experiments, femtosecond transient absorption measurements were conducted by using 670 nm photoexcitation. For ZnPc (Fig. 3) the singlet excited state, which is formed essentially right after the laser pulse, comprises a broad transient maximum at 490 nm followed by bleaching between 610 nm and 685 nm, which corresponds nicely to the singlet–singlet absorptions of ZnPc. This transient decays rather slowly on the time scale of our instrumental detection (i.e., up to 3000 ps) to intersystem cross to the corresponding triplet manifold. In fact, a lifetime of about 3.0 ns was determined for that process. The main spectral feature of the latter is a broad transient with a notable maximum at 500 nm and with two minima at 350 and 680 nm. In the absence of molecular oxygen, the ZnPc triplet excited state returns to the singlet ground state over the course of several microseconds with a rate constant of 3.5 × 104 s–1 in THF. Moreover, the nature of the triplet excited state was confirmed by quenching with molecular oxygen, which happens to be close to diffusion controlled.
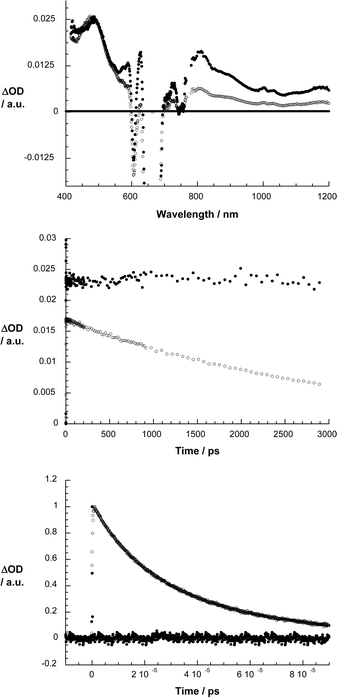 | ||
| Fig. 3 Upper part: differential absorption spectra (visible and near-infrared) obtained upon femtosecond flash photolysis (670 nm) of ZnPc in argon saturated THF with several time delays (i.e., 100 ps – filled circles; 2900 ps – open circles) at room temperature. Center part: time–absorption profiles of the spectra shown above at 500 (●) and 800 nm (○), monitoring the intersystem crossing. Lower part: time–absorption profile of the ZnPc triplet at 500 nm, monitoring the recovery of the singlet ground state in the absence of molecular oxygen (black) and in the presence of molecular oxygen (grey). | ||
As Fig. 4 and 5 illustrate, the same ZnPc singlet excited characteristics—despite the presence of the electron accepting C60—were registered upon 670 nm photoexcitation of 2 and 1. This confirms the successful excitation of the ZnPc component. In contrast to the slow intersystem crossing, which was noted for the ZnPc reference, the ZnPc singlet excited state features now decay rather quickly. The rate constants, which depend on the linkage (i.e., ZnPc–phenylene vinylene–C60 > ZnPc–paracyclophane–C60) and/or the solvent (i.e., toluene < anisole < THF < benzonitrile), reflect the global trends seen in the fluorescence experiments. Simultaneously with the ZnPc singlet excited state decay, the formation of a new transient species evolves with distinct maxima at 520, 840 and 1000 nm. While the maxima at 520 and 840 nm are attributes of the one-electron oxidized ZnPc radical cation,7h,12 the maximum at 1000 nm matches the absorption of the one-electron reduced fullerene radical anion.13 In other words, the transient spectrum features all the fingerprints of an intramolecularly formed radical ion pair state. The quantum yields of charge separation are typically quite high with values of 0.8 and 0.71 for 1 and 2 in THF, respectively.14
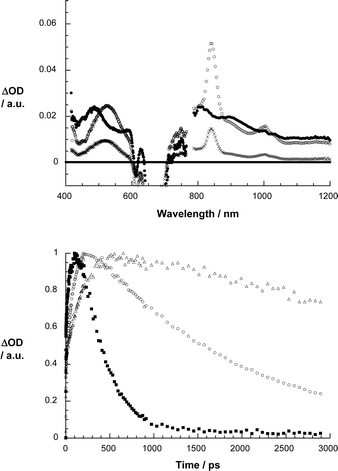 | ||
| Fig. 4 Upper part: differential absorption spectra (visible and near-infrared) obtained upon femtosecond flash photolysis (670 nm) of 1 in argon saturated THF with several time delays (i.e., 1 ps – filled circles; 102 ps – open circles; 2892 ps – crosses) at room temperature. Lower part: time–absorption profiles of the spectra shown above at 845 nm, monitoring the charge separation and charge recombination in anisole (△), THF (○) and benzonitrile (■). | ||
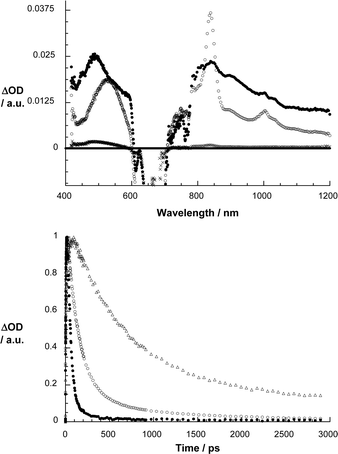 | ||
| Fig. 5 Upper part: differential absorption spectra (visible and near-infrared) obtained upon femtosecond flash photolysis (670 nm) of 2 in argon saturated THF with several time delays (i.e., 1 ps – filled circles; 102 ps – open circles; 2892 ps – crosses) at room temperature. Lower part: time–absorption profiles of the spectra shown above at 845 nm, monitoring the charge separation and charge recombination in anisole (△), THF (○) and benzonitrile (■). | ||
Somewhat lower are the quantum yields in anisole (i.e., 0.62 for ZnPc–paracyclophane–C60; 0.83 for ZnPc–phenylene vinylene–C60) and benzonitrile (i.e., 0.39 for ZnPc–paracyclophane–C60; 0.42 for ZnPc–phenylene vinylene–C60).
Importantly, on our instrumentation femtoseconds time scale of our, namely, up to maximal 3000 ps, both features of the radical ion pair state decay. Again, the impact that the linkages and the solvent polarity exerts on the decay of the radical ion pair state is reflected in Fig. 4 and 5 showing the time profiles of the ZnPc radical cation absorption at 840 nm. Firstly, the lifetime of the radical ion pair state is much longer in the non-polar solvents than in the polar solvents: benzonitrile = 330 ps; THF = 2400 ps; anisole = 26![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 ps; toluene = 458000 ps, for the ZnPc–paracyclophane–C60 dyad.15
000 ps; toluene = 458000 ps, for the ZnPc–paracyclophane–C60 dyad.15
Conclusions
In conclusion, we have demonstrated with success the benefits of integrating a [2.2]paracyclophane buliding block into novel donor–acceptor conjugates. Moreover, we have contrasted the charge transfer features with that of a simpler conjugate, that is, a phenylene vinylene bridged analogue. On one hand, [2.2]paracyclophane ensures the efficient and rapid formation of the radical ion pair state—nearly comparable to that found for phenylene vinylene. On the other hand—and this should be stressed—it exerts an appreciable stabilization of the radical ion pair state before it converts to the singlet ground state.Experimental
Synthesis
The synthesis of dyad 2 has already been reported.16![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C), 1330, 1090, 747. UV–vis (CHCl3), λmax (nm) (log ε): 693 (5.07), 620 (4.36), 351 (4.82), 293 (4.59). MS (MALDI, dithranol), m/z: 1004.3 [M+], 872.2 [M – CHO – C8H8]+, 772.3 [M – CHO – pCP]+.
C), 1487 (C–N), 1089, 747, 527. UV–Vis (CHCl3), λmax (nm) (log ε): 692 (5.16), 621 (4.47), 435 (4.32), 347 (5.04), 256 (5.23). MS (HR-MALDI, dithranol), m/z (C125H61N9Zn): calculated, 1751.4298; found, 1751.4336.
C), 1330, 1090, 747. UV–vis (CHCl3), λmax (nm) (log ε): 693 (5.07), 620 (4.36), 351 (4.82), 293 (4.59). MS (MALDI, dithranol), m/z: 1004.3 [M+], 872.2 [M – CHO – C8H8]+, 772.3 [M – CHO – pCP]+.
C), 1487 (C–N), 1089, 747, 527. UV–Vis (CHCl3), λmax (nm) (log ε): 692 (5.16), 621 (4.47), 435 (4.32), 347 (5.04), 256 (5.23). MS (HR-MALDI, dithranol), m/z (C125H61N9Zn): calculated, 1751.4298; found, 1751.4336.
Photophysics
Optical absorptions were measured with a Cary 5000 UV–vis–NIR spectrometer (Varian). Such measurements, with the exception of the femtosecond and nanosecond photolysis experiments, were carried out using quartz cells with a 1 cm optical path length. Steady-state fluorescence measurements were performed by using a Fluoromax 3 (Horiba Jobin Yvon). Transient absorption experiments—based on nanosecond laser photolysis—were performed with the output of the third harmonics (i.e., 355 nm) coming from a Nd:YAG laser (Brilliant, Quantel). Pulse widths of <5 ns with an energy of 10 mJ were selected. The optical detection is based on a pulsed xenon lamp (XBO 450, Osram), a monochromator (Spectra Pro 2300i, Acton Research), a R928 photomultiplier tube (Hamamatsu Photonics) and a 1 GHz digital oscilloscope (WavePro7100, LeCroy). The laser power of every laser pulse was registered using a bypath with a fast silicon photodiode. The solvents were always of spectroscopic grade. The ns-laser photolysis experiments were performed using 1 cm quartz cells and the solutions were saturated with argon if no other gas saturation is indicated. The femtosecond transient absorption measurements were carried out with a CPA-2001 femtosecond laser (Clark MXR). The excitation wavelength was generated with a NOPA (Clark MXR). The laser output energy was 240 nJ in anisole, 420 nJ in THF and 240 nJ in benzonitrile.Acknowledgements
This work has been supported by the Spanish MEC (CTQ-2005-08933-BQU), the Comunidad de Madrid (S-0505/PPQ/000225), the ESF-MEC (MAT2006-28180-E, SOHYDS), the SFB 583, DFG (GU 517/4-1), FCI, and the Office of Basic Energy Sciences of the U.S. Department of Energy.References
- M. N. Paddon-Row, in Stimulating Concepts in Chemistry, ed. F. Vögtle, J. F. Stoddart and M. Shibasaki, Wiley-VCH, Weinheim, 2000, pp. 267–292 Search PubMed.
- (a) C. R. Treadway, M. G. Hill and J. K. Barton, Chem. Phys., 2002, 281, 409 CrossRef CAS; (b) J. W. Hong, H. Y. Woo, B. Liu and G. C. Bazan, J. Am. Chem. Soc., 2005, 127, 7435 CrossRef CAS.
- (a) Cyclophanes, ed. P. M. Keehn and S. M. Rosenfeld, Academic Press, New York, 1983, vol. 1 and 2 Search PubMed; (b) Cyclophane Chemistry, ed. F. Vogtle, Wiley, New York, 1983 Search PubMed; (c) F. Diederich Cyclophanes, Monographs in Supramolecular Chemistry, ed. J. F. Stoddart, The Royal Society of Chemistry, London, 1991, vol. 2 Search PubMed.
- (a) S. E. Gibson and J. D. Knight, Org. Biomol. Chem., 2003, 1, 1256 RSC; (b) V. Rozenberg, E. Sergeeva and H. Hopf, in Modern Cyclophane Chemistry, ed. R. Gleiter and H. Hopf, Wiley-VCH, Weinheim, 2004, p. 435 Search PubMed; (c) T.-Z. Zhang, L.-X. Dai and X.-L. Hou, Tetrahedron: Asymmetry, 2007, 18, 251 CrossRef CAS; (d) M. Kreis, M. Nieger and S. Braese, J. Organomet. Chem., 2006, 691, 2171 CrossRef CAS.
- (a) J. Zyss, I. Ledoux, S. Volkov, V. Chernyak, S. Mukamel, G. P. Bartholomew and G. C. Bazan, J. Am. Chem. Soc., 2000, 122, 11956 CrossRef CAS; (b) J. B. Maddox, U. Harbola, N. Liu, C. Silien, W. Ho, G. C. Bazan and S. Mukamel, J. Phys. Chem. A, 2006, 110, 6329 CrossRef CAS.
- (a) G. de la Torre, C. G. Claessens and T. Torres, Chem. Commun., 2007, 2000 RSC; (b) Phthalocyanines: Properties and Applications, ed. C. C. Leznoff and A. B. P. Lever, VCH, Weinheim, 1989, 1993, 1996, vol. 1–4 Search PubMed; (c) The Porphyrin Handbook, ed. K. M. Kadish, K. M. Smith and R. Guilard, Academic Press, San Diego, 2003, vol. 15–20 Search PubMed.
- (a) A. Sastre, A. Gouloumis, P. Vazquez, T. Torres, V. Doan, B. J. Schwartz, F. Wudl, L. Echegoyen and J. Rivera, Org. Lett., 1999, 1, 1807 CrossRef CAS; (b) D. M. Guldi, A. Gouloumis, P. Vázquez and T. Torres, Chem. Commun., 2002, 2056 RSC; (c) M. A. Loi, H. Neugebauer, P. Denk, C. J. Brabec, N. S. Sariciftci, A. Gouloumis, P. Vázquez and T. Torres, J. Mater. Chem., 2003, 13, 700 RSC; (d) D. M. Guldi, J. Ramey, M. V. Martínez-Díaz, A. de la Escosura, T. Torres, T. Da Ros and M. Prato, Chem. Commun., 2002, 2774 RSC; (e) M. V. Martínez-Díaz, N. S. Fender, M. S. Rodríguez-Morgade, M. Gómez-López, F. Diederich, L. Echegoyen, J. F. Stoddart and T. Torres, J. Mater. Chem., 2002, 12, 2095 RSC; (f) D. M. Guldi, I. Zilbermann, A. Gouloumis, P. Vázquez and T. Torres, J. Phys. Chem. B, 2004, 108, 18485 CrossRef CAS; (g) A. de la Escosura, M. V. Martínez-Díaz, D. M. Guldi and T. Torres, J. Am. Chem. Soc., 2006, 128, 4112 CrossRef CAS; (h) M. E. El-Khouly, O. Ito, P. M. Smith and F. D'Souza, J. Photochem. Photobiol., C, 2004, 5, 79 CrossRef CAS; (i) D. M. Guldi, A. Gouloumis, P. Vázquez, T. Torres, V. Georgakilas and M. Prato, J. Am. Chem. Soc., 2005, 127, 5811 CrossRef CAS; (j) J. L. Sessler, J. Jayawickramarajah, A. Gouloumis, T. Torres, D. M. Guldi, S. Maldonado and K. J. Stevenson, Chem. Commun., 2005, 1892 RSC; (k) B. Ballesteros, G. de la Torre, T. Torres, G. L. Hug, G. M. A. Rahman and D. M. Guldi, Tetrahedron, 2006, 62, 2097 CrossRef CAS; (l) T. Torres, A. Gouloumis, D. Sanchez-Garcia, J. Jayawickramarajah, W. Seitz, D. M. Guldi and J. L. Sessler, Chem. Commun., 2007, 292 RSC; (m) B. Ballesteros, G. de la Torre, C. Ehli, G. M. A. Rahman, F. Agulló-Rueda, D. M. Guldi and T. Torres, J. Am. Chem. Soc., 2007, 129, 5061 CrossRef CAS.
- E. M. Maya, P. Vázquez and T. Torres, Chem.–Eur. J., 1999, 5, 2004 CrossRef CAS.
- T. H. Tran-Thi, Coord. Chem. Rev., 1997, 160, 53 CrossRef CAS.
- S. H. Gallagher, R. S. Armstrong, P. A. Lay and C. A. Reed, J. Phys. Chem., 1995, 99, 5817 CrossRef CAS.
- P. S. Vincett, E. M. Voigt and K. E. Rieckhoff, J. Chem. Phys., 1971, 55, 4131 CrossRef CAS.
- T. Nyokong, Z. Gasyna and M. J. Stillman, Inorg. Chem., 1987, 26, 548 CrossRef.
- Z. Gasyna, L. Andrews and P. N. Schatz, J. Phys. Chem., 1992, 96, 1525 CrossRef CAS.
- The quantum yield for the charge separation was calculated form the fs-transient absorption experiments taking the ZnPc triplet (Φ = 0.7, ε = 28900 M–1 cm–1 [ I. Carmichael, W. P. Helman and G. L. Hug, J. Phys. Chem. Ref. Data, 1987, 16(2), 239 Search PubMed ]) in THF and the methylene blue chloride triplet in methanol (Φ = 0.5 [ C. Tanielian and C. Wolff, J. Phys. Chem., 1995, 99(24), 9831 CAS ], ε = 14400 M–1 cm–1 [ I. Carmichael, W. P. Helman and G. L. Hug, J. Phys. Chem. Ref. Data, 1987, 16(2), 239 Search PubMed ]) as reference. The extinction coefficient of the one-electron oxidized ZnPc (29000 M–1 cm–1) were taken from Ohno et al. [ T. Ohno, S. Kato and N. N. Lichtin, Bull. Chem. Soc. Jpn., 1982, 55(9), 2753 CrossRef CAS ].
- The lifetime of the radical ion pair state in toluene and anisole was determined in complementary nanosecond experiments. Secondly, the cyclophane linker helps to suppress the charge recombination when compared to the phenylene vinylene linker: ZnPc–phenylene vinylene–C60 = 35000 ps; ZnPc–paracyclophane–C60 = 458000 ps for toluene.
- A. de la Escosura, M. V. Martínez-Díaz, J. Barberá and T. Torres, submitted.
| This journal is © The Royal Society of Chemistry 2008 |