Structural control of the monolayer stability of water-soluble gold nanoparticles†
Sarit S.
Agasti
,
Chang-Cheng
You
,
Palaniappan
Arumugam
and
Vincent M.
Rotello
*
Department of Chemistry, University of Massachusetts, 710 North Pleasant Street, Amherst, MA 01003, USA. E-mail: rotello@chem.umass.edu; Fax: +1-413-5454490; Tel: +1-413-5452058
First published on 15th October 2007
Abstract
The thermodynamic and kinetic stability of three structurally related monolayer-protected gold clusters have been systematically investigated, revealing that the nanoparticles display significantly different stability against thermo- and cyanide-induced decomposition and external thiol agents.
Monolayer protected clusters (MPCs) represent hybrid systems intermediate between molecular systems and macroscopic materials and surfaces. In these systems, the nanometre-scale metal cores of polyhedral shape are covered by a self-assembled monolayer of organic ligands. Stimulated by the pioneering work of Brust et al. in 1994,1thiol -protected gold clusters (Au-MPCs) have evolved to the most widely studied particle–ligand systems so far.2 The optical, electronic and bioinert properties of the gold core combined with the versatile surface functionalization facilitate the applications of this kind of material in both fundamental research and industrial development.3 These functional nanomaterials have shown promising potential in electronics,4 catalysis,5 biosensing,6 and nanomedicine.7
The functionalization of gold clusters is commonly achieved through Brust–Schiffrin reduction1 followed by Murray place-exchange reaction8 with appropriate functional ligands. The incorporation of multiple functionality within the scaffold of a single cluster coupled with a scale comparable with biomacromolecules (e.g.proteins and DNA) provides multiple applications at the interface of biotechnology and nanomaterials. For instance, some recent studies have shown that proper surface functionalization holds great promise in disrupting protein –protein interaction,9 regulating DNA transcription,10 gene transfection,11etc.
Particle stability is of crucial importance for biological applications. On one hand, surface functionality of particles needs to be preserved for proper function, e.g. imaging agents. Under some circumstances, however, the MPC ligands need to be readily replaceable by other ligands for the transformation of surface functionality, for example in drug and gene delivery where the carrier releasing can be triggered by certain cellular agents (e.g.glutathione).12 In this regard, the modulation of monolayer stability of MPCs is central to their environmentally dependent applications.
Densely packed organic ligands confer stability to MPC monolayers. Murray et al. have differentiated the cluster stability by varying the chain length of the surrounding alkanethiol ligands.13 Various peptide ligands14 and dendritic ligands15 have also been used to tune the MPC stability. In a recent study, we have investigated the in situ place-exchange reactions of gold MPCs with a variety of alkanethiols and found that the reaction kinetics are critically dependent on the structure of the external alkanethiols.16 From this result, we envision that minor alterations of ligand structures should allow tuning of MPC stability diversity. To address the issues regarding the monolayer stability in terms of subtle changes in the ligand structure, we prepared three structurally related MPCs (Fig. 1) and investigated their thermodynamic and kinetic stability. We have demonstrated that the steric arrangement of substituents as well as the interaction between adjacent ligands govern the MPC stability.
 | ||
| Fig. 1 Monolayer structure of three structurally related Au-MPCs. | ||
The three nanoparticles were prepared through a single-phase reduction of chloroauric acid by sodium borohydride in the presence of the corresponding thiol ligands according to a procedure described by Huang et al.17Transmission electron microscopy (TEM) revealed that the three Au-MPCs have the same average core diameter of ∼3 nm (see ESI† ). As shown in Fig. 1, the three nanoparticles are capped with the respective thiol ligands of the same backbone, where a three-carbon alkyl chain is followed by a tri(ethylene glycol) segment. The tri(ethylene glycol) moiety endows the resultant nanoparticles with good water solubility. For the three ligands, the only structural difference lies in the presence/absence of a methyl group on the alkyl chain and its substituted position. A small amount of L-tryptophan moieties (ca. 10%) were introduced onto the terminus of the thiol ligands to facilitate monitoring of the ligand-exchange reactions by harnessing their inherent fluorescence properties (for the fluorescence spectra, see Fig. S1, S2 and S3 in ESI† ).
The thermal stability of the MPCs was studied by using thermogravimetric analysis (TGA). The mass change was plotted against temperature to generate the thermogravimetric combustion profiles. Such mass loss reflects the decomposition of thiol ligands from the gold surface. Fig. 2 shows the derivative curves of TGA mass loss for the three MPCs. As can be seen, three MPCs exhibit different thermal decomposition profiles, indicating the diverse thermal stability provided by the surface ligands.18 From the positions of the derivative peaks,‡ it is clear that the well-defined mass loss step for the cluster protected with iso-thiol (NP_Iso) is comparatively higher than its linear analog (NP_Nor). By contrast, NP_Sec shows the least thermal stability although it differs from NP_Iso only by the position of the methyl substituent. These results are consistent with our previous studies on alkyl-functionalized particles,16 indicating that the ligand substituent position distinctly affects the packing performance of ligands on the gold surface and, thereby, the thermal stability of the resulting MPCs.
 | ||
| Fig. 2 Derivative thermogravimetric curves of three MPCs with mixed monolayers. The particles were heated from room temperature (ca. 25 °C) to 500 °C at a rate of 10 °C min–1 under a nitrogen atmosphere and the mass change was recorded as a function of temperature. The inset shows the weight loss versus the temperature. | ||
The chemical stability of the clusters in water was further tested by cyanide-induced decomposition of the gold core. Previous studies on self-assembled monolayers (SAMs) of thiols on a 2D gold surface have shown that the gold underlayer can be corroded by cyanide anions.19 In a similar fashion, thiol ligand protected 3D gold clusters can also be decomposed into a colourless mixture comprising an Au(CN)2– complex, dialkyl disulfides and alkyl cyanides.13 As the accessibility of cyanide anions to the gold core is dependent on the protection of SAMs, the decay rate of gold clusters in the presence of cyanide is correlated with the particle stability.
In our experiments, the nanoparticle surface plasmon band decreased gradually and the colour of the nanoparticle solution changed from red-brown to colourless (for UV/vis spectral changes, see inset in Fig. 3). The absorption changes of 0.2 µmol dm–3 MPCs at 520 nm were monitored over time in the presence of an excess amount of sodium cyanide (3 mmol dm–3). Representative absorption change profiles for three MPCs are depicted in Fig. 3. As the cyanide concentration is 15 times higher than that of gold atoms and is essentially constant during the reaction course, the absorption change data was fitted to a general first-order reaction function. As can be seen in Fig. 3, excellent curve-fitting results are obtained for all three MPC systems, confirming the first-order kinetics. The kinetic constants, including pseudo-first-order and second-order rate constants, are compiled in Table 1.
 | ||
| Fig. 3 UV/vis absorption changes of phosphate buffered saline (PBS) solution of MPCs (0.2 µmol dm–3) at 520 nm in the presence of potassium cyanide (3 mmol dm–3) at 37 °C. The lines represent the best fitting of the data according to the kinetics of first-order reactions. The inset shows the UV/vis spectrum change of NP_Iso during the time course of cyanide decomposition. | ||
| Au-MPCs | k 1/10–3 s–1 | k 2/dm3 mol–1 s–1 |
|---|---|---|
| NP_Sec | 3.58 ± 0.59 | 1.19 ± 0.19 |
| NP_Nor | 1.84 ± 0.20 | 0.61 ± 0.06 |
| NP_Iso | 0.84 ± 0.13 | 0.28 ± 0.04 |
The rate constants in Table 1 reveal that the decomposition rates of MPCs increase in the order of NP_Iso < NP_Nor < NP_Sec. Accordingly, the chemical stability of the MPCs follows an inverse order, which is governed by the protection of the gold core against cyanide attack. Such a chemical stability sequence displays the same trend as that of MPC thermal stability (vide supra). As cyanide anions need first to access the surface gold atoms to destroy the MPCs, the chemical stability of MPCs is obviously related to the organization of ligands on the cluster surface. In the case of NP_Sec, the methyl group adjacent to the mercapto group provides steric hindrance upon interaction with the nanoparticle surface, resulting in the relatively loose packing of ligands within the monolayer.20 Indeed, TGA studies have revealed that NP_Sec possesses the lowest ligand loading among the three MPCs, while NP_Nor and NP_Iso have comparable ligand loadings (see inset in Fig. 2).§ Therefore, the surface gold atoms in NP_Sec are the most accessible by cyanide anions and its decomposion rate is twice that of its linear analog. Interestingly, in comparison with NP_Nor that is covered by linear ligands, the branched ligand-functionalized NP_Iso shows additional stabilization and its decomposition is only half that of NP_Nor. This protection effect should arise from the methyl group in the alkyl chain. It has been well demonstrated that a splay angle exists between the adjacent ligands on spherical nanoclusters.20,21 The methyl group in NP_Iso is expected to block such a splaying space between the adjacent ligands. The methyl group is separated from the mercapto group by two carbon atoms and thereby the steric hindrance as in NP_Sec is significantly reduced. Moreover, the methyl groups in the adjacent ligands can produce favourable van der Waals and hydrophobic interactions, which further improve the chemical stability of NP_Iso. In this context, it is not surprising that NP_Iso shows the lowest cyanide decomposition rate.
The above studies establish the stability of MPCs in both solid and solution states by regarding the favorable and unfavorable steric arrangement of the substituents. Another important issue that needs to be addressed is the MPC stability in the presence of external thiol ligands, which is central to the applications of such kinds of MPCs in biological systems. Therefore, we further explored the in situ place-exchange reaction of the MPCs with dithiothreitol (DTT), dihydrolipoic acid (DHLA), and glutathione (GSH). The latter two compounds are particularly attractive since they are the most abundant thiols in living cells. In the ligand exchange experiments, the reaction kinetics were followed by using a fluorophore displacement approach.22 The tryptophan fluorescence is initially quenched by gold clusters through energy/electron transfer, while fluorescence recovery occurs once the ligands are replaced by other thiols . To avoid the possible interference from precipitation of the clusters or aggregation of hydrophobic HSTrp in aqueous solution, a two-phase system was employed to extract the released tryptophan-containing ligands into the organic phase for signal detection (Fig. 4a).
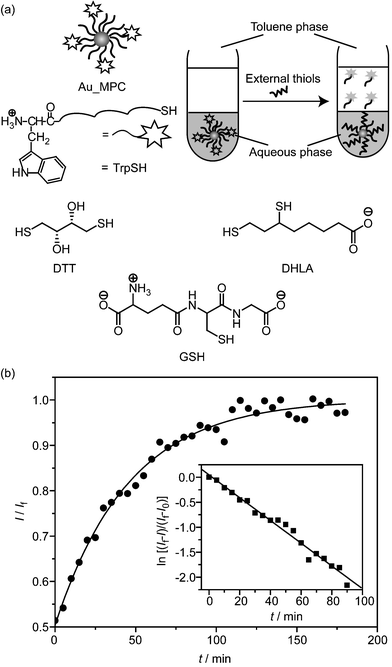 | ||
| Fig. 4 (a) Schematic depiction of surface monolayer exchange experiments in a two-phase system and chemical structure of thiol ligands used in ligand exchange. The released fluorophores were transferred into the toluene phase and the fluorescence intensity of the toluene phase was recorded over time. (b) The normalized fluorescence intensity at 350 nm (λex = 295 nm) for the ligand exchange of NP_Sec (0.2 µmol dm–3) with DHLA (1 mmol dm–3) in PBS at 37 °C. The inset shows the logarithm of fluorescence intensity changes versus time and corresponding linear regression plot (see ESI† for details). | ||
Fig. 4b illustrates a representative fluorescence recovery curve of NP_Sec in the presence of DHLA. As can be seen, the fluorescence intensity at 350 nm increases exponentially overtime. Moreover, the kinetic analysis of the parameter of ln[(If – I)/(If – I0)] as a function of time shows a linear dependence on time (inset in Fig. 4b), indicating the first-order kinetics. The pseudo-first-order and second-order rate constants obtained are listed in Table 2. While not as marked as for the cyanide decomposition, these rates increase substantially in the order of NP_Iso < NP_Nor < NP_Sec. In other words, the kinetics of the ligand displacement correlates again with the monolayer structure of MPCs. As the ligand exchange reaction takes place first at the edges and corners in the clusters,21 this result indicates that the branched ligand in NP_Iso provides also a certain level of protection over these regions.
| Au-MPC | DTT | DHLA | GSH | |||
|---|---|---|---|---|---|---|
| k 1/10–2 min–1 | k 2/dm3 mol–1 min–1 | k 1/10–2 min–1 | k 2/dm3 mol–1 min–1 | k 1/10–2 min–1 | k 2/dm3 mol–1 min–1 | |
| a [DTT] = [DHLA] = 1 mmol dm–3, [GSH] = 5 mmol dm–3. | ||||||
| NP_Sec | 2.86 ± 0.25 | 28.6 ± 2.5 | 2.19 ± 0.02 | 21.9 ± 0.2 | 0.33 ± 0.02 | 0.66 ± 0.04 |
| NP_Nor | 2.38 ± 0.13 | 23.8 ± 1.3 | 1.96 ± 0.11 | 19.6 ± 1.1 | 0.18 ± 0.02 | 0.36 ± 0.04 |
| NP_Iso | 1.76 ± 0.09 | 17.6 ± 0.9 | 1.57 ± 0.07 | 15.7 ± 0.7 | 0.11 ± 0.04 | 0.22 ± 0.08 |
It is interesting to compare the impact of incoming thiols on the place-exchange kinetics. In the reaction with MPCs, the reaction rates decrease in the order of DTT > DHLA ≫ GSH. Structurally, DTT and DHLA carry two mercapto groups but GSH possesses only one. The interaction of the former two thiols with the target gold surface can be stabilized by synergetic bonding, or namely a chelation effect. Therefore, DTT and DHLA show higher activity against ligand exchange. On the other hand, the steric hindrance in close proximity to the thiol group in the case of GSH drastically reduces its activity in ligand-exchange reactions. DTT displays slightly higher activity over DHLA. One plausible explanation is that the molecular size of the former is smaller and it can penetrate the monolayer more easily during the ligand-exchange reaction. Taken together, the three MPCs show interesting activity and/or inertness against ligand exchange, confirming the important role of monolayer packing in cluster stability.
In summary, we have explored the effect of subtle structural changes of surface ligands on the cluster stability. These systematic investigations on thermodynamic and kinetic stability of three structurally related Au-MPCs reveal the modulation of surface ligand packing through the introduction of substituents into the side chain of ligands. The control over monolayer stability by properly tuning ligand structures represents an important step towards successful design of similar systems for future nanotechnology applications. The kinetic studies have demonstrated that the ligand displacement rates by intracellularthiols (e.g.DHLA and GSH) are critically dependent on the monolayer composition. Such activity diversity can be utilized to achieve temporal release of drug molecules from MPC-based drug delivery systems. Tunable access of MPCs through chemical modification of monolayer structures and incorporation of targeting functionalities will further lead to controlled delivery in vivo.
Acknowledgements
This research is supported by the Center for Hierarchical Manufacturing (NSEC, DMI-0531171), MRSEC facilities, and National Institute of Health (NIH, GM077173).Notes and references
- M. Brust, M. Walker, D. Bethell, D. J. Schiffrin and R. Whyman, J. Chem. Soc., Chem. Commun., 1994, 801 RSC.
- M.-C. Daniel and D. Astruc, Chem. Rev., 2004, 104, 293–346 CrossRef CAS.
- (a) K. G. Thomas and P. V. Kamat, Acc. Chem. Res., 2003, 36, 888–898 CrossRef CAS; (b) L. Pasquato, P. Pengo and P. Scrimin, Supramol. Chem., 2005, 17, 163–171 CrossRef CAS; (c) S. Eustis and M. A. El-Sayed, Chem. Soc. Rev., 2006, 35, 209–217 RSC; (d) C.-C. You, A. Verma and V. M. Rotello, Soft Matter, 2006, 2, 190–204 RSC.
- (a) D. I. Gittins, D. Bethell, D. J. Schiffrin and R. J. Nichols, Nature, 2000, 408, 67–69 CrossRef CAS.
- (a) H. Li, Y. Y. Luk and M. Mrksich, Langmuir, 1999, 15, 4957–4959 CrossRef CAS; (b) P. Pengo, L. Baltzer, L. Pasquato and P. Scrimin, Angew. Chem., Int. Ed., 2007, 46, 400–404 CrossRef CAS.
- C.-C. You, O. R. Miranda, B. Gider, P. S. Ghosh, I. B. Kim, B. Erdogan, S. A. Krovi, U. H. F. Bunz and V. M. Rotello, Nat. Nanotechnol., 2007, 2, 318–323 Search PubMed.
- (a) I. H. El-Sayed, X. H. Huang and M. A. El-Sayed, Nano Lett., 2005, 5, 829–834 CrossRef CAS; (b) P. K. Jain, I. H. El-Sayed and M. A. El-Sayed, Nano Today, 2007, 2, 18–29 Search PubMed.
- A. C. Templeton, M. P. Wuelfing and R. W. Murray, Acc. Chem. Res., 2000, 33, 27–36 CrossRef CAS.
- H. Bayraktar, P. S. Ghosh, V. M. Rotello and M. J. Knapp, Chem. Commun., 2006, 1390–1392 RSC.
- (a) C. M. McIntosh, E. A. Esposito, A. K. Boal, J. M. Simard, C. T. Martin and V. M. Rotello, J. Am. Chem. Soc., 2001, 123, 7626–7629 CrossRef CAS; (b) G. Han, C.-C. You, B. J. Kim, R. S. Turingan, N. S. Forbes, C. T. Martin and V. M. Rotello, Angew. Chem., Int. Ed., 2006, 45, 3165–3169 CrossRef CAS; (c) C. Agbasi-Proter, J. Ryman-Rasmussen, S. Franzen and D. Feldheim, Bioconjugate Chem., 2006, 17, 1178–1183 CrossRef.
- (a) K. K. Sandhu, C. M. McIntosh, J. M. Simard, S. W. Smith and V. M. Rotello, Bioconjugate Chem., 2002, 13, 3–6 CrossRef CAS; (b) N. L. Rosi, D. A. Giljohann, C. S. Thaxton, A. K. R. Lytton-Jean, M. S. Han and C. A. Mirkin, Science, 2006, 312, 1027–1030 CrossRef CAS.
- (a) G. Han, N. S. Chari, A. Verma, R. Hong, C. T. Martin and V. M. Rotello, Bioconjugate Chem., 2005, 16, 1356–1359 CrossRef CAS; (b) R. Hong, G. Han, J. M. Fernandez, B. J. Kim, N. S. Forbes and V. M. Rotello, J. Am. Chem. Soc., 2006, 128, 1078–1079 CrossRef CAS.
- A. C. Templeton, M. J. Hostetler, C. T. Kraft and R. W. Murray, J. Am. Chem. Soc., 1998, 120, 1906–1911 CrossRef CAS.
- L. Fabris, S. Antonello, L. Armelao, R. L. Donkers, F. Polo, C. Toniolo and F. Maran, J. Am. Chem. Soc., 2006, 128, 326–336 CrossRef CAS.
- C. S. Love, I. Ashworth, C. Brennan, V. Chechik and D. K. Smith, J. Colloid Interface Sci., 2006, 302, 178–186 CrossRef CAS.
- R. Hong, J. M. Fernández, H. Nakade, R. Arvizo, T. Emrick and V. M. Rotello, Chem. Commun., 2006, 2347–2349 RSC.
- M. Zheng, F. Davidson and X. Y. Huang, J. Am. Chem. Soc., 2003, 125, 7790–7791 CrossRef.
- R. H. Terrill, T. A. Postlethwaite, C. H. Chen, C. D. Poon, A. Terzis, A. D. Chen, J. E. Hutchison, M. R. Clark, G. Wignall, J. D. Londono, R. Superfine, M. Falvo, C. S. Johnson, E. T. Samulski and R. W. Murray, J. Am. Chem. Soc., 1995, 117, 12537–12548 CrossRef CAS.
- (a) A. Kumar and G. M. Whitesides, Appl. Phys. Lett., 1993, 63, 2002–2004 CrossRef; (b) C. S. Weisbecker, M. V. Merritt and G. M. Whitesides, Langmuir, 1996, 12, 3763–3772 CrossRef CAS.
- J. C. Love, L. A. Estroff, J. K. Kriebel, R. G. Nuzzo and G. M. Whitesides, Chem. Rev., 2005, 105, 1103–1170 CrossRef CAS.
- A. M. Jackson, Y. Hu, P. J. Silva and F. Stellacci, J. Am. Chem. Soc., 2006, 128, 11135–11149 CrossRef CAS.
- (a) M. J. Hostetler, A. C. Templeton and R. W. Murray, Langmuir, 1999, 15, 3782–3789 CrossRef CAS; (b) M. Montalti, L. Prodi, N. Zaccheroni, R. Baxter, G. Teobaldi and F. Zerbetto, Langmuir, 2003, 19, 5172–5174 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Synthesis and characterization of the ligands, the preparation of nanoparticles, and experimental procedures. See DOI: 10.1039/b711434f |
| ‡ Repeated experiments indicate a reproducibility of ±3 °C. |
| § According to the TGA and TEM results, it is estimated that ligand densities of NP_Sec, NP_Nor and NP_Iso are 3.1, 4.5, and 4.3 ligand nm–2, respectively. The relatively lower ligand density of NP_Sec is not surprising as the steric repulsion between the methyl groups adjacent to the surface bound sulfur atom would lead to a bigger footprint of the ligands. |
| This journal is © The Royal Society of Chemistry 2008 |