A simple combined sample–standard bracketing and inter-element correction procedure for accurate mass bias correction and precise Zn and Cu isotope ratio measurements
Kate
Peel
a,
Dominik
Weiss
*ab,
John
Chapman
a,
Tim
Arnold
a and
Barry
Coles
ab
aEarth Science and Engineering, Imperial College London, London, UK SW7 2AZ. E-mail: d.weiss@imperial.ac.uk; Fax: +44 (0)207 594 7747; Tel: +44 (0)207 594 6383
bMineralogy, The Natural History Museum, London, UK SW7 5PD
First published on 24th August 2007
Abstract
The modified sample–standard bracketing method (m-SSB) combines a sample–standard bracketing and an inter-element correction procedure to account for instrumental mass fractionation during multi-collector ICP-MS measurements. Precisions for Cu and Zn isotopes in plant and experimental granite leachate samples are in line with those obtained using other mass bias correction techniques. In addition, the inherent temporal drift of mass bias during the analytical session and the empirical linear relationship between dopant and analyte are used to apply independent correction schemes that rigorously check the accuracy of mass bias correction using m-SSB. Consequently, a very robust isotope data set is obtained. We further suggest the use of a matrix-element spike in inter-element doped standards to increase the mass bias variability. This improves the quality of the empirical relationship between dopant and analyte and enables cross-checking of the m-SSB method when instrumental mass bias is stable.
1. Introduction
The application of Zn and Cu stable isotope ratio analysis has great promise for addressing fundamental problems in many scientific disciplines.1 The key analytical technique is multi-collector inductively-coupled plasma mass spectrometry (MC-ICP-MS) as it combines simultaneous collection of the ion beams of the different isotopes with a high temperature plasma source, therefore overcoming problems associated with the high first ionization energy of these two d-block elements.Fundamental to any application are robust analytical techniques, with mass bias being arguably the prime obstacle to precise and accurate isotope ratio determination.2 Mass bias varies significantly on a temporal scale of seconds to days3,4 and incorporates instrumental mass discrimination and non-spectral matrix effects. The causes of this phenomenon are not fully understood, but probably arise from a combination of supersonic expansion of the neutral plasma into the vacuum between the sample and skimmer cones5 and space-charge effects in the wake of the skimmer cone.6
There is no current consensus on how best to deal with the problem of mass bias,7 and various correction methods are used including double spike techniques, direct sample–standard bracketing (SSB), or the use of an internal standard element. A double spike method has been developed effectively for Zn analysis,8 but as four isotopes are required it is unsuitable for Cu analysis. The direct standard–sample bracketing method (hereafter termed d-SSB) involves the measurement of the isotope ratio of the analyte element in standard solutions run between samples and has been successfully applied to the isotopic analysis of Cu and Zn in simple matrices such as pure mineral digests or industrial standards.9 However, it does not quantify the fractionation effect itself and matrix-induced mass bias cannot be corrected for. This problem is addressed by doping both sample and standard using an element with isotopes of similar mass. Using the known or assumed isotopic composition of the dopant and the relationship fdopant/fanalyte, derived from plotting the ratios in natural log spaces, the mass bias of the analyte can be quantified using the exponential law. Corrected analyte isotope ratios in samples and standards are then used with the SSB method. This approach, termed en-SSB hereafter, has been applied widely for Zn and Cu isotope measurements.10–12 Alternatively, the intercepts of linear regression lines of analyte and dopant ratios of standards and samples in ln–ln space are determined. The gradient for both samples and standards is identical, while the difference in intercept values is the difference in isotopic composition between samples and standard. This ‘empirical external normalization’ method, hereafter termed EEN, has been developed by Maréchal and co-workers.3 Baxter and co-workers recently developed a revised exponential model for mass bias correction using an internal standard.7
Problems with the en-SSB and EEN methods arise as they depend on various assumptions: first, that the mass bias relationship (fdopant/fanalyte) is constant over the analytical session; second, that the relationship, established from measurements of standards, also holds for samples (i.e., (fCu/fZn)standard ≈ (fCu/fZn)sample); and third, that the variation of mass bias of the standards has enough spread that a good linear regression can be calculated. All of these assumptions can break down during an analytical session,13 potentially leading to inaccurate and low precision analyses. To address the latter, Archer and Vance (2004) proposed the addition of a matrix element to induce mass bias variation and thus the spread of the linear regression line that defines the mass bias relationships.14 This technique has previously been applied to various isotope systems.11,15,16
In 2004 Mason et al.4 developed the so-called modified sample–standard bracketing technique (m-SSB) to account for changes in mass bias that are not adequately quantified by the d-SSB. The m-SSB technique is a combined sample–standard bracketing and inter-element correction procedure, whereby samples and standards are doped and the δ-values (deviation of the isotope ratio of the sample relative to that of a reference standard expressed as parts per mil, see below) calculated for the dopant are subtracted from the measured δ-values of the analyte, using the assumption that fCu ≈ fZn. Using a suite of industrial standards, they showed that calculated δ-values using the EEN and m-SSB techniques agreed well within error and that the precision on industrial standards improved from ±0.38‰ to ±0.049‰ (2SD), providing empirical evidence that the modification was successful, even though fCu ≠ fZn.
The aim of this paper is two-fold. First, we compare m-SSB calculated δ-values with δ-values derived from the same analytical session using the en-SSB and EEN approaches. This comparison with a second and third independent mass bias correction scheme is an effective way to assure data quality. In this way we show that the m-SSB method produces precise and accurate δ-values for Zn and Cu for a suite of materials with complex environmental matrices, derived from experiments conducted in our laboratory. Second, as en-SSB and EEN depend on the establishment of a significant mass bias spread, not always guaranteed under dry plasma conditions, we show that the mass bias spread within an analytical session can be increased by spiking doped standards with Pb as a matrix element. Thus, bracketing samples and standards with a set of Pb-spiked standards during an analytical session enables combination of m-SSB with en-SSB or EEN, even if the instrumental mass bias is very stable, without significant loss of sample throughput.
2. Experimental
2.1. Instrumentation
All isotopic measurements were made using the IsoProbe MC-ICP-MS (Thermo Scientific, Manchester, UK) connected to a Cetac Aridus desolvating nebuliser (Cetac Technologies, Omaha, USA). Operational settings are given in Table 1. The IsoProbe was run in ‘soft extraction’ mode,14 eliminating instrumental Ni interferences. Instrumental background and acid matrix blank corrections were performed using on-peak blank measurements taken before every sample and standard. Sample and standard measurements were made by taking 50 five second integrations. The internal precision for each measurement was better than 20 ppm (SE at 95% confidence level) for all ratios. A full description of the analytical protocol development is given elsewhere.4,17| Instrument parameters | |
| Coolant Ar flow | 14 l min–1 |
| Auxiliary Ar flow | 1.0–1.4 l min–1 |
| Nebuliser Ar flow | 0.9–1.1 l min–1 |
| Collision cell Ar flow | 1.2–2.0 ml min–1 |
| Extraction voltage (soft) | +10–20 V |
| Torch power | 1336 W |
| Cone material | Ni |
| Aridus parameters | |
| Spray chamber temperature | +70 °C |
| Desolvator temperature | +160 °C |
| Ar sweep gas flow | 2.5–3.5 l min–1 |
| Sample uptake rate | ca. 70 µl min–1 |
| Sensitivity | Typically 7 V µg–1 ml–1 for Cu and Zn |
2.2. Samples and sample preparation
All solutions for MC-ICP-MS measurements were prepared in 0.1 M HNO3 using >18.2 MΩ cm–1 H2O. In-house standards, named IMP Cu and IMP Zn, were prepared by digesting Johnson–Matthey Purotronic Cu (batch W1508) and Zn (batch NH27040) metal foil using concentrated Supra Pure HNO3 (Merck). Industrial single element solutions used as samples (denoted as Romil Cu and Romil Zn) were made up from single element solutions (Romil Ltd, Cambridge, UK). Plant samples used were Ryegrass BCR 281, Peach Leaves GBW 08501 and an in-house standard HRM-14. Geological samples used were leachates of a biotite granite using 0.5 M HCl and 5 µM oxalic acid over a period of 1–168 hours. The plants were digested using a HF–HNO3–H2O2 acid mixture and microwave oven. Copper and Zn were separated from the matrices using anion exchange chromatography methods.18Strontium and Pb plasma emission standards (BDH) and Spec-pure U ICP-MS standard (Alfa Aesar) were used to spike the solutions.2.3. Experimental set-up
Samples and standard solutions were concentration matched to within ±10% at approximately 2 µg ml–1 and spiked with the dopant (Cu if Zn was the analyte or Zn if Cu was the analyte). The final dopant concentration was matched to sample concentration giving an element/dopant ratio of 1. The dopant concentration and isotopic composition was thus identical in the samples and standards. An analytical session comprised duplicate analyses (denoted as run A and B) of 12 samples and 12 standards measured alternately. The session lasted approximately 10 hours (5 h per run).To develop the chemically induced mass bias, we investigated first the effect of element and concentration, using Sr, U and Pb as spikes in a concentration series of 0, 3, 15, 30, 45 and 60 µg ml–1. After the initial experiments a series of six standards were spiked with Pb to concentrations of 0, 3, 10, 15, 25 and 40 µg ml–1 and the ‘calibration standards’ were measured three times during an analytical session: at the beginning (series 1), half way through (series 2) and at the end (series 3). Series 1 and 3 were thus bracketing all standards and samples used for the m-SSB method. The long-term reproducibility on our IsoProbe is estimated at ±0.1‰ for δ66Zn and δ65Cu from repeated measurements of Romil Zn and Romil Cu over a period of three years.
2.4. Corrections for mass bias
For the m-SSB method, we calculated first the δ-value of the analyte using the ratio measurements of samples and the two standards measured immediately before and after the sample. Using Zn as the analyte: | (1) |
| δ66Zntrue = δ66Znmeasured – δ65Cumeasured | (2) |
 | (3) |
 | (4) |
 | (5) |
Finally, for the EEN method we plotted the natural logarithms of the measured isotope ratios of analyte and dopant of the standards and determined graphically the gradient and intercept (c1) of the regression line. As gradients are identical for samples and standards, i.e., (fZn/fCu)standard = (fZn/fCu)sample, the intercept of each sample (c2) was calculated using the gradient of the standard regression line. The difference in the intercepts, Δc = c1 – c2, is a function of the difference in isotopic composition between the sample and the standard. The δ-values are then calculated using:
| δ66Zn = 1000(eΔc – 1) | (6) |
3. Results and discussion
3.1. Initial observations
Fig. 1 shows Cu isotope ratios (as analyte) taken during a SSB measurement session of twelve samples: sample 1 was an industrial single element standard (Romil Cu) and the remaining samples were leachates of granite (with HCl or oxalic acid). The calculated SSB δ65Cu values are given in Table 2. All the solutions were doped with IMP Zn for the inter-element corrections. Sample 1 shows no change in the Zn dopant isotope ratio between sample and bracketing standards (δ66Zn within error of zero). Other samples show at times a significant change in Zn isotope ratio (up to 1.0‰ for δ66Zn in sample 6). This pattern is reproduced during runs A and B, suggesting a sample-specific matrix induced mass bias effect. Mass scans of the solutions prior to the measurements did not indicate the presence of any isobaric or polyatomic interferences. Sample specific shifts are superimposed over a systematic drift of mass bias over the analytical session (difference in ratio between the first and last measured standards is 2.6‰).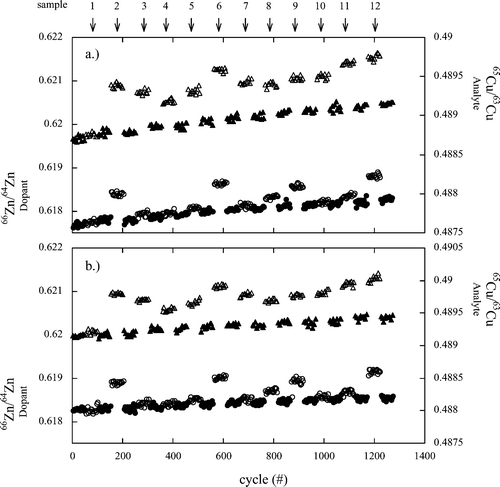 | ||
| Fig. 1 Copper isotope ratios (analyte, shown as triangles in upper sections of plots (a) and (b) and Zn isotope ratios (dopant, circles in lower sections of plots a and b) of samples (open symbols) and bracketing standards (closed symbols) measured in a SSB session consisting of twelve samples. Plot (a) shows run A and plot (b) shows run B (i.e., the replicate). Sample 1 is an industrial single element standard (Romil Cu) and samples 2 to 12 are granite leachates. | ||
| Sample type | f Zn | f Cu | δ 65Cud-SSB | δ 66Zn | δ 65Cum-SSB | δ 65Cuen-SSB | δ 65CuEEN | Δ65Cum-SSB – dSSB | Δ65Cum-SSB – enSSB | Δ65Cum-SSB – EEN | |
|---|---|---|---|---|---|---|---|---|---|---|---|
| Run A | |||||||||||
| 1 | Industrial standard (Romil Cu) | 2.404 | 2.304 | 0.021 | 0.012 | 0.009 | 0.008 | 0.066 | –0.01 | 0.00 | –0.06 |
| 2 | Granite leachate A 96 h1 | 2.438 | 2.336 | 1.258 | 0.975 | 0.282 | 0.192 | 0.275 | –0.98 | 0.09 | 0.01 |
| 3 | Granite leachate G 168 h | 2.414 | 2.312 | 1.002 | 0.152 | 0.850 | 0.836 | 0.900 | –0.15 | 0.01 | –0.05 |
| 4 | Granite leachate A 1 h | 2.415 | 2.314 | 0.645 | 0.114 | 0.531 | 0.521 | 0.577 | –0.11 | 0.01 | –0.05 |
| 5 | Granite leachate A 48 h | 2.421 | 2.320 | 0.828 | 0.195 | 0.633 | 0.615 | 0.648 | –0.20 | 0.02 | –0.01 |
| 6 | Granite leachate H 96 h | 2.450 | 2.348 | 1.286 | 0.999 | 0.287 | 0.194 | 0.268 | –1.00 | 0.09 | 0.02 |
| 7 | Granite leachate A 2 h | 2.424 | 2.322 | 0.939 | 0.129 | 0.810 | 0.798 | 0.843 | –0.13 | 0.01 | –0.03 |
| 8 | Granite leachate A 168 h | 2.435 | 2.333 | 0.771 | 0.413 | 0.358 | 0.320 | 0.382 | –0.41 | 0.04 | –0.02 |
| 9 | Granite leachate H 168 h | 2.447 | 2.345 | 0.840 | 0.721 | 0.118 | 0.052 | 0.130 | –0.72 | 0.07 | –0.01 |
| 10 | Granite leachate A 10 h | 2.428 | 2.326 | 0.868 | 0.074 | 0.793 | 0.786 | 0.833 | –0.07 | 0.01 | –0.04 |
| 11 | Granite leachate B 168 h | 2.436 | 2.334 | 1.154 | 0.263 | 0.891 | 0.866 | 0.906 | –0.26 | 0.02 | –0.01 |
| 12 | Granite leachate A 96 h 2 | 2.460 | 2.357 | 1.265 | 0.926 | 0.339 | 0.253 | 0.289 | –0.93 | 0.09 | 0.05 |
| Run B | |||||||||||
| 1 | Industrial standard (Romil Cu) | 2.432 | 2.330 | 0.112 | 0.011 | 0.101 | 0.100 | 0.095 | –0.01 | 0.00 | 0.01 |
| 2 | Granite leachate A 96 h 1 | 2.464 | 2.361 | 1.278 | 0.968 | 0.310 | 0.221 | 0.257 | –0.97 | 0.09 | 0.05 |
| 3 | Granite leachate G 168 h | 2.439 | 2.337 | 0.998 | 0.154 | 0.844 | 0.829 | 0.872 | –0.15 | 0.01 | –0.03 |
| 4 | Granite leachate A 1 h | 2.439 | 2.336 | 0.625 | 0.083 | 0.541 | 0.534 | 0.592 | –0.08 | 0.01 | –0.05 |
| 5 | Granite leachate A 48 h | 2.444 | 2.342 | 0.793 | 0.206 | 0.587 | 0.568 | 0.626 | –0.21 | 0.02 | –0.04 |
| 6 | Granite leachate H 96 h | 2.471 | 2.367 | 1.233 | 0.982 | 0.252 | 0.161 | 0.256 | –0.98 | 0.09 | 0.00 |
| 7 | Granite leachate A 2 h | 2.445 | 2.343 | 0.943 | 0.147 | 0.795 | 0.782 | 0.833 | –0.15 | 0.01 | –0.04 |
| 8 | Granite leachate A 168 h | 2.454 | 2.351 | 0.704 | 0.378 | 0.326 | 0.291 | 0.346 | –0.38 | 0.03 | –0.02 |
| 9 | Granite leachate H 168 h | 2.466 | 2.363 | 0.845 | 0.718 | 0.127 | 0.061 | 0.128 | –0.72 | 0.07 | 0.00 |
| 10 | Granite leachate A 10 h | 2.447 | 2.345 | 0.891 | 0.103 | 0.788 | 0.779 | 0.825 | –0.10 | 0.01 | –0.04 |
| 11 | Granite leachate B 168 h | 2.446 | 2.343 | 1.133 | 0.024 | 1.109 | 1.107 | 1.182 | –0.02 | 0.00 | –0.07 |
| 12 | Granite leachate A 96 h 2 | 2.478 | 2.374 | 1.225 | 0.984 | 0.242 | 0.151 | 0.269 | –0.98 | 0.09 | –0.03 |
3.2. Modified sample–standard bracketing: accuracy of mass bias correction and precision
Fig. 2 shows Cu and Zn isotope ratios in ln–ln space, measured in the bracketing standard solutions during Cu isotope measurements of granite leachates (same samples in Fig. 2(b) and Fig. 1) and Zn isotope measurements of plant samples (Fig. 2(a)). Two important observations are made: first, the spread of ln(66Zn/64Zn) and ln(65Cu/63Cu) is sufficient to achieve a good linear correlation, and second, the relationship of fCu/fZn (eqn (3)) is constant during each analytical session though significantly different between the analytical sessions and from the theoretical slope of 0.9840 for the exponential law. | ||
| Fig. 2 ln(65Cu/63Cu) versus ln(66Zn/64Zn) for doped standards measured during δ66Zn determinations of plant digests (plot (a)) and δ65Cu determinations of granite leachates (plot (b)). The mass bias relationship fCu/fZn is determined using a least-squares regression of each data set. | ||
Table 2 shows the calculated mass bias factors fCu and fZn, δ66Zn relative to IMP Zn, and δ65Cu of leachates relative to IMP Cu, calculated using the various mass bias correction approaches. Also calculated is the difference between the sample δ65Cu ratios derived from the different mass bias correction approaches, using Δ65Cum-SSB –x = δ65Cum-SSB – x – δ65Cux, where x is d-SSB, EEN or en-SSB. δ65Cu values calculated using the m-SSB, en-SSB and EEN approaches are identical within the long-term precision of ±0.1‰. Any inaccuracies associated with the assumptions made using the m-SSB (i.e., fCu ≈ fZn), EEN (i.e., (fCu/fZn)standards ≈ (fCu/fZn)samples) and en-SSB (that the exponential law is applicable) are insignificant relative to the levels of reproducibility attained with present day MC-ICP-MS instruments. This observation is also true of δ66Zn values measured in plant samples (Table 3).
| Sample | Type | δ 65Cud-SSB | ±2σ | δ 65Cum-SSB | ±2σ | δ 65Cuen-SSB | ±2σ | δ 65CuEEN | ±2σ |
|---|---|---|---|---|---|---|---|---|---|
| Granite acid leachates | |||||||||
| 1 | Industrial standard (Romil Cu) | 0.07 | 0.13 | 0.05 | 0.13 | 0.06 | 0.13 | 0.08 | 0.04 |
| 2 | Granite leachate A 96 h 1 | 1.27 | 0.03 | 0.21 | 0.04 | 0.32 | 0.04 | 0.27 | 0.03 |
| 3 | Granite leachate G 168 h | 1.00 | 0.01 | 0.83 | 0.01 | 0.85 | 0.01 | 0.89 | 0.04 |
| 4 | Granite leachate A 1 h | 0.63 | 0.03 | 0.53 | 0.02 | 0.54 | 0.01 | 0.58 | 0.02 |
| 5 | Granite leachate A 48 h | 0.81 | 0.05 | 0.59 | 0.06 | 0.62 | 0.06 | 0.64 | 0.03 |
| 6 | Granite leachate H 96 h | 1.26 | 0.07 | 0.18 | 0.05 | 0.30 | 0.05 | 0.26 | 0.02 |
| 7 | Granite leachate A 2 h | 0.94 | 0.00 | 0.79 | 0.02 | 0.81 | 0.02 | 0.84 | 0.01 |
| 8 | Granite leachate A 168 h | 0.74 | 0.09 | 0.31 | 0.04 | 0.35 | 0.05 | 0.36 | 0.05 |
| 9 | Granite leachate H 168 h | 0.84 | 0.01 | 0.06 | 0.01 | 0.14 | 0.01 | 0.13 | 0.00 |
| 10 | Granite leachate A 10 h | 0.88 | 0.03 | 0.78 | 0.01 | 0.79 | 0.01 | 0.83 | 0.01 |
| 11 | Granite leachate B 168 h | 1.14 | 0.03 | 0.99 | 0.33 | 1.00 | 0.30 | 1.04 | 0.38 |
| 12 | Granite leachate A 96 h 2 | 1.24 | 0.06 | 0.20 | 0.14 | 0.32 | 0.14 | 0.28 | 0.03 |
| Average | 0.05 | 0.07 | 0.07 | 0.06 | |||||
| Plant samples (δ66Zn) | |||||||||
| 1 | Peach leaves GBW 08501 | 1.23 | 0.08 | 0.91 | 0.03 | 0.93 | 0.06 | 0.91 | 0.06 |
| 8 | Peach leaves GBW 08501 | 1.18 | 0.23 | 1.08 | 0.10 | 1.07 | 0.12 | 1.05 | 0.13 |
| 3 | Peach leaves GBW 08501 | 1.32 | 0.15 | 1.46 | 0.09 | 1.47 | 0.10 | 1.43 | 0.05 |
| 5 | Peach leaves GBW 08501 | 1.26 | 0.2 | 1.29 | 0.12 | 1.27 | 0.10 | 1.22 | 0.11 |
| 2 | Ryegrass BCR 281 | 0.90 | 0.17 | 0.74 | 0.05 | 0.73 | 0.05 | 0.68 | 0.10 |
| 10 | Ryegrass BCR 281 | 1.09 | 0.19 | 0.83 | 0.18 | 0.82 | 0.18 | 0.81 | 0.10 |
| 11 | Ryegrass BCR 281 | 0.90 | 0.08 | 0.69 | 0.10 | 0.68 | 0.09 | 0.65 | 0.13 |
| 4 | In-house HRM-14 | 1.14 | 0.16 | 0.80 | 0.21 | 0.77 | 0.23 | 0.73 | 0.24 |
| 6 | In-house HRM-14 | 0.85 | 0.27 | 0.74 | 0.23 | 0.71 | 0.25 | 0.65 | 0.18 |
| 7 | In-house HRM-14 | 0.88 | 0.18 | 0.64 | 0.08 | 0.64 | 0.09 | 0.60 | 0.05 |
| 9 | In-house HRM-14 | 0.95 | 0.02 | 0.84 | 0.17 | 0.84 | 0.17 | 0.83 | 0.10 |
| Average | 0.16 | 0.12 | 0.13 | 0.11 | |||||
Table 3 shows the calculated ±2σ from replicate analyses (i.e., runs A and B) of sample aliquots during δ65Cu and δ66Zn determinations of granite leachates and plants, respectively. For the leachates, mean precision is ±0.07‰ or better with all correction methods. For plant samples, the mean precision improves slightly for the δ66Zn using m-SSB, en-SSB and EEN compared to d-SSB: however, it is poorer than for the leachates. This likely reflects the complex plant matrix affecting anion-exchange separation procedures and mass spectrometry.21 The variation of δ66Zn between the four GBW and the four in-house plant samples likely reflects natural fractionation within the plant and the quality of milling and homogenisation of the original sample material. Precisions achieved are in line with reports from other laboratories and/or different instruments3,4,12,14 and the error is at least 20 times less than natural variability.22
The mass fractionation coefficients measured on the IsoProbe (2.34 ± 0.02 for fCu and 2.44 ± 0.02 for fZn) are similar to those measured on another MC-ICP-MS instrument, the Plasma 54 (2.1 ± 0.1).3 We also find that fZn is systematically and significantly higher than fCu (i.e., fCu ≠ fZn).
3.3. Generation of variable mass bias for the fCu/fZn calibration: identifying the best spike element
Fig. 3 shows the effect of spiking the Cu/Zn calibration standards with Sr, Pb or U. When spiked with Sr the range of ln(65Cu/63Cu) values is approximately doubled from the typical range obtained with ‘pure’ standards. The range of ln(66Zn/64Zn) values increased approximately five-fold. However, in contrast to previous observations,14 Sr-spiked measurements do not form a correlated linear trend and the relationship between the mass bias factors for Cu and Zn breaks down (R2 = 0.135). This might be explained by the difference in instrumental settings.23 Calibration standards spiked with U or Pb form well-correlated linear arrays, with R2 = 0.989 and 0.993, respectively. The range of mass bias increases by approximately 25-fold for ln(65Cu/63Cu) and 12-fold for ln(66Zn/64Zn), for both U and Pb. The effects of the Pb spike on the mass bias of the calibration standard series were confirmed with other industrial single element Cu and Zn standards (data not shown). | ||
| Fig. 3 ln(65Cu/63Cu) versus ln(66Zn/64Zn) for two series of six IMP Cu/Zn standards (2 µg ml–1) spiked with Sr (closed circles), U (open circles) and Pb (open diamonds) at a range of concentrations from 0 to 60 µg ml–1. With the Sr-spike, the spread of data points is increased with respect to pure standards but does not produce a correlated linear trend. Uranium and Pb increase the spread of data further and form a linear relationship. | ||
Fig. 4 shows the effect of matrix concentration on the extent of mass bias variation, expressed in per mil (‰), relative to the unspiked standards. The Pb spike causes the largest deviation for both Cu and Zn, with increases of up to ∼8‰ for δ65Cu and ∼9‰ for δ66Zn at Pb concentrations of 60 µg ml–1. The U spike causes an increase of 4–5‰ at 60 µg ml–1, whereas the Sr matrix effect appears to be comparatively small at ∼1‰.
 | ||
| Fig. 4 Effect of concentration of Sr (closed circles), Pb (open diamonds) and U (open circles) spikes in IMP Cu/Zn standards (2 µg ml–1) on measured 65Cu/63Cu (plot (a)) and 66Zn/64Zn (plot (b)) expressed as per mil relative to the unspiked standards. | ||
We note a trend between ionisation energy of the spike element and induced mass bias per spike concentration (gradient of the linear regression in Figs. 4 (a) and (b)), given the first ionisation energies of Pb (715.5 kJ mol–1), U (584 kJ mol–1) and Sr (549.5 kJ mol–1). This suggests that the dominant mass bias effect is caused in the plasma rather than in the ion beam, as space-charge effects would result in the heaviest element, U, causing the strongest effect on mass bias.
3.4. Generation of variable mass bias for the fCu/fZn calibration: using Pb to define the fCu/fZn relationship during an analytical session
Fig. 5 shows ln(65Cu/63Cu) versus ln(66Zn/64Zn) of the three series of standards Pb-spiked at concentrations between 1 and 40 µg ml–1. The correlation factor of R2 = 0.991 for the combined data set is similar to that for standards without any spike element (Fig. 2). Thus, spiking doped standards with Pb at a range of concentrations significantly increases the variation of mass bias (spread of 13‰ for 65Cu/63Cu and 11‰ for 66Zn/64Zn between highest and lowest ratios) whilst achieving similar correlations. These findings are in line with previous work on the Cu–Zn isotope system conducted by Archer and Vance.14 Consequently, bracketing an analytical session with series of Pb-spiked standards will allow definition of a dopant/analyte relationship even if the instrumental mass bias is stable. | ||
| Fig. 5 ln(65Cu/63Cu) versus ln(66Zn/64Zn) for three series of six IMP Cu/Zn (2 µg ml–1) calibration standards spiked with Pb matrix at 1–40 µg ml–1 measured during an analytical session (∼10 hours). The spread of mass bias is significantly greater than for pure standards, covering a range of 11‰ for Cu and 13‰ for Zn between the highest and lowest points. The data set is a combination of all three series measured during an analytical session. | ||
4. Conclusions
Independent mass bias correction schemes applied to a single sample–standard bracketing analytical session assure the accuracy of isotope ratio measurements and act as a solid quality control. This cross-checking of mass bias corrected isotope ratios has been successfully applied to δ65Cu and δ66Zn determinations in plant and geological samples. The δ66Zn and δ65Cu values obtained using the m-SSB method agree with values obtained using the EEN and en-SSB methods well within the long-term reproducibility achieved on the IsoProbe MC-ICP-MS. Bracketing the SSB analytical session with a series of calibration standards with varying concentrations of Pb-spike leads to increased variability of mass bias effect, which in turn allows the fCu/fZn relationship to be defined even when the instrumental mass bias is very stable. This allows the application of the EEN or en-SSB methods and consequently the same solid quality control can be achieved.Acknowledgements
This work was funded by a Natural Environment Research Council PhD studentship to Kate Peel (NER/S/A/2004/12141), Imperial College London and The Natural History Museum. The authors are grateful to the staff involved in the running and maintenance of the IC/NHM Joint Analytical Facility IsoProbe and, in particular, to the Isotope Geochemistry Group with Mark Rehkämper, Maria Schönbächler, Richard Baker, Simone Gioia, Andrew Berry and Jamie Wilkinson. We also acknowledge three excellent reviews and the editorial help.References
- F. Albarède, in Geochemistry of non traditional stable isotopes: Reviews in Mineralogy, eds. C. M. Johnson, B. L. Beard and F. Albarède, Mineralogical Society of America, 2004, vol. 55, pp. 409–427 Search PubMed.
- C. P. Ingle, B. L. Sharp, M. S. A. Horstwood, R. R. Parrish and D. J. Lewis, J. Anal. At. Spectrom., 2003, 18, 219–229 RSC.
- C. N. Maréchal, P. Télouk and F. Albarède, Chem. Geol., 1999, 156, 251–273 CrossRef CAS.
- T. F. D. Mason, D. J. Weiss, M. Horstwood, R. R. Parrish, S. S. Russell, E. Mullane and B. J. Coles, J. Anal. At. Spectrom., 2004, 19, 218–226 RSC.
- K. G. Heumann, S. M. Gallus, G. Radlinger and J. Vogl, J. Anal. At. Spectrom., 1998, 13, 1001–1008 RSC.
- G. R. Gillson, D. J. Douglas, J. E. Fulford, K. W. Halligan and S. D. Tanner, Anal. Chem., 1988, 60, 1472–1474 CrossRef.
- D. C. Baxter, I. Rodushkin, E. Engström and D. Malinovsky, J. Anal. At. Spectrom., 2006, 21, 427–430 RSC.
- J. Bermin, D. Vance, C. Archer and P. J. Statham, Chem. Geol., 2006, 226, 280–297 CrossRef CAS.
- X. K. Zhu, R. K. O’Nions, Y. Guo, N. S. Belshaw and D. Rickard, Chem. Geol., 2000, 163, 139–149 CrossRef CAS.
- T. Ohno, A. Shinohhara, M. Chiba and T. Hirata, Anal. Sci., 2005, 21, 425–427 CrossRef CAS.
- A. Stenberg, H. Andrén, S. Malinovsky, E. Engström, I. Rodushkin and D. C. Baxter, Anal. Chem., 2004, 76, 3971–3978 CrossRef CAS.
- S. Ehrlich, I. Butler, L. Halicz, D. Rickard, A. Oldroyd and A. Matthews, Chem. Geol., 2004, 209, 259–269 CrossRef CAS.
- M. Rehkämper and K. Mezger, J. Anal. At. Spectrom., 2000, 15, 1451–1460 RSC.
- C. Archer and D. Vance, J. Anal. At. Spectrom., 2004, 19, 656–665 RSC.
- J. D. Woodhead, J. Anal. At. Spectrom., 2002, 17, 1381–1385 RSC.
- A. S. Al-Ammar and R. M. Barnes, J. Anal. At. Spectrom., 2001, 16, 327–332 RSC.
- T. F. D. Mason, D. J. Weiss, M. Horstwood, R. R. Parrish, S. S. Russell, E. Mullane and B. J. Coles, J. Anal. At. Spectrom., 2004, 19, 209–217 RSC.
- J. Chapman, T. F. D. Mason, D. J. Weiss, B. J. Coles and J. J. Wilkinson, Geostand. Geoanal. Res., 2006, 30, 5–16 Search PubMed.
- W. R. Shields, S. S. Goldich, E. L. Garner and T. J. Murphy, J. Geophys. Res., 1965, 70, 479–491 CrossRef CAS.
- M. Tanimizu, Y. Asada and T. Hirata, Anal. Chem., 2002, 74, 5814–5819 CrossRef CAS.
- R. Schoenberg and F. von Blankenburg, Int. J. Mass Spectrom., 2005, 242, 257–272 CrossRef CAS.
- C. Cloquet, J. Carignan and G. Libourel, Environ. Sci. Technol., 2006, 40, 6552–6600.
- H. Andrén, I. Rodushkin, A. Stenberg, S. Malinovsky and D. C. Baxter, J. Anal. At. Spectrom., 2004, 19, 1217–1224 RSC.
| This journal is © The Royal Society of Chemistry 2008 |