Assessment of plutonium measurement in the femtogram range by ICP-MS; correction from interfering polyatomic species
Fabien
Pointurier
a,
Philippe
Hémet
b and
Amélie
Hubert
b
aCEA/DAM-DIF/DASE/SRCE, PO Box 12, 91680, Bruyères-le-Châtel, France. E-mail: fabien.pointurier@cea.fr; Fax: +33 1 69 26 70 65; Tel: +33 1 68 26 49 17
bCommissariat à l’Energie Atomique, DAM-DIF/DASE/SRCE, 91680, Bruyères-le-Châtel, France
First published on 14th September 2007
Abstract
In the frame of the low level plutonium measurements performed by ICP-MS on environmental samples in support of the International Atomic Energy Agency’s safeguards program, we observe that background in the actinide mass range for the sample solutions is always higher than for blanks made of deionised water and ultra-pure grade nitric acid. This additional background is due to polyatomic species including heavy metals like Pb, Hg, Ir, etc., which remain in sample solutions despite the purification chemistry that is systematically carried out. As Pu content in the samples may be extremely low—in the femtogram (10–15 g) range—it is of uttermost importance to estimate detection limits as accurately as possible and to minimize risks of false detections. We propose an approach that consists of systematically correcting for contributions of polyatomic species to the background. To accomplish this, it is necessary to estimate all the formation rates of polyatomic species consisting of a “heavy metal” (from Hf to Bi) and one or more atoms, such as O, N, Ar, Cl, etc., that have total masses corresponding to those of Pu isotopes. Although the formation rates can vary largely with settings and analytical conditions, this study allows identification of the higher formation rates. The analytical procedure must also include, in addition to Pu and background masses, the measurement of one isotope for each “heavy metal”. Starting from examples of real sample measurements, we compare various calculation models for background and detection limits and prove that the one taking into account corrections from polyatomic interferences gives the most accurate results and minimises the risk of false detection for ultra low-level Pu measurements in the fg range.
Introduction
Environmental sampling is considered to be one of the key tools in safeguards for the detection of undeclared activities. Inspectors from the IAEA (International Atomic Energy Agency) collect small amounts of nuclear material (uranium, plutonium, fission products, etc.) by wiping various surfaces inside or around nuclear facilities, using small pieces of cotton cloth, referred to as “swipe samples”. These samples are sent for analysis to a few laboratories that are members of the Network of Analytical Laboratories (NWAL) of the IAEA in support of the safeguards. When “U–Pu bulk analysis” is requested by the IAEA, the whole “swipe sample” is reduced to ashes, digested, U and Pu are purified from the matrix and from various impurities, and U and Pu content as well as isotopic ratios are measured using thermal ionisation mass spectrometry (TIMS) or inductively coupled plasma-mass spectrometry (ICP-MS). Our laboratory is currently involved in an environmental survey of French nuclear facilities and in “bulk analysis” of environmental samples as a member of the IAEA’s NWAL. Plutonium contents in the samples analysed by our laboratory are typically in the femtogram to picogram range. For those purposes, we routinely use quadrupole-based and double-focusing sector-field ICP-MS.Due to its high sensitivity, ICP-MS seems to be the technique of choice for low level plutonium measurements. As a matter of fact, ICP-MS with single or multiple ion-counting detectors has been used to measure plutonium isotopes in environmental materials. In particular, numerous examples can be found in the literature about ICP-MS with outstanding sensitivity, typically several billion counts s–1 per µg g–1 of analyte, with best detection limits for plutonium isotopes in the femtogram range.1–16 However, it is also well known that difficulties arise when real samples are analysed. These difficulties come from preparative chemistry (contamination issuing from atmosphere, glassware, chemical reagents, etc.), introduction system (memory effect, clogging of capillaries, etc.), plasma (matrix effects, interferences due to polyatomic ions, etc.) and analyser stage of the mass spectrometer itself (mass bias, abundance sensitivity, dead time, limitation of the electron multiplier range of acquisition, etc.). At the high end of the mass range, the actinides have typically been considered as far removed from the notorious polyatomic interferences that tend to plague the lighter elements. This is not true for new generation high sensitivity ICP-MS that may present significantly higher background values in the actinide mass range (1 count s–1 or more) with real samples than with deionised water and ultra-pure grade nitric acid (∼0.2 counts s–1). This additional background is due to interferences that are combinations of one heavy element (Pb, Hg, W, Ir, Pt, etc.) with some of the most abundant atoms in the plasma (O, N, H, Cl, Ar). Those interferences can lead, if not identified and corrected, to false detection of plutonium isotopes.
Surprisingly, with the exception of 238U hydrides at mass 239, the possible occurrence of interferences due to polyatomic species with total masses equal to masses of actinide isotopes is rarely mentioned,17–20 and, to our knowledge, no extensive study has ever been published. Given that ICP-MS detection limits in the femtogram or even sub-femtogram are now attainable, all types of potential interferences must receive additional consideration. Even when sample matrices are reasonably clean and care has been taken to minimize oxides during performance optimisation, measurements performed near the instrumental detection limits are especially susceptible to overestimation due to polyatomic interferences.
In this paper, we first describe briefly the instrumental performance of the double focusing sector-field ICP-MS VG “Axiom” that we use in our laboratory for low-level Pu measurement of environmental samples, in terms of sensitivity, background and instrumental detection limits for actinide isotopes like 239Pu and 240Pu. We also describe the purification procedure used for IAEA cotton “swipe samples” that we routinely analyse in support of the IAEA’s safeguards program. Then, we focus on the formation of polyatomic ions: after some considerations about calculations of detection limits using different approaches with and without correction from polyatomic interferences, we compare formation ratios of interferences due to the studied “heavy elements” (Hf, Ta, W, Re, Ir, Au, Hg, Tl, Pb, Bi) that may affect the background for Pu isotopes. Then, we propose modifications of the analytical methodology and we compare the results obtained for real samples with and without taking into account correction from polyatomic interferences.
Experimental
Instrumentation
For trace analyses of long-lived actinides, we optimized and routinely used a double-focusing sector field ICP-MS (“Axiom SC”, VG Elemental, Winsford, Cheshire, UK) already described in the literature.7–9 The separation of the isotope ion beams is carried out through a double-focusing arrangement, providing focusing both in angle and energy, consisting of an electrostatic sector followed by a magnetic sector (so-called “Nier–Johnson” geometry). This instrument is equipped with the S-option (an additional primary pump that decreases vacuum in the interface) which provides an increase in sensitivity by a factor of three compared to a non-S option. The performance of the instrument is optimised daily with respect to sensitivity, short term stability (10 minutes) and background. The instrument is operated in electric scanning by varying the accelerating voltage, with the intensity of the magnetic field set to a fixed value. A Teflon micro-concentric nebuliser (“PFA200”, Elemental Scientific, Inc. Omaha, NE, USA) was used for sample introduction. The nebuliser was routinely operated in combination with two on-line spray chambers (“cyclonic” and “impact bead”, Thermo-Fisher, Les Ulis, France) cooled to 12 °C. The “Axiom” exhibits very high sensitivity (around 3 × 106 counts s–1 per µg l–1 of 238U) and low background (about 0.3 ± 0.2 counts s–1 in the actinide mass range). The instrumental detection limits for Pu isotopes calculated according to the 3σ criteria based on background obtained with de-ionised water and ultra-pure grade HNO3 at 2% is about 0.2 fg ml–1.Sample preparation and purification procedure
All the acids used for sample preparation and purification are of the ultra-pure grade (Merck, Darmstadt, Germany). 233U and 242Pu were used as isotopic dilution tracers for U and Pu quantitative analysis. The standard solutions were diluted (by weight) to obtain accurate stock solutions between 0.2–2 ng ml–1.For each analysis, a preparative chemistry based on ion-exchange resins is performed on the samples (see Fig. 1). At first, “swipe samples” are transferred to a Pyrex beaker, and 10 ml of concentrated HNO3 are added. The mixture is boiled on a hot plate (120–150 °C) for at least 2 h. During heating, the beaker is covered with a watch glass to prevent significant evaporation. The mixture is evaporated to dryness and the sample is then reduced to ash at 550 °C for 12 h in an electric furnace to decompose organic matter. Dissolution of the ashes is obtained by repetition of acid digestions: 20 ml of aqua regia, evaporation to dryness, 10 ml of concentrated HNO3 + 1 ml of H2O2, evaporation to dryness, 10 ml of concentrate HCl and evaporation to dryness.
 | ||
| Fig. 1 Procedure for U and Pu separation from swipe samples. | ||
233U and 242Pu are added in appropriate quantities (respectively about 0.1 and 0.01 ng) to the sample solutions before separations. Spiked samples are evaporated to dryness and 20 ml of concentrated nitric acid are added. Adjustment and redox state stabilisation of plutonium are performed by addition of nitrite followed by evaporation. 20 ml of 8 N HNO3 are added and the solution is passed through the conditioned ion-exchange resin columns. The first plutonium purification is performed with a 20 ml column filled with Dowex AG1X8 anion-exchange resin (10 ml of 50/100 mesh resin at the bottom and 10 ml of 100/200 mesh resin on the top). The plutonium fraction is eluted with HCl–NH4I. Further U–Pu purification is obtained by using a 2 ml column filled with Dowex AG1X4 anion-exchange resin for Pu or AG1X8 100/200 mesh for U. These resins are washed with 8 N HNO3 (U fraction), 10 N HCl (Th fraction) and, finally, NH4I (1.5%)–12 N HCl solution is added to elute the Pu fraction. The final solution is evaporated to dryness and recovered with 5 ml of de-ionised water acidified by HNO3 at 2%. In addition, one or more process blanks are prepared in the same conditions as the samples for each series of samples.
Experiments for the determination of interferences in the actinide mass range
We focus only on the heaviest elements below the actinides: Hf, Ta, Re, W, Ir, Pt, Au, Hg, Tl, Pb, Bi, but not Os which is not taken into account due to its low abundance. We consider that we don’t need to study the lighter elements because their combination with some abundant elements of the plasma to equal the masses of plutonium isotopes would be too complicated and, thus, very unlikely. All potential polyatomic interferences arising from the aforementioned heavy elements are listed in Table 1. We prepared a set of standard solutions with increasing concentrations from 100 ng ml–1 to 5 µg ml–1. Dilutions were made from commercial standards (Spex, Longjumeau, France) using a calibrated micro-pipette. Each series of standards contained only one heavy element. For each standard, background measurements were performed for masses 233 to 247. In most of the cases, count rates for isotopes of the heavy elements themselves could not be measured directly and had to be inferred from count rates measured with a more dilute solution (1 ng ml–1). Then, the formation rates, defined as the ratio of the count rate at a given background mass in the actinide mass range to the count rate of a selected isotope of a given heavy metal, can be calculated.| Mass | Polyatomic species |
|---|---|
| 233 | 185ReO3, 193Ir40Ar, 197Au36Ar, 198Hg35Cl, 201HgO2, 200HgO2H 203TlNO |
| 234 | 186WO3, 185ReO3H, 193Ir40ArH, 194Pt40Ar, 198Pt36Ar, 197Au37Cl, 199Hg35Cl, 202HgO2, 201HgO2H, 198Hg36Ar, 203TlNOH, 204PbNO |
| 235 | 186WO3H, 187ReO3, 195Pt40Ar, 194Pt40ArH, 200Hg35Cl, 198Hg37Cl, 202HgO2H, 199Hg36Ar, 203TlO2, 205TlNO, 204PbNOH |
| 236 | 187ReO3H, 196Pt40Ar, 195Pt40ArH, 201Hg35Cl, 199Hg37Cl, 204HgO2, 200Hg36Ar, 203TlO2H, 205TlNOH, 206PbNO, 204PbO2 |
| 237 | 176HfNO3, 191IrNO2, 196Pt40ArH, 197Au40Ar, 202Hg35Cl, 200Hg37Cl, 204HgO2H, 201Hg36Ar, 205TlO2, 207PbNO, 206PbNOH, 204PbO2H |
| 238 | 198Pt40Ar, 201Hg37Cl, 198Hg40Ar, 202Hg36Ar, 205TlO2H, 203Tl35Cl, 208PbNO, 207PbNOH, 206PbO2 |
| 239 | 177HfNO3, 176HfNO3H, 191IrO3, 193IrNO2, 198Pt40ArH, 204Hg35Cl, 202Hg37Cl, 199Hg40Ar, 203Tl36Ar, 208PbNOH, 207PbO2, 204Pb35Cl, 209BiNO |
| 240 | 178HfNO3, 177HfNO3H, 191IrO3H, 193IrNO2H, 194PtNO2, 200Hg40Ar, 205Tl35Cl, 203Tl37Cl, 208PbO2, 207PbO2H, 204Pb36Ar, 209BiNOH |
| 241 | 179HfNO3, 178HfNO3H, 193IrO3, 195PtNO2, 194PtNO2H, 204Hg37Cl, 201Hg40Ar, 205Tl36Ar, 208PbO2H, 204Pb37Cl, 206Pb35Cl, 209BiO2 |
| 242 | 180HfNO3, 179HfNO3H, 193IrO3H, 196PtNO2, 195PtNO2H, 194PtO3, 202Hg40Ar, 205Tl37Cl, 207Pb35Cl, 206Pb36Ar, 209BiO2H |
| 243 | 180HfNO3H, 181TaNO3, 196PtNO2H, 195PtO3, 194PtO3H, 197AuNO2, 203Tl40Ar, 208Pb35Cl, 206Pb37Cl, 207Pb36Ar |
| 244 | 181TaNO3H, 182WNO3, 198PtNO2, 196PtO3, 195PtO3H, 197AuNO2H, 204Hg40Ar, 198HgNO2, 203Tl40ArH, 207Pb37Cl, 204Pb40Ar, 208Pb36Ar, 209Bi35Cl |
| 245 | 183WNO3, 182WNO3H, 198PtNO2H, 196PtO3H, 197AuO3, 199HgNO2, 198HgNO2H, 205Tl40Ar, 208Pb37Cl, 204Pb40ArH |
| 246 | 184WNO3, 183WNO3H, 198PtO3, 197AuO3H, 200HgNO2, 199HgNO2H, 198HgO3, 205Tl40ArH, 206Pb40Ar, 209Bi37Cl |
| 247 | 184WNO3H, 185ReNO3, 198PtO3H, 201HgNO2, 200HgNO2H, 199HgO3, 198HgO3H, 207Pb40Ar, 206Pb40ArH |
Great care must be taken with homogenization and conservation of the solutions. Some elements are not stable in a nitric acid medium and cannot be stored more than a few days. For elements like Hg, Ir, W, great attention must also be paid to contamination problems and to memory effects. It may be necessary to rinse for a very long time (1 h or more) with various acids to return to the normal background. In the case of mercury, glassware must be cleaned.
Results and discussion
Interferences
Typical examples of background count rates obtained with the double focusing sector-field ICP-MS VG “Axiom” in the actinide mass range from mass 236 to 247 are shown in Fig. 2. In this figure, we compare background values between a real sample that contains a very low amount of Pu in the fg range and its associated rinsing blank (de-ionized water acidified with ultra-pure grade HNO3 at 2%) and process blank. It can be seen that background is globally higher for the process blank than for the rinsing blank, especially in the mass range 243 to 247, and that the background for the samples is higher than the background for the process blank. Actually, despite final purification of the solutions using anionic exchange resins, signals can sometimes reach more than 10 counts s–1 for some background masses in the actinide mass range. | ||
| Fig. 2 Mass spectra from 233 to 247 atomic mass units obtained with the double-focusing single-collector “Axiom” ICP-MS for an IAEA’s “swipe sample” number 07/009/24 (graph c) and its associated process blank (graph b) and rinsing blank (graph a) that is de-ionised water acidified with ultra-pure grade nitric acid at 2%. Analytical conditions are: 100 ms dwell time, 5 points per peak and 100 sweeps. | ||
This phenomenon was already mentioned by Taylor et al.17 They observed minor but significant background peaks at masses 239 to 244. The peak at mass 239 can be explained by 238U hydrides. Peaks at other masses were considerably decreased after cleaning of the glassware and cones (remaining background between 5 and 20 counts s–1 at masses 240 to 242). However, no explanation is provided about the origin of the high background observed at these masses before cleaning. The authors evaluated possible influence of polyatomic species including lead but found no influence of such species at the aforementioned masses. Wyse et al.18 also observed occurrence of non-hydride polyatomic interferences in the Pu isotopes mass range. They first analysed samples in the low resolution mode for maximum sensitivity. Then, the acquired peaks are assessed for the presence of polyatomic interferences evidenced by a measurable negative shift in mass position of the isotope of interest. If, based on this assessment, a polyatomic interference is suspected, medium resolution mode is employed for unequivocal resolution of the analyte peak. Use of medium to high resolution (2000 to 4000) is possible with a sector-field instrument, albeit at a significant sacrifice of sensitivity. This approach is not valid if the signals are not significantly higher than detection limits. In such a case, the peak may not be detected in the medium resolution and, if it is henceforth detected, its position would not be measured with enough precision. Therefore, we consider that this approach is not applicable for our trace analysis of environmental samples whose Pu content is fairly low. Magara et al.19 mentioned occurrence of 207PbO2+ and 208PbO2+ interferences at masses 239 and 240, and gave estimates of the corresponding formation rates for their high sensitivity ICP-MS (∼3 × 10–8).
Considerations about calculation of the detection limits
Traditionally, the detection limits for plutonium isotopes is defined as three times the standard deviation over background value at the mass of interest. In addition to background variations, this standard deviation also includes contribution of standard deviations over U hydride, abundance sensitivity, and impurities from the tracer corrections. However, the elevated variations of the background from one mass to the other lead to an important question: how to define properly the detection limit (DL) for plutonium isotopes (239Pu and 240Pu)? The first possibility, referred as the “calculation model 1” in the continuation of this paper, is to use background values at the mass of interest for the process blank or for the rinsing blank. Therefore, the classical DL formula for 239Pu based on the standard deviation of the signal measured at 239 amu for the blank is given by: | (1) |
Instead of eqn (1), we can use a more realistic calculation for background, referred as “calculation model 2” in the continuation, that is the average of the background values for selected neighbouring masses (including 233, 236, 237, 243–247) acquired for the sample solution itself. For this model, the DL formula for 239Pu takes into account the standard deviation over count rates of these selected neighbouring masses acquired during the measurements of the samples, instead of the standard deviation of the signal measured at 239 amu for the blank:
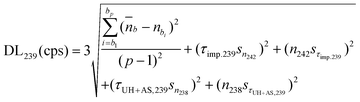 | (2) |
Use of this calculation for background and DL decreases the risk of false plutonium detection, because it provides higher DL and, according to our experience, because background values at masses 239 and 240 are generally lower than the ones at other masses in the actinide mass range. This calculation also results in analytical detection limits that can be higher than the “instrumental” ones. Nevertheless, we used the calculation model 2 in our laboratory for several years.21 In Fig. 3, we gathered all the analytical limits of detection for 239Pu and 240Pu obtained by the laboratory using the calculation model 2 for years 2003–2005. All the measurements were performed on various types of environmental samples (biological, water, soil, swipe samples), that previously underwent a preparative chemistry.
 | ||
| Fig. 3 Distribution of 239Pu and 240Pu analytical limits of detection for real samples analysed using the sector-field ICP-MS “Axiom” for years 2003, 2004 and 2005, calculated according to the 3σ criteria, using calculation model 2 (see text). Instrumental detection limit determined with ultra-pure HNO3 2% and deionised water is around 0.2 fg ml–1. | ||
It can be seen from Fig. 3 that these analytical detection limits range from less than 0.2 to about 5 fg ml–1 and are most of the time higher than the instrumental detection limits obtained with pure reagents (about 0.2 fg ml–1). This is due mainly to higher background (and then higher standard deviation over background) but also, for some analysis, to a loss of sensitivity or to contributions of the corrections from 238U hydrides, abundance sensitivity and isotopic impurities from 242Pu isotopic dilution tracer. Nevertheless, these analytical detection limits remain in the fg ml–1 range, i.e. lower than the best detection limits obtained in our laboratory with alpha-spectrometry and quadrupole-based ICP-MS. However, this method has two drawbacks. Firstly, it may lead to an overestimation of the background and, thus, to a poor accuracy of the results in the fg range. Secondly, a significant polyatomic interference at mass 239 or 240 due to an outstanding concentration of a heavy metal in the sample solution would not be detected and corrected, and this would result in a false detection of Pu.
The 3rd approach consists in calculating contribution of polyatomic interferences to background, and in integrating these corrections in the DL calculation. This is the “calculation model 3” for which the DL for 239Pu is given by the following formula:
 | (3) |
Formation rates of interfering species in the Pu mass range
The contribution of the polyatomic species at each mass of the background in the actinide mass range is given by the following matrix multiplication: | (4) |
Of course, the most important formation rates in this context are those that contribute to the background at masses 239–242 and so interfere directly with plutonium isotopes. Nevertheless, the other masses can be used to check that after subtraction of the polyatomic interferences the background is close to the electronic noise (about 0.3 count s–1). Examples of calculated formation rates of interferences with masses of 239, 240, 241 and 242 and the associated uncertainties obtained at the beginning of 2007 are given in Table 2.
| Isotope of reference for each element | 239 a.m.u. | 240 a.m.u. | 241 a.m.u. | 242 a.m.u. |
|---|---|---|---|---|
| 180Hf | (5.0 ± 0.5) × 10–9 | (7.3 ± 0.7) × 10–9 | (4.1 ± 0.8) × 10–11 | (9.0 ± 0.5) × 10–10 |
| 181Ta | (2.2 ± 0.3) × 10–9 | (9.9 ± 1.1) × 10–9 | (9.0 ± 0.6) × 10–9 | (6.2 ± 2.6) × 10–10 |
| 184W | (1.0 ± 0.5) × 10–9 | (1.8 ± 0.4) × 10–9 | (3.2 ± 2.6) × 10–10 | (1.4 ± 0.4) × 10–9 |
| 187Re | (1.5 ± 0.2) × 10–10 | (1.0 ± 0.2) × 10–9 | (3.3 ± 0.2) × 10–10 | (1.4 ± 0.5) × 10–10 |
| 193Ir | (3.1 ± 0.2) × 10–6 | (3.9 ± 1.4) × 10–7 | (6.3 ± 0.6) × 10–6 | (4.1 ± 1.0) × 10–7 |
| 195Pt | (5.7 ± 1.4) × 10–9 | (5.9 ± 1.5) × 10–9 | (1.1 ± 0.6) × 10–9 | (3.0 ± 0.1) × 10–8 |
| 197Au | <10–9 | (1.3 ± 0.3) × 10–9 | (1.4 ± 0.3) × 10–8 | <10–9 |
| 202Hg | (6.0 ± 0.9) × 10–6 | (9.3 ± 0.8) × 10–6 | (6.5 ± 1.4) × 10–6 | (1.2 ± 0.1) × 10–5 |
| 205Tl | (7.5 ± 0.2) × 10–9 | (7.8 ± 0.6) × 10–10 | (1.6 ± 0.1) × 10–8 | (3.3 ± 0.3) × 10–8 |
| 208Pb | (2.2 ± 0.1) × 10–8 | (5.1 ± 0.4) × 10–8 | (7.2 ± 0.6) × 10–9 | (1.2 ± 0.1) × 10–8 |
| 209Bi | (2.0 ± 0.4) × 10–10 | (8.6 ± 1.5) × 10–11 | (1.1 ± 0.3) × 10–10 | (8.5 ± 2.2) × 10–10 |
Most of the ratios are very low, around 10–9 or even less. This means that, taking into account the very high sensitivity of our sector-field ICP-MS, concentrations in the µg ml–1 range (ppm) are necessary to obtain a significant interfering signal at masses that correspond to plutonium isotopes. Finding these concentrations in sample solutions after a chemical purification is highly unlikely. However, these data also show that higher formation rates at masses 239, 240, 241, and 242 are in the range 10–6 to 10–5, through species IrO3+ and HgAr+. As a consequence, moderate concentrations, between the pg ml–1 and the ng ml–1 range can result in significant increases of the background at masses that correspond to plutonium isotopes. Even after chemical purification, it is not unlikely to observe such concentration levels in sample solution, depending on the purity of reagents, various sources of contamination and, above all, of the content of the “swipe samples”. As a matter of fact, experience shows that such concentrations are obtained in a lot of sample solutions after chemical treatment of “swipe samples”. An example of a spectra from 180 to 210 atomic mass units measured for a sample is given in Fig. 4.
 | ||
| Fig. 4 Example of spectra from 180 to 210 atomic mass units measured for an IAEA QC sample number 06/056/449 after chemical purification using the double-focusing sector-field ICP-MS “Axiom”. | ||
Identification of the polyatomic species that corresponds to a given interference is not always easy. Identification is somewhat easier when the heavy element that is part of the polyatomic species has several abundant isotopes. It should be noted that Pb is of special interest for us as this element is particularly abundant in nuclear installations, and can potentially be at the origin of polyatomic interferences with Pu isotopes. Moreover, even if Pb can be largely eliminated after a chemical purification, it can easily contaminate solutions prior to measurements. However, influence of polyatomic species containing Pb at masses 239 to 242 is low, as can be seen from Table 2. Formation rates are in the 10–9 to the 10–8 range, thus 2 to 4 orders of magnitude below Ir and Hg. Taking into account the sensitivity of our ICP-MS, Pb concentrations in the sample solutions must be in the µg ml–1 range to induce a few counts s–1 at masses 239 to 242. It should also be noted that the formation rate for PbO2+ is close to the value of 3.0 × 10–8 announced by Magara et al.19
Lastly, all these formation rates can vary largely depending on the settings of the instrument (gas flow rates, plasma power, high voltages for ionic lenses, etc.). Furthermore, strong variations of the PbCl+ interference are expected due to changes in the chloride concentration in the sample solutions. Thus, the formation rates for the heavy elements whose concentrations in the sample solutions may result in a significant influence on the masses of the plutonium isotopes must be measured for each analysis. These formation rate measurements must be performed with the same settings of the instrument and the same analytical conditions. It is also expected that these ratios should vary largely from one instrument to another, and probably from one injection device to another. For instance, addition of a desolvating membrane may largely alter formation ratios.
Modification of the analytical methodology
It is also necessary to modify the analytical methodology: each sample solution should not be measured only for Pu isotopes and neighbouring masses (233, 236, 237, 243 to 247) but also for at least one isotope of all the heavy metals Hf, Ta, W, Re, Ir, Pt, Au, Hg, Tl, Pb, Bi. We performed two separate measurements. The first analysis was carried out with one isotope of all heavy elements, with Pu isotopes (masses 239 to 242), and with background masses in the actinide mass range (masses 233 to 238 and 243 to 247). The selected isotopes for heavy elements were: 180Hf, 181Ta, 184W, 187Re, 193Ir, 195Pt, 197Au, 202Hg, 205Tl, 208Pb and 209Bi. When an element has several isotopes, we chose the most abundant one. All these isotopes were measured in a single analysis, with a resolution of 430 (low resolution mode), 1 point per peak, 5 replicates per sample solution, 20 sweeps per replicate, a dwell time of 400 ms for Pu isotopes and background masses, and a dwell time of 10 ms for “heavy element” isotopes. Total analysis time was about 10 min, including a 90 s sample introduction period. Solution is taken up by the nebuliser at a rate of 200 µl min–1, meaning that about 2 ml is consumed during the Pu analysis. This first measurement allows identification of the most abundant heavy elements that may significantly contribute to background at masses 239 and 240. Then, a second measurement is performed using the same analytical conditions with series of synthetic solutions, each one containing an appropriate concentration of one of the heavy metals that were identified as possible sources of background for Pu isotopes. This second measurement lasts about two hours, including preparation of the series of synthetic solutions. It allows calculation of up-to-date formation rates, obtained with the same settings and analytical conditions as the first measurements.Results from these two measurements are obtained thanks to home-made calculation software, based on MS Excel™. Corrections from various bias and interferences (U hydrides, impurities of the isotopic dilution tracer, but also polyatomic interferences) along with calculations of uncertainties and detection limits are automatically performed. Contribution of each heavy element to each mass of the background is calculated according to the formation rates. Limits of detection and concentrations for Pu isotopes are calculated according to the corrected values of the background and the other sources of errors.
Application to real samples
We illustrated this work by the measurement of 3 series of “swipe samples” from the International Atomic Energy Agency containing low amounts of plutonium, in the fg to pg range, or no plutonium at all. We have gathered in Table 3 all the detection limits and results for 239Pu, using the three possible calculations described above for estimation of background and detection limit: (i) using background at mass 239 for blank (calculation model 1), (ii) using background values of neighbouring masses (calculation model 2), and (iii) calculating contribution from polyatomic interferences (calculation model 3). In the case of calculation model 2, the selected neighbouring masses are 236, 241, 243, 244 and 245. Masses 233, 237, 246 and 247 are excluded because they exhibit particularly high background values. Sample solutions of the first series of samples are relatively “clean”, meaning that they do not contain very high concentrations of “heavy metals”, however their influence on the background is not negligible. For this first series, DLs using calculation models 1 and 3 are quite similar (about 1.5 fg per “swipe sample”), whereas conservative calculations with model 2 result in largely higher DLs, by about a factor of 5. On the contrary, sample solutions of the second series of samples contain larger concentrations of “heavy metals”, especially Ir (between 3 × 105 and 2 × 106 counts s–1 at mass 193) and Hg (between 1.3 × 105 and 1.7 × 105 counts s–1 at mass 202) that result in a significantly higher background in the actinide mass range, including masses 239 and 240. The origin of these relatively high concentrations is not known although we suspect that they may be due to contaminations during the prurification procedure. For this second series, DLs calculated according to model 1 remain very low (about 1 fg). On the contrary, larger values are obtained according to models 2 (between 4 and 8 fg per sample) and 3, with DLs between 3.5 and 8.1 fg, except in the case of the process blank (DL of 24.5 fg) that was obviously contaminated with heavy metals (Ir) during sample processing. For the 3rd series, results show a similar trend to the results from the first series, with lower DLs using models 1 and 3 (about 2 fg per sample) and higher results than the ones obtained using model 2.| Sample id. | Background correction and DL calculation using blank count rates at 239 amu (model 1) | Background correction and DL calculation using count rates at neighbouring masses (model 2) | Background correction and DL calculation using interference corrections (model 3) | ||||||
|---|---|---|---|---|---|---|---|---|---|
| DL/fg | Results/fg | Decision | DL/fg | Results/fg | Decision | DL/fg | Results/fg | Decision | |
| Process blank 07/009 | 1.6 | 1.1 ± 2.0 | <DL | 6.9 | –2.7 ± 5.0 | <DL | 1.6 | 0.4 ± 2.4 | <DL |
| Blank swipe 07/009 | 1.5 | 1.9 ± 1.7 | Detected | 7.7 | –2.1 ± 5.4 | <DL | 1.6 | 1.1 ± 2.6 | <DL |
| IAEA 07/009/24 | 1.5 | 13.5 ± 4.3 | Detected | 6.9 | 9.3 ± 6.3 | Detected | 1.7 | 12.6 ± 4.2 | Detected |
| IAEA 07/009/25 | 1.4 | 22.6 ± 2.1 | Detected | 5.7 | 16.9 ± 4.3 | Detected | 1.9 | 21.8 ± 5.0 | Detected |
| IAEA 07/009/26 | 1.5 | 19.6 ± 4.9 | Detected | 7.0 | 14.0 ± 6.8 | Detected | 1.6 | 18.9 ± 4.6 | Detected |
| IAEA 07/009/27 | 1.5 | 831 ± 24 | Detected | 10.5 | 824 ± 24 | Detected | 1.6 | 830 ± 34 | Detected |
| IAEA 07/009/28 | 1.4 | 99.0 ± 7.3 | Detected | 10.7 | 93.1 ± 10.2 | Detected | 1.5 | 98.1 ± 9.6 | Detected |
| Process blank 06/121 | 1.1 | 13.6 ± 13.0 | Detected | 8.0 | 11.0 ± 7.4 | Detected | 24.5 | –1.4 ± 17.9 | <DL |
| Blank swipe 06/121 | 0.9 | 3.3 ± 3.3 | Detected | 5.9 | 1.2 ± 5.2 | <DL | 3.6 | –0.8 ± 2.6 | <DL |
| IAEA 06/121/1239 | 1.0 | 12.5 ± 8.8 | Detected | 4.0 | 10.4 ± 9.2 | Detected | 8.1 | 6.1 ± 6.6 | <DL |
| IAEA 06/121/1240 | 0.9 | 7.7 ± 1.9 | Detected | 6.1 | 5.4 ± 4.6 | <DL | 4.3 | 3.5 ± 3.2 | <DL |
| IAEA 06/121/1241 | 0.9 | 5.6 ± 3.5 | Detected | 5.4 | 3.9 ± 5.1 | <DL | 5.2 | 0.9 ± 3.7 | <DL |
| IAEA 06/121/1242 | 0.9 | 690 ± 37 | Detected | 3.8 | 688 ± 37 | Detected | 3.5 | 685 ± 18 | Detected |
| Process blank 06/056 1 | 1.8 | –0.4 ± 3.3 | <DL | 5.2 | –3.8 ± 4.8 | <DL | 2.2 | –0.5 ± 3.4 | <DL |
| Process blank 06/056 2 | 1.7 | –0.6 ± 4.0 | <DL | 11.1 | –7.1 ± 8.5 | <DL | 2.1 | –0.8 ± 4.1 | <DL |
| Process blank 06/056 3 | 1.6 | 0.7 ± 4.5 | <DL | 10.1 | –4.2 ± 8.2 | <DL | 2.0 | 0.6 ± 4.6 | <DL |
| IAEA 06/056/447 | 1.7 | 925 ± 52 | Detected | 10.3 | 919 ± 53 | Detected | 2.1 | 925 ± 52 | Detected |
| IAEA 06/056/448 | 1.7 | 90.5 ± 11.4 | Detected | 16.5 | 83.9 ± 16.0 | Detected | 2.1 | 90.3 ± 11.4 | Detected |
| IAEA 06/056/449 | 1.7 | 13.5 ± 4.0 | Detected | 15.8 | 4.3 ± 11.4 | <DL | 2.1 | 9.7 ± 4.1 | Detected |
It is clear from these results that when the concentrations of “heavy metals” in the sample solutions are moderate (as in the 1st and 3rd series of samples), the influence of polyatomic interferences on the background at masses 239 and 240 is low. In such a case, the background and DL obtained using model 3 are very close to the ones calculated according to model 1. However, the background and DL obtained using model 2, that take into account background values at neighbouring masses, can be largely higher than with the two other models and therefore may lead to background overcorrection (negative net results for process blanks and blank swipes of the 1st and 3rd series) and to DL overestimation (Pu is not detected in sample 06/056/149). On the contrary, when concentrations of “heavy metals” in the sample solutions are higher (as in the 2nd series of samples), the background and DL calculated according to model 3 become higher than the ones obtained using model 1. Consequently, the background and DL calculations of model 1 that do not take into account polyatomic interference are clearly underestimated and may lead to false detection of plutonium, as it is observed for the process blank and the blank swipe of the 2nd series of samples. The background and DL calculated according to model 2 are also higher than the ones obtained with model 1, but—depending on the neighbouring masses taken into account—can be lower or higher than the background and DL of model 3. As a consequence, use of model 2 leads on the one hand to a probable false detection (process blank 06/121) and on the other hand results in missing two detections (samples 06/121/1240 and 06/121/1241).
Samples from the 3rd series are quality control “swipe samples”, i.e. cotton cloth similar to the regular samples but spiked with target masses of Pu (unknown at the time of the measurement). No heavy elements are theoretically present in these samples. For this series, comparison of the various results with the target values for 239+240Pu are given in Table 4. It appears that results obtained with the samples 06/056/447 and 06/056/448 whose 239+240Pu contents (respectively 1 pg and 100 fg) are largely above the DL are in good accordance with the target values, whatever the calculation model for the background and DL. However, for the 3rd QC sample (06/056/449) whose 239Pu + 240Pu content (10 fg) is close to the DL, we observe that the best agreement is obtained with calculation model 3, whereas results obtained using calculation model 1 are clearly overestimated and the results obtained using model 2 are underestimated with regards to the target values. Thus, model 3 that takes into account correction from polyatomic species seems to be more accurate and more robust in relation to false detection than other more or less conservative calculation models that do not take into consideration the interferences that affect masses of Pu isotopes.
| Calculation model 1 239Pu + 240Pu/fg | Calculation model 2 239Pu + 240Pu/fg | Calculation model 3 239Pu + 240Pu/fg | Target values 239Pu + 240Pu/fg | |
|---|---|---|---|---|
| IAEA 06/056/447 | 1020 ± 29 | 1010 ± 29 | 1010 ± 29 | 1000 |
| IAEA 06/056/448 | 99.6 ± 6.5 | 86.3 ± 10.3 | 96.1 ± 6.5 | 100 |
| IAEA 06/056/449 | 15.1 ± 3.4 | –1.6 ± 8.3 | 11.4 ± 3.4 | 10 |
For the sample 06/121/1241, we compared the contribution of the different sources of errors (see Fig. 5) that contribute to the count rate measured at mass 239. For this sample, the major contributor to the background is the polyatomic interference (1.97 ± 0.46 counts s–1 over a total background of 3.65 ± 0.71 counts s–1), with 193Ir and 202Hg count rates of, respectively, 1.35 × 105 counts s–1 (about 0.3 ng ml–1) and 6.2 × 105 counts s–1 (about 5 ng ml–1). The high sum of polyatomic interferences with respect to net count rate results in a larger uncertainty and much higher DL.
 | ||
| Fig. 5 Example of breaking down of the raw count rate measured at mass 239 for sample 06/121/1241 between different contributions. All values are given in counts s–1 and uncertainties are given with a coverage factor of 2. Raw count rate for this sample at mass 239 is 3.65 ± 0.71 counts s–1. | ||
Further work
It should be noted that the chemical purification theoretically removes most of all the heavy elements. Nevertheless, removal of impurities by chromatographic separation is never complete and some heavy metals can also be released by the resins during the final Pu elution, by the glassware used during the treatment, or can be contained in the various reagents. Moreover, the initial content of elements like Pb, Ir, Hg, etc., in the original samples is of course never known, especially in the case of “swipe samples” collected in nuclear industrial and research facilities where a large range of materials and reagents can be present. Besides, slight recontamination after or at the end of the chemical treatment can never be dismissed (releases by the anionic resins, presence in reagents and/or in the glassware, etc.). These concentrations are sufficient to induce count rates on the order of one count s–1 at masses 239, 240, 241, and 242, i.e. above electronic background for most of the double-focusing sector-field ICP-MS. Therefore, an extensive study about the origin of the heavy metals can be very useful, to limit the source of such elements and, thus, to have analytical detection limits very close to the instrumental ones.Furthermore, it would be necessary to evaluate more precisely the variability of the formation rates thanks to repeated measurements. It will provide a better estimation of the uncertainty, and to determine influence of introduction devices, of plasma parameters, etc. These repeated experiments will also help to define the best analytical conditions that would minimise the formation rates of the interfering species. Another useful work will be of course to identify the origin of heavy elements (from the sample itself or from the chemistry: reagents, resin, glassware, etc.) and, if possible, to reduce the content of these elements in sample solutions.
Lastly, similar corrections must also be applied to minor 234U and 236U isotopes of uranium, to avoid overestimation of 234U/238U and 236U/238U ratios. 236U is an excellent fingerprint for irradiated uranium and, thus, for human nuclear activities, but may be present at a very low abundance in environmental samples. So, considering the interest of the 236U/238U ratio in the frame of safeguards, it is of uttermost importance to be certain that a low signal at mass 236 cannot be due to polyatomic species.
Conclusion
Experiments have been performed to determine the formation rates of polyatomic interferences that consist of one atom of a heavy element, such as Hf, Ta, W, Re, Ir, Pt, Au, Hg, Tl, Pb and Bi, with some of the most abundant atoms in the plasma (O, N, C, H, Cl, Ar), and have total masses in the actinide mass range (from 233 to 247 atomic mass unit). We observed that most of these formation rates are quite low (below 10–8) so that relatively high concentrations of the corresponding heavy metals (typically in the µg ml–1 range) would be necessary to induce a significant increase at masses 239 and 240. These concentrations are unlikely in sample solutions that are obtained after chemical purification. However, we also noticed that some formation rates with Hg and Ir are higher, so that tiny amounts of such elements can be the origin of significant increases of background at masses 239 to 242 through polyatomic species IrO3+ and HgAr+. For these elements, significant concentrations are in the pg ml–1 to the ng ml–1 range, meaning that the sample solution contains no more than a few pg to a few ng of such elements.Polyatomic interferences can be neglected for Pu measurements when Pu content is well above the instrumental detection limits of high sensitivity ICP-MS. However, when Pu content is closer to the detection limits in the fg range, correction from polyatomic interferences deserves much attention. To validate detection of ultra-trace of Pu, it is at least necessary to check that polyatomic interferences are negligible. If they are not, calculations can be performed to correct for these interferences and to adjust detection limits. To provide the most accurate and precise correction, it is necessary to measure systematically the formation rates of interferences that potentially interfere with Pu isotopes just after the analysis. Studying and taking into account polyatomic interferences that increase background in the actinide mass range is the price to pay to optimize exploitation of the amazing performance of the new generation high sensitive ICP-MS.
More work has to be done, especially on the chemical preparation of the samples, by developing an ultra-pure chemistry with appropriate reagents and handling of samples not only to limit contamination by uranium and to avoid cross-contamination, but above all to limit contamination by all the elements that can be at the origin of interferences in the actinide mass range.
References
- S. Stürup, H. Dahlgaard and S. C. Nielsen, J. Anal. At. Spectrom., 1998, 13, 1321–1326 RSC.
- J. S. Becker and H. J. Dietze, J. Anal. At. Spectrom., 1999, 14, 1493–1500 RSC.
- R. Chiappini, F. Pointurier, J. C. Milliès-Lacroix, G. Lepetit and P. Hémet, Sci. Total Environ., 1999, 237/238, 269–276 CrossRef.
- Y. Muramatsu, S. Uchida, K. Tagami, S. Yoshida and T. Fujikawa, J. Anal. At. Spectrom., 1999, 14, 859–865 RSC.
- S. E. Boulyga, C. Testa, D. Desideri and J. S. Becker, J. Anal. At. Spectrom., 2001, 16, 1283–1289 RSC.
- J. S. Becker, Spectrosc. Eur., 2002, 14(6), 8–16 CAS.
- F. Pointurier, N. Baglan and P. Hémet, Appl. Radiat. Isot., 2003, 60, 561–566.
- B. C. Ting, R. S. Pappas and D. C. Paschal, J. Anal. At. Spectrom., 2003, 18, 795–797 RSC.
- N. Baglan, P. Hémet, F. Pointurier and R. Chiappini, J. Radioanal. Nucl. Chem., 2004, 261, 609–617 CrossRef CAS.
- R. S. Pappas, B. G. Ting and D. C. Paschal, J. Anal. At. Spectrom., 2004, 19, 762–766 RSC.
- J. S. Becker, M. Zoriy, L. Hallicz, N. Tepliakov, Chr. Müller, I. Segal, C. Pickhardt and I. T. Platzner, J. Anal. At. Spectrom., 2004, 19, 1257–1261 RSC.
- M. E. Ketterer, K. M. Hafer, C. L. Link, D. Kolwaite, J. Wilson and J. W. Mietelski, J. Anal. At. Spectrom., 2004, 19, 241–245 RSC.
- Z. L. Wang and M. Yamada, Earth Planet. Sci. Lett., 2005, 233, 441–453 CrossRef CAS.
- J. Zheng and M. Yamada, J. Radioanal. Nucl. Chem., 2006, 267(1), 73–83 CAS.
- J. Zheng and M. Yamada, Talanta, 2006, 69, 1246–1253 CrossRef CAS.
- K. Norisuye, K. Okamura, Y. Sohrin, H. Hasegawa and T. Nakanishi, J. Radioanal. Nucl. Chem., 2006, 267(1), 183–193 CAS.
- R. N. Taylor, T. Warneke, J. A. Milton, I. W. Croudace, P. E. Warwick and R. W. Nesbitt, J. Anal. At. Spectrom., 2001, 16, 279–284 RSC.
- E. J. Wyse, S. H. Lee, J. La Rosa, P. Povinec and S. J. De Mora, J. Anal. At. Spectrom., 2001, 16, 1107–1111 RSC.
- M. Magara, S. Ichimura, M. Takahashi, S. Kurosawa, Y. Saito, F. Esaka, K. Yasuda, S. Sakurai, J. Chai, K. Watanabe and S. Usuda, “Study of polyatomic ions in isotope ratio measurement of actinides by ICP-MS”, poster presented at Winter Conference on Plasma Spectrochemistry, Fort Lauderdale, Florida, January 5–10, 2004.
- T. Kishimoto, T. Sanada, K. Sato and H. Higuchi, J. Radioanal. Nucl. Chem., 2004, 252, 395–398.
- F. Pointurier, N. Baglan and P. Hémet, in Plasma source mass spectrometry—current trends and future developments, ed. G. Holland and D. R. Bandura, RSC Publishing, Cambridge, UK, 2005, pp. 130–142 Search PubMed.
| This journal is © The Royal Society of Chemistry 2008 |