Green synthesis of carbamates from CO2, amines and alcohols
Angelica
Ion
a,
Charlie Van
Doorslaer
a,
Vasile
Parvulescu
b,
Pierre
Jacobs
a and
Dirk De
Vos
*a
aCatholic University of Leuven, Centre for Surface Science and Catalysis, Kasteelpark Arenberg 23, 3001, Leuven, Belgium. E-mail: dirk.devos@biw.kuleuven.be; Fax: 32 1632 1998; Tel: 32 1632 1639
bUniversity of Bucharest, Department of Catalysis, B-dul R. Elisabeta 4-12, 030016, Bucharest, Romania. E-mail: v_parvulescu@chem.unibuc.ro; Fax: 40 2131 59249; Tel: 40 2141 00241
First published on 20th November 2007
Abstract
Various carbamates can be prepared in a halogen-free way starting from cheap and readily available reagents such as amines, alcohols and carbon dioxide. Basic catalysts were able to convert both linear and branched aliphatic amines to the corresponding carbamates with good yields, in mild reaction conditions (2.5 MPa CO2) and even in the absence of dehydrating agents.
Introduction
The continuous increase of carbon dioxide concentrations in the atmosphere is of worldwide concern due to the impact on global warming. Thus, utilization and transformation of CO2 into useful chemicals is an area of major interest. Among the few major industrial processes using CO2, urea synthesis is the most important one, with a production of about 100 million tonnes per year.1 Other organic compounds such as salicylic acid and its derivatives, and cyclic carbonates are produced in smaller amounts.1,2 One of the most promising fields for CO2 utilization is the synthesis of carbamates, since the carbon atom in CO2 does not need to be reduced in this reaction. Carbamates are important raw materials for the manufacture of a variety of polymers used in foams, coatings, adhesives, plastics and fibres.3 Current commercial processes for production of carbamates are alcoholysis of isocyanates or aminolysis of chloroformates.4 The preparation of isocyanates and chloroformates requires use of the highly toxic and corrosive phosgene (COCl2). Alternative routes imply the utilization of poisonous carbon monoxide,5 or rather expensive dialkyl carbonates.6Use of CO2 in carbamate synthesis is particularly attractive since CO2 is a non-toxic, non-corrosive, inflammable, abundant and cheap C1 source. Despite its stability, it easily combines with amines at ambient temperature and atmospheric pressure to form the corresponding carbamic acids.7 However, when alcohols are used as the alkyl source, the subsequent dehydrative condensation to the carbamate proceeds much less easily, partly because the reaction is equilibrium limited. As one solution, alkyl halides have been used as the electrophilic alkyl source. Even if several catalysts exist for this reaction,8 the co-production of HCl remains a drawback. Thus, the use of alcohols would be a major advance, since alcohols are cheap and would allow to make the process completely halogen-free.
Few reports exist on direct synthesis of carbamates from CO2, amines and alcohols.9 The known catalytic systems contain a homogeneous metal catalyst (Sn, Ni) and reactions are preferentially performed at very high CO2 pressures (e.g. 30 MPa of CO2). An excess of a dehydrating ketal is mostly used to overcome the equilibrium limitations. Even then, the only reactions reported concern sterically congested amines, such as tert-butylamine or cyclohexylamine. In another approach, it has been reported that 1,2-aminoalcohols react with CO2 at 150–180 °C to form the cyclic carbamate products, either in the presence of Sn10 or Sb11 or even in absence of a catalyst.12 However, the cyclic nature of the oxazolidinone product has a decisive influence on the reaction, and the same reaction conditions fail when it is attempted to make non-cyclic carbamates.
Clearly, there is a lack of knowledge regarding the one-step production of carbamates from preferred reagents, viz. amines, alcohols and carbon dioxide. In the present paper we report that basic catalysts are suitable to convert a broad variety of amines and alcohols into the corresponding carbamates using carbon dioxide as a carbonyl source (Scheme 1).
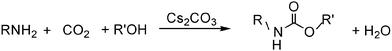 | ||
| Scheme 1 | ||
Results and discussion
Catalysts for the synthesis of carbamates
In previous work, Cs+ compounds were shown to be very active in the catalytic activation of carbon dioxide and amines to give symmetric and asymmetric urea derivatives selectively.13 Therefore, we decided to investigate the activity of these catalysts for the synthesis of carbamates starting from amines, alcohols and CO2. A series of basic catalysts were tested for two model reactions, viz. n-octylamine/n-PrOH and n-butylamine/MeOH (Table 1). In the absence of a catalyst, almost no reaction occurred and carbamate yields were negligible (entries 1 and 7). For the couple n-octylamine/n-PrOH, the selectivity to carbamate increases in going from K+ to Cs+ in the series of alkali carbonates, reaching 67% for Cs2CO3 after 24 h (entries 2–4). The identity of the carbamates was confirmed by GC-MS (see Experimental section). The main side-products were the corresponding symmetric urea derivatives. Note that the reactions proceed to appreciable conversion even in the absence of dehydrating agents. The conversions are also far higher than usually achieved in the synthesis of organic carbonates from alcohols and CO2.14| Entry | Catalyst | Amine | Alcohol | X b (%) | S c c (%) | S u d (%) | Y c e (%) |
|---|---|---|---|---|---|---|---|
| a Conditions: 50 mmol alcohol (molar ratio amine : alcohol = 1 : 10), 0.1 g catalyst, 200 °C, 24 h, 2.5 MPa CO2. b X = amine conversion. c S c = carbamate selectivity. d S u = urea selectivity. e Y c = carbamate yield. f DMAP = 4-dimethylaminopyridine. g TBD = 1,5,7-triazabicyclo[4.4.0]dec-5-ene. | |||||||
| 1 | — | n-OctNH2 | n-PrOH | 2 | — | 100 | — |
| 2 | K2CO3 | n-OctNH2 | n-PrOH | 48 | 52 | 48 | 25 |
| 3 | Rb2CO3 | n-OctNH2 | n-PrOH | 37 | 66 | 34 | 25 |
| 4 | Cs2CO3 | n-OctNH2 | n-PrOH | 51 | 67 | 33 | 34 |
| 5 | CsF | n-OctNH2 | n-PrOH | 57 | 63 | 37 | 36 |
| 6 | CH3COOCs | n-OctNH2 | n-PrOH | 60 | 46 | 34 | 28 |
| 7 | — | n-BuNH2 | MeOH | 5 | 22 | 78 | 1 |
| 8 | K2CO3 | n-BuNH2 | MeOH | 55 | 22 | 78 | 12 |
| 9 | Rb2CO3 | n-BuNH2 | MeOH | 45 | 28 | 72 | 13 |
| 10 | Cs2CO3 | n-BuNH2 | MeOH | 48 | 26 | 74 | 13 |
| 11 | MgO | n-BuNH2 | MeOH | 4 | 23 | 77 | 1 |
| 12 | DMAP f | n-BuNH2 | MeOH | 27 | 30 | 70 | 8 |
| 13 | TBD g | n-BuNH2 | MeOH | 24 | 14 | 86 | 3 |
| 14 | KF/Al2O3 | n-BuNH2 | MeOH | 41 | 24 | 76 | 10 |
Concerning the amine conversion, Cs2CO3 proved again to be the most active of the alkali carbonates. The counter-anions of Cs+ have strong effects as well. With F– or CH3COO– salts, the amine conversion increases but the carbamate selectivity decreases, especially for Cs acetate (entries 5–6). Even if the couple n-butylamine/MeOH is considerable less reactive than n-octylamine/n-PrOH, a more than 10-fold increase of the amine conversion was observed when alkali carbonates were used as catalysts (entries 7–10). The presence of such catalysts also slightly enhances the selectivity to carbamate. Traces of alkylated amines were observed, but only when MeOH was used as the alcohol.
Other strongly basic compounds such as MgO, 4-dimethylaminopyridine (DMAP) or 1,5,7-triazabicyclo[4.4.0]dec-5-ene (TBD) generated carbamates in poor yields (entries 11–13). Employment of a heterogeneous basic catalyst such as KF/Al2O3 did not improve the yield of carbamate either. Summarizing, alkali-metal compounds seem to be the best choice for this reaction.
Reaction conditions
In order to select the optimum reaction conditions, experiments focused on the reaction temperature, time, amount of catalyst, amine to alcohol ratio and type of alcohol. Influences of the reaction temperature were investigated using the n-octylamine/n-PrOH system and Cs2CO3 as a catalyst. As shown in Table 2 for reactions of 24 h, a temperature of 200 °C seems the optimum to reach the highest carbamate yields (entries 1–3). Lower temperatures were unfavorable, likely because the reaction is simply too slow. At a higher temperature of 220 °C there was again a decrease of the conversion. This probably indicates that the reaction is exothermic, as was previously found for the formation of urea compounds from amines and CO2.13 Hence, the equilibrium shifts to the left with increasing temperature, and the conversion decreases.| Entry | T/°C | Catalyst/g | Alcohol | Amine : alcohol (molar ratio) | X b (%) | S c c (%) | S u d (%) | Y c e (%) |
|---|---|---|---|---|---|---|---|---|
| a Conditions: 50 mmol alcohol, Cs2CO3, 24 h, 2.5 MPa CO2. b X = amine conversion. c S c = carbamate selectivity. d S u = urea selectivity. e Y c = carbamate yield. f Alkylated compounds were formed as side products (∼1%). | ||||||||
| 1 | 170 | 0.1 | n-PrOH | 1 : 10 | 15 | 14 | 86 | 2 |
| 2 | 200 | 0.1 | n-PrOH | 1 : 10 | 51 | 67 | 33 | 34 |
| 3 | 220 | 0.1 | n-PrOH | 1 : 10 | 37 | 62 | 38 | 23 |
| 4 | 200 | 0.05 | n-PrOH | 1 : 10 | 42 | 28 | 72 | 12 |
| 5 | 200 | 0.15 | n-PrOH | 1 : 10 | 44 | 64 | 36 | 28 |
| 6 | 200 | 0.25 | n-PrOH | 1 : 10 | 48 | 62 | 38 | 30 |
| 7 | 200 | 0.1 | n-PrOH | 1 : 8 | 44 | 34 | 66 | 15 |
| 8 | 200 | 0.1 | n-PrOH | 1 : 15 | 56 | 79 | 21 | 44 |
| 9f | 200 | 0.1 | MeOH | 1 : 15 | 35 | 58 | 41 | 20 |
| 10 | 200 | 0.1 | EtOH | 1 : 15 | 51 | 58 | 42 | 30 |
| 11 | 200 | 0.1 | n-BuOH | 1 : 15 | 51 | 60 | 40 | 30 |
| 12 | 200 | 0.1 | n-HexOH | 1 : 15 | 55 | 72 | 28 | 42 |
| 13 | 200 | 0.1 | 2-PrOH | 1 : 15 | 44 | 43 | 57 | 19 |
| 14 | 200 | 0.1 | 2-BuOH | 1 : 15 | 55 | 49 | 51 | 27 |
Next, the effect of the amount of the catalyst on the reaction was examined. 0.1 g of Cs2CO3 was found to be necessary to achieve good yields of carbamates (entry 2). This most likely corresponds to a solubility maximum of cesium carbonate in the alcoholic reaction mixture, as confirmed by further tests with a higher amount of catalyst (entries 5, 6). In the presence of a smaller amount of catalyst (e.g. 0.05 g) the selectivity to carbamate was quite poor (entry 4). Variation of the amine : alcohol ratio showed that a larger alcohol excess promoted the reaction (entries 2, 7, 8). Both conversion and selectivity for carbamate increased with increasing the excess of alcohol, reaching their highest values for a ratio of amine to alcohol of 1 : 15.
In order to study the influence of the alcohol type, n-octylamine was subjected to reactions with n-alcohols of varying chain length. The reactivity of the alcohols was lowest for MeOH, and increased with the alkyl chain length (entries 8–12). Clearly, as the acidity of the alcohol decreases,15 the reaction rate increases. A simple explanation is that basicity of the Cs2CO3 catalyst is least affected by long chain aliphatic alcohols, while it is weakened by a large excess of the more acidic methanol. In the case of reactions in methanol, this even results in formation of by-products by alkylation of the amine, which is normally an acid-catalyzed reaction. The fact that a high yield is reached for n-hexanol is also in line with a nucleophilic substitution mechanism, and with the increased nucleophilicity of alcohols with larger alkyl substituents. Secondary alcohols are in this reaction less reactive than their primary homologs, which is ascribed to increased steric hindrance of the nucleophile's attack (entries 13, 14). Again a similar order of reactivity is observed, with 2-BuOH giving higher yields than isopropanol.
In order to follow the evolution of the product distribution in time, reactions were performed at 4, 8, 12, 24 and 48 h (Fig. 1). While in the first stage of the reaction mainly dialkylurea is formed, the selectivity shifts towards the formation of carbamate for longer reaction times. The isocyanate is initially detected in small concentration as well, but its concentration becomes negligible after 24 h. While the carbamate selectivity increases continuously, the conversion reaches a maximum value at 24 h. A further increase of the reaction time to 48 h produced no change in conversion, while the selectivity and yield of carbamate marginally increased.
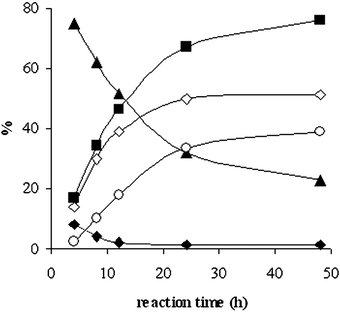 | ||
| Fig. 1 Change of product distribution in time: (◇) amine conversion, (■) selectivity for carbamate, (▲) selectivity for urea derivative, (◆) isocyanate selectivity, (○) yield of carbamate. Conditions: 50 mmol n-PrOH, n-OctNH2 : n-PrOH = 1 : 10, 0.1 g Cs2CO3, 200 °C, 2.5 MPa CO2. | ||
Reaction scope
With the best conditions in hand, other carbamates were successfully synthesized in considerable yields as shown in Table 3. Experiments performed at different amine : alcohol ratios reconfirmed the necessity of a high alcohol excess in the reaction mixture to achieve good yields of carbamates (entries 2–4). Compared with the n-amines, internal amines such as 2-heptylamine reacted more selectively, most probably due to the steric hindrance. However, they were considerably less reactive than the linear amines (entries 5–7).| Entry | Amine | Alcohol | Amine : alcohol (molar ratio) | X b (%) | S c c (%) | S u d (%) | Y c e (%) |
|---|---|---|---|---|---|---|---|
| a Conditions: 50 mmol alcohol, 0.1 g Cs2CO3, 24 h, 200 °C, 2.5 MPa CO2. b X = amine conversion. c S c = carbamate selectivity. d S u = urea selectivity. e Y c = carbamate yield. | |||||||
| 1 | n-BuNH2 | n-PrOH | 1 : 15 | 40 | 50 | 50 | 20 |
| 2 | n-HexNH2 | n-PrOH | 1 : 8 | 38 | 37 | 63 | 14 |
| 3 | n-HexNH2 | n-PrOH | 1 : 10 | 45 | 43 | 57 | 19 |
| 4 | n-HexNH2 | n-PrOH | 1 : 15 | 51 | 61 | 39 | 31 |
| 5 | 2-HeptNH2 | n-PrOH | 1 : 10 | 26 | 70 | 30 | 18 |
| 6 | 2-HeptNH2 | n-PrOH | 1 : 15 | 31 | 73 | 27 | 23 |
| 7 | 2-OctNH2 | n-PrOH | 1 : 15 | 30 | 76 | 24 | 23 |
Reactions in the presence of a dehydrating agent
As the carbamate formation produces water,9 it can be attempted to shift the equilibrium of the reaction to the right by withdrawing water from the medium. Besides, an excess of water could also cause catalyst deactivation. Acetals such as dimethoxypropane (DMP) or diethoxypropane (DEP), molecular sieves and urea were used as dehydrating agents to selectively remove water from the reaction system. Acetals react with water to produce a ketone and two alcohols (Scheme 2). | ||
| Scheme 2 | ||
The influence of the dehydrating agents on the catalytic performance is shown in Table 4. The acetals bring about a significant increase of the amine conversion and of the carbamate yield (compare Table 4, entries 2, 3 with Table 4, entry 1 and Table 2, entry 10). A clear disadvantage, however, is that imines are formed from the side reaction between acetone and the amines. Moreover, in the presence of the basic catalysts, a series of other side-products is formed from acetone, such as mesityl oxide, isophorone and 4-methoxy-4-methyl-2-pentanone.
| Entry | Amine | Alcohol | Amine : alcohol (molar ratio) | Catalyst/g | Dehydrating agent/g | X b (%) | S c c (%) | S u d (%) | Y c e (%) |
|---|---|---|---|---|---|---|---|---|---|
| a Conditions: 50 mmol alcohol, molar ratio amine : DMP/DEP = 1 : 2, 24 h, 200 °C, 2.5 MPa CO2. b X = amine conversion. c S c = carbamate selectivity. d S u = urea selectivity. e Y c = carbamate yield. f Zeolite 3A in the K-form. g Zeolite L in the K-form. h 3.3 mmol urea. | |||||||||
| 1 | n-OctNH2 | MeOH | 1 : 10 | Cs2CO3 (0.1) | — | 56 | 30 | 70 | 17 |
| 2 | n-OctNH2 | MeOH | 1 : 10 | Cs2CO3 (0.1) | DMP | 68 | 37 | 14 | 25 |
| 3 | n-OctNH2 | EtOH | 1 : 15 | Cs2CO3 (0.1) | DEP | 82 | 56 | 5 | 46 |
| 4 | n-OctNH2 | n-PrOH | 1 : 15 | Cs2CO3 (0.25) | 3A (0.2)f | 57 | 60 | 40 | 34 |
| 5 | n-OctNH2 | n-PrOH | 1 : 15 | Rb2CO3 (0.25) | — | 64 | 67 | 33 | 43 |
| 6 | n-OctNH2 | n-PrOH | 1 : 15 | Rb2CO3 (0.25) | 3A (0.2)f | 47 | 57 | 43 | 27 |
| 7 | n-OctNH2 | n-PrOH | 1 : 15 | Rb2CO3 (0.25) | L (0.2)g | 63 | 64 | 36 | 40 |
| 8 | n-OctNH2 | n-PrOH | 1 : 15 | Cs2CO3 (0.1) | Urea h | 82 | 84 | 16 | 69 |
Since zeolites have a well known water absorption capacity, and since potassium salts catalyze the reaction, commercially available K-zeolites such as molecular sieves 3A and L were tested as dehydrating agents. In each case, the reactions were compared to those catalysed by alkali carbonates in absence of a zeolite. Unfortunately, both conversion and selectivity of carbamate decreased upon zeolite addition (Table 4), probably because part of the Cs+ or Rb+ ions were exchanged on the molecular sieves. On the other hand, one may expect that the uptake of water by these sieves is limited at the reaction temperature of 200 °C. Much better results were obtained in the presence of urea, with a 69% yield of propyl N-octylcarbamate (Table 4, entry 8). However, further tests performed in nitrogen instead of carbon dioxide atmosphere revealed that rather than CO2, urea is employed as the carbonyl source in this case.
The use of dehydrating agents appeared particularly effective when sterically hindered amines were used as starting compounds for the carbamate synthesis. As an example, tert-butylamine was investigated (Table 5). No reaction took place in the absence of a catalyst (entry 2). Using cesium carbonate as a catalyst, and in the absence of a dehydrating agent, the amine conversion was very poor, even if the carbamate was almost exclusively obtained (entry 1). With Cs2CO3 and the DMP dehydrating agent combined, a dramatic increase in conversion was observed, with preservation of the >99% carbamate selectivity (entry 3). A higher amount of catalyst produced higher yields of carbamate (entry 4). This was expected since the total reaction volume has increased due to the presence of acetals; hence a higher amount of catalyst can be solubilized in the reaction mixture. In the presence of methanol, traces (<1%) of alkylated compounds and urea were occasionally observed. Side products derived from acetone such as mesityl oxide were also present. In agreement with the results obtained for the linear amines, the EtOH/DEP couple proved to be slightly more effective than the MeOH/DMP system (entry 5).
| Entry | Catalyst/g | Alcohol | t-BuNH2 : alcohol (molar ratio) | Dehydrating agent | t/h | X b (%) | S c c (%) | Y c d (%) |
|---|---|---|---|---|---|---|---|---|
| a Conditions: 50 mmol alcohol, 10 mmol DMP/DEP or 5 mmol urea, Cs2CO3 as catalyst, 200 °C, 2.5 MPa CO2. b X = amine conversion. c S c = carbamate selectivity. d Y c = carbamate yield. e 0.2 g zeolite 3A (K-form). f N2 instead of CO2. g Selectivity of urea derivative = 28%, yield of urea = 8%. | ||||||||
| 1 | 0.1 | MeOH | 1 : 10 | — | 24 | 4 | >99 | 4 |
| 2 | — | MeOH | 1 : 10 | DMP | 24 | — | — | — |
| 3 | 0.1 | MeOH | 1 : 10 | DMP | 24 | 36 | >99 | 36 |
| 4 | 0.25 | MeOH | 1 : 10 | DMP | 24 | 47 | >99 | 47 |
| 5 | 0.25 | EtOH | 1 : 10 | DEP | 24 | 52 | >99 | 52 |
| 6 | 0.25 | EtOH | 1 : 10 | DEP | 48 | 62 | >99 | 62 |
| 7 | 0.25 | EtOH | 1 : 11.5 | DEP | 48 | 71 | >99 | 71 |
| 8 | 0.25 | EtOH | 1 : 15 | DEP | 48 | 68 | >99 | 68 |
| 9 | 0.25 | EtOH | 1 : 11.5 | DEP + 3Ae | 48 | 64 | >99 | 64 |
| 10 | 0.25 | EtOH | 1 : 11.5 | Urea | 24 | 37 | 90 | 33 |
| 11f | 0.25 | EtOH | 1 : 11.5 | Urea | 24 | 33 | 90 | 30 |
| 12g | 0.25 | t-BuOH | 1 : 10 | — | 24 | 28 | 54 | 15 |
While in reactions without dehydrating agent a ceiling conversion is attained after 24 h due to the presence of water (see Fig. 1), this is not the case when DMP or DEP are added, as water is efficiently eliminated from the reaction mixture. By allowing the reaction to proceed for 48 h and by adjusting the amine : alcohol ratio, a 71% yield in carbamate was attained with Cs2CO3 in 48 h using only 2.5 MPa of carbon dioxide (entries 6–8). A combination of acetals with molecular sieves did not further improve the carbamate yield (entry 9). t-BuNH2 reacts selectively and in acceptable yield when urea is used instead of an acetal, but it was again proven that urea itself, rather than CO2, is the source of the inserted carbonyl group (entries 10 and 11).
Mechanism of carbamate formation over Cs catalysts
As can be observed from Fig. 1, mainly dialkylurea is formed in the first stage of the reaction of n-alkylamines. Indeed, the urea formation is much faster in comparison with the reaction of an alcohol, amine and carbon dioxide towards a carbamate.13 The strong initial increase in conversion is mainly due to urea formation. The decrease of urea selectivity with time and the increase of carbamate selectivity suggest that the carbamates may be produced viaurea alcoholysis, with the amine as a coproduct (Scheme 3). The sharp dependence of carbamate selectivity on the concentration of the base catalyst suggests that the alcoholysis step is speeded up by the base (Table 2, entries 4 vs. 5). In order to gather more information concerning this reaction pathway, the alcoholysis of N,N′-dibutylurea (DBU) with n-PrOH was conducted under the same experimental conditions as the initial reaction. Tests were performed at various reaction times and at DBU concentrations corresponding to 5, 10 and 15% conversion of amine in the initial reaction. The data presented in Fig. 2 suggest that indeed N,N′-dialkylureas are gradually transformed into the corresponding carbamates. The main by-product detected was n-propyl butyrate. Although this compound was formed in significant amounts in the N,N′-dibutylurea alcoholysis, only traces of this product were observed in the reaction of n-BuNH2, n-PrOH and carbon dioxide.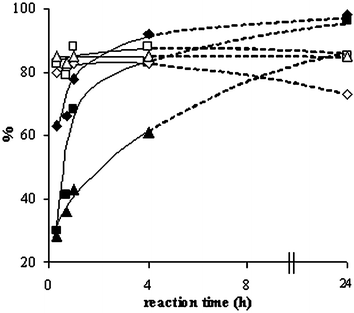 | ||
| Fig. 2 N,N′-Dibutylurea (DBU) alcoholysis with n-PrOH: (◆) conversion and (◇) carbamate selectivity when 0.125 mmol DBU were used, (■) conversion and (□) carbamate selectivity when 0.25 mmol DBU were used, (▲) conversion and (△) carbamate selectivity when 0.375 mmol DBU were used. Other conditions: 50 mmol n-PrOH, 0.1 g Cs2CO3, 200 °C, 2.5 MPa CO2. | ||
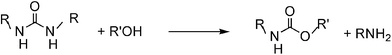 | ||
| Scheme 3 | ||
A second possibility for carbamate formation is that in the high-temperature reaction conditions, the unstable carbamic acid undergoes a thermal dehydration to form the corresponding isocyanate (Scheme 4, eqn (2)), which can further condense with an alcohol or an amine (Scheme 4, eqn (3) and (4)) to form the carbamate or urea derivative respectively. Evidence for this mechanism is supplied by the presence of small amounts of isocyanates in the reaction mixtures, particularly at short reaction times (see Fig. 1). An isocyanate pathway could also explain the lack of reactivity of secondary amines. Isocyanates may well intervene at several places in the reaction network, since they have also been proposed as intermediates in urea alcoholysis.16
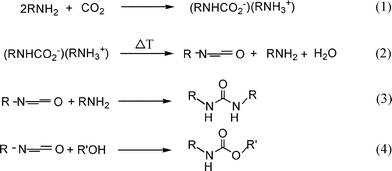 | ||
| Scheme 4 | ||
While evidence for both routes is available for n-alkylamines, the mechanism is less clear for t-BuNH2. The urea alcoholysis pathway is improbable for a sterically hindered amine such as t-BuNH2. It has indeed been demonstrated that the base-catalyzed urea formation from t-BuNH2 is poorly effective;13 in the present experiments (Table 5), the N,N′-di-tert-butylurea is only detected in trace amounts, except in the reaction of t-BuNH2 with t-BuOH, where it was formed in 8% yield (Table 5, entry 12).
Remarkably, isocyanate concentrations were below the detection limit in the reactions of t-BuNH2 with various alcohols. For these reactions, indirect evidence for an isocyanate route comes from the strong promotion of the carbamate formation by the addition of an acetal (Table 5, entries 1 vs. 3); indeed, the formation of the isocyanate is a dehydration. The reaction of t-BuNH2 with t-BuOH was the only reaction of t-BuNH2 that gave an appreciable carbamate yield even in the absence of a dehydrating agent (Table 5, entry 12 vs. entry 1); this may well be ascribed to the high reactivity of t-BuOH.
In conclusion, there are two, most probably parallel routes that lead to carbamates using this catalytic system, either viaureas or via isocyanates. Their relative importance likely depends on the amine structure.
Conclusions
In summary, this is the first systematic study proving that basic catalysts can promote the transformation of a large variety of amines and alcohols into carbamates in appreciable yields. The most active among the catalysts tested were Cs2CO3 and Rb2CO3. The proposed catalytic system was able to convert both linear and branched aliphatic amines to their corresponding carbamates in mild reaction conditions (i.e. 2.5 MPa CO2), even in the absence of dehydrating agents. Only when a sterically hindered amine such as tert-butylamine was involved, a dehydrating agent was necessary. Mechanistic investigations revealed two main pathways for carbamate formation, viz. a direct pathway with isocyanates as reaction intermediates, and an indirect one viaurea alcoholysis.Experimental
All reactions were conducted under pressurized CO2 in small stainless steel autoclaves (10 ml inner volume). In a typical experiment, the amine (5 mmol), alcohol (50 mmol) and the catalyst (0.1 g) were charged in the reactor, which was then saturated with CO2 under a pressure of 2.5 MPa at room temperature. The mixture was magnetically stirred and heated to 170–220 °C in a temperature-controlled electrical heater for 4–48 h. After heating, the reactor was cooled to room temperature (0 °C in the case of the more volatile amines) and carefully depressurized. The liquid reaction mixture was diluted in MeOH and analyzed with a Shimadzu 2014 gas chromatograph (flame ionization detector) with CP-Sil5 CB capillary column (50 m × 0.25 µm). The reaction products were identified by GC-MS using an Agilent 5973 Network Mass Selective Detector coupled to an Agilent 6890N GC with HP5MS capillary column (30 m × 0.25 mm). The reaction products could easily be separated from the catalyst by extraction with ethyl acetate from an aqueous solution of the reaction mixture. For product isolation, the reaction mixture was poured into distilled water (8 ml) and then extracted thrice with 6 ml ethyl acetate. The GC analysis of both organic and water phases after extraction confirmed the complete uptake of the carbamate by the organic layer. For urea alcoholysis, N,N′-dibutylurea (0.125–0.375 mmol), n-PrOH (50 mmol) and Cs2CO3 (0.1 g) were charged in the reactor after which the same procedure as for the initial reaction was applied. Spectroscopic data for the synthesized carbamates are given below.Methyl N-butylcarbamate
GC-MS, m/z: 57 (10%), 88 (100), 102 (3), 116 (3), 131 (8).Methyl N-octylcarbamate
GC-MS, m/z: 59 (10%), 76 (19), 88 (100), 116 (4), 130 (4), 144 (3), 172 (3), 187 (4).Ethyl N-octylcarbamate
GC-MS, m/z: 57 (14%), 71 (6), 85 (8), 99 ( 24), 116 (2), 130 (3), 144 (2), 172 (8), 201 (2).Propyl N-octylcarbamate
GC-MS, m/z: 57 (16%), 71 (9), 85 (5), 99 (12), 113 (2), 116 (100), 130 (5), 144 (3), 158 (5), 172 (18), 186 (1), 215 (5).Isopropyl N-octylcarbamate
GC-MS, m/z: 57 (46%), 74 (45), 116 (100), 130 (7), 144 (3), 172 (26), 200 (1), 215 (3).Butyl N-octylcarbamate
GC-MS, m/z: 57 (100%), 74 (28), 118 (17), 130 (83), 174 (30), 186 (4), 229 (6).2-Butyl N-octylcarbamate
GC-MS, m/z: 57 (100%), 130 (21), 156 (14), 174 (30), 200 (1), 214 (1), 229 (2).Hexyl N-octylcarbamate
GC-MS, m/z: 57 (46%), 74 (30), 85 (26), 116 (6), 130 (6), 158 ( 40), 174 (100), 186 (3), 200 (3), 214 (2), 257 (2).Propyl N-hexylcarbamate
GC-MS, m/z: 57 (6%), 85 (11), 99 (4), 116 (100), 144 (13), 158 (4), 172 (1), 187 (7).Propyl N-(2 heptyl)carbamate
GC-MS, m/z: 57 (18%), 70 (14), 88 (9), 99 (7), 113 (8), 130 (100), 186 (3).Methyl N-tert-butylcarbamate
GC-MS, m/z: 57 (37%), 72 (24), 84 (21), 116 (100), 131 (1).Hexyl isocyanate
GC-MS, m/z: 56 (100%), 69 (12), 85 (32), 99 (100), 112 (20).Ethyl N-tert-butylcarbamate
GC-MS, m/z: 58 (100%), 86 (11), 130 (64), 145 (1).Butyl isocyanate
GC-MS, m/z: 56 (100%), 70 (16), 98 (26).Octyl isocyanate
GC-MS, m/z: 56 (55%), 70 (30), 85 (31), 99 (100), 113 (31), 126 (14), 140 (3), 154 (3).Acknowledgements
Angelica Ion is grateful to the K.U. Leuven Research Fund for a doctoral fellowship. The authors appreciate funding of this work in the frame of a Bilateral Agreement Flanders/Romania and of IAP, GOA and CECAT projects.References
- I. Omae, Catal. Today, 2006, 115, 33 CrossRef CAS.
- M. Shi and Y. Shen, Curr. Org. Chem., 2003, 7, 737 CAS.
- R. Srivastava, M. D. Manju, D. Srinivas and P. Ratnasamy, Catal. Lett., 2004, 97, 41 CrossRef CAS.
- Ullmann's Encyclopedia of Industrial Chemistry, 6th edn, electronic version, 2000 Search PubMed.
- (a) F. Shi, J. Peng and Y. Deng, J. Catal., 2003, 219, 372 CrossRef CAS; (b) B. Chen and S. S. C. Chuang, J. Mol. Catal. A: Chem., 2003, 195, 37 CrossRef CAS; (c) B. Chen and S. S. C. Chuang, Green Chem., 2003, 5, 484 RSC.
- (a) Z. H. Fu and Y. Ono, J. Mol. Catal., 1994, 91, 399 CrossRef; (b) S. Carloni, D. De Vos, P. A. Jacobs, R. Maggi, G. Sartori and R. Sartorio, J. Catal., 2002, 205, 199 CrossRef CAS; (c) N. Katada, H. Fujinaga, Y. Nakamura, K. Okumura, K. Nishigaki and M. Niwa, Catal. Lett., 2002, 80, 47 CrossRef CAS.
- E. M. Hampe and D. M. Rudkevich, Tetrahedron, 2003, 59, 9619 CrossRef CAS.
- (a) R. N. Salvatore, F. Chu, A. S. Nagle, E. A. Kapxhiu, R. M. Cross and K. W. Jung, Tetrahedron, 2002, 58, 3329 CrossRef CAS; (b) W. D. McGhee, Y. Pan and D. P. Riley, J. Chem. Soc., Chem. Commun., 1994, 699 RSC; (c) M. Yoshida, N. Hara and S. Okuyama, Chem. Commun., 2000, 151 RSC; (d) R. Sristava, D. Srinivas and P. Ratnasamy, J. Catal., 2005, 233, 1 CrossRef CAS.
- (a) M. Abla, J. C. Choi and T. Sakakura, Chem. Commun., 2001, 2238 RSC; (b) M. Abla, J. C. Choi and T. Sakakura, Green Chem., 2004, 6, 524 RSC.
- K.-I. Tominaga and Y. Sasaki, Synlett, 2002, 2, 307 CrossRef.
- R. Nomura, M. Yamamoto and H. Matsuda, Ind. Eng. Chem. Res., 1987, 26, 1056 CrossRef CAS.
- B. M. Bhanage, S. Fujita, Y. Ikushima and M. Arai, Green Chem., 2003, 5, 340 RSC.
- A. Ion, V. Parvulescu, P. Jacobs and D. De Vos, Green Chem., 2007, 9, 158 RSC.
- (a) T. Sakakura, J. Choi, Y. Saito and T. Sako, Polyhedron, 1999, 19, 573; (b) C. Jiang, Y. Guo, C. Wang, C. Hu, Y. Wu and E. Wang, Appl. Catal., A, 2003, 256, 203 CrossRef CAS; (c) M. Aresta, A. Dibenedetto, C. Pastore, I. Papai and G. Schubert, Top. Catal., 2006, 40, 71 CrossRef CAS.
- (a) W. M. Olmstead, Z. Margolin and F. G. Bordwell, J. Org. Chem., 1980, 45, 3295 CrossRef CAS; (b) W. L. Mock and J. Z. Zhang, Tetrahedron Lett., 1990, 31, 5687 CrossRef CAS.
- Q. Li, N. Zhao, W. Wei and Y. Sun, J. Mol. Catal., 2007, 270, 44 Search PubMed.
| This journal is © The Royal Society of Chemistry 2008 |