The continuous synthesis of ε-caprolactam from 6-aminocapronitrile in high-temperature water
Chong
Yan
a,
Joan
Fraga-Dubreuil
a,
Eduardo
Garcia-Verdugo
a,
Paul A.
Hamley
a,
Martyn
Poliakoff
*a,
Ian
Pearson
b and
A. Stuart
Coote
b
aThe School of Chemistry, The University of Nottingham, Nottingham, UK NG7 2RD. E-mail: martyn.poliakoff@nottingham.ac.uk
bINVISTA Performance Technologies, Wilton, Cleveland, UK TS10 4XX
First published on 15th November 2007
Abstract
Caprolactam (CPL) is a widely used chemical intermediate for the production of Nylon-6. However, existing synthetic routes in industry have severe drawbacks. The development on the synthesis of CPL from 6-aminocapronitrile (ACN), using near- and supercritical water as the solvent, reactant and catalyst, is described in this paper. The two-step reaction (hydrolysis and cyclization) to produce CPL is combined in a single process, by using a continuous-flow system. Effects of pressure, temperature, residence time and the concentration of ACN were studied. The high-temperature high-pressure environment possesses unique properties which result in very efficient catalysis. The overall CPL yield reaches 90% within a short residence time (<2 min).
Background
The production of ε-caprolactam (CPL), the primary use of which is in the production of Nylon-6, is a most industrially important process. About 90% of the CPL is manufactured in processes which are based on (or partially based on) the cyclohexanone route.1 However, two main drawbacks make the process costly and environmentally harmful. Firstly, cyclohexanone is made by the aerobic oxidation of cyclohexane, where the conversion must be kept at only 3–6% to maintain a high selectivity.1 Secondly, although the total yield of CPL from cyclohexanone can approach 98%, the major product generated by the process is actually ammonium sulfate, with CPL as a mere “by-product” in weight terms. Ammonium sulfate is sold as a fertilizer, but it has a limited market as it is only useful in sulfur-deficient soils, and the H2SO4 formed on decomposition acidifies the soil.Most process improvements are focussed on avoiding the production of (NH4)2SO4. Many of these efforts have been introduced.2,3 Some of these developments have been introduced industrially. There are three ammonium sulfate free methods which might be commercialized in the near future:4 (1) ammoximation, (2) vapour phase Beckmann rearrangement and (3) butadiene-based processes. The catalysts involved are often the key factor in these three processes. Both the ammoximation and the vapour phase Beckmann processes are based on the existing reaction route from benzene or toluene, and a 60![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 tonnes year–1 plant was commissioned in 2003 in Japan5 to commercialise the technology which combines a proprietary vapour-phase Beckmann rearrangement6,7 with Italian company EniChem's direct ammoximation method.8 The only by-product is H2O and no ammonium sulfate is co-produced. However, the butadiene-based processes not only avoid co-production of ammonium sulfate, but also decrease the monetary cost by utilising butadiene as a raw material, which is less expensive than benzene or toluene. Also, problems associated with cyclohexane oxidation9,10 can be avoided by using butadiene feedstock. Such processes involve the formation of ACN viaadiponitrile.
000 tonnes year–1 plant was commissioned in 2003 in Japan5 to commercialise the technology which combines a proprietary vapour-phase Beckmann rearrangement6,7 with Italian company EniChem's direct ammoximation method.8 The only by-product is H2O and no ammonium sulfate is co-produced. However, the butadiene-based processes not only avoid co-production of ammonium sulfate, but also decrease the monetary cost by utilising butadiene as a raw material, which is less expensive than benzene or toluene. Also, problems associated with cyclohexane oxidation9,10 can be avoided by using butadiene feedstock. Such processes involve the formation of ACN viaadiponitrile.
Our work describes the preparation of CPL and NH3 from ACN in a continuous-flow reactor, using high-temperature water (HTW) as an alternative, environmentally benign solvent (Scheme 1). The patent literature has several reports of this reaction being carried out catalytically in organic solvents.11,12 The reaction of ACN to form CPL in the absence of catalysts and organic solvent is quite rare. In one patent from BASF,13 a solution of 10% by weight of ACN in H2O was heated to 300 °C in a tube reactor (volume 300 mL) with an average residence time of 1 h. All of the ACN was converted but some components with high boiling point were produced during this process and needed to be recycled; thus a yield of 93% CPL could be achieved. Also, Vogel et al. described results from this reaction in supercritical water in their reviews;14,15 an ACN conversion of ca. 70% and a selectivity of CPL of ca. 66% (i.e., a CPL yield of less than 50%) was obtained at 350 °C and 250 bar with a residence time of 240 s. Compared to these studies, our method for CPL production from ACN, using near- and supercritical water as the solvent, reactant and catalyst, provides advantages of efficiency, productivity and green technology.
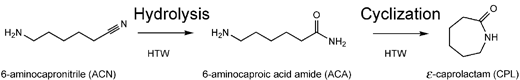 | ||
| Scheme 1 Hydrolysis of ACN followed by cyclization of ACA to CPL in HTW. | ||
From an industrial aspect, the use of supercritical water can be advantageous. H2O is abundant, inexpensive, non-flammable and non-toxic, but the use of ambient water is limited by its high polarity and the consequent poor solubility of organic molecules. However, water becomes a tunable solvent for organics under high-temperature and high-pressure conditions. Two important properties—adjustable static dielectric constant (ε) and ionic product (Kw)—make supercritical water a unique reaction medium. As the temperature is increased, the dielectric constant of water becomes successively comparable to those of conventional organic solvents at ambient conditions (e.g., methanol, ethanol, acetone, and hexane).16 Thus, small organic compounds and most gases are highly soluble in near- and supercritical water. At a constant pressure of 250 bar, the ionic product of water first increases with temperature until it reaches a maximum (10–11) at 250 °C, and then decreases to 10–19 at 400 °C and 10–22 at 500 °C. This not only allows acid- and base-catalysed reactions to be performed in HTW without additional catalyst, but also allows the medium to be varied between ionic and radical environments. Our group has reported the hydrolysis of esters17 and nitriles,18 the reduction of nitroarenes and subsequent cyclization to quinolines,19 the partial oxidation of p-xylene (to terephthalic acid) and other xylenes,20–22 the use of water as a reactant in hydrogen exchange reactions,23 and synthesis of benzimidazoles.24 In these reactions, water plays an important role as a solvent, reactant, catalyst, or a combination of these.
Experimental
Caution: these reactions involve high pressures and should only be carried out in an apparatus with the appropriate pressure rating at the reaction temperature. A thorough safety assessment should be made. All experiments were conducted using a tubular continuous-flow reactor, Fig. 1. Generally, an aqueous solution of feedstock is pumped through the pre-heater to the reactor. After reaction, the mixture is cooled, filtered and released via the back pressure regulator (BPR) which maintains the system pressure. The pre-heater and the reactor are similar in construction, consisting of 1/16 inch stainless steel tubing coiled around an aluminium block. A cartridge heater and a band heater are used to supply heat. The temperature of the fluid is measured by thermocouples secured in a Swagelok T-piece with the tip of the thermocouple in the flow. The reason for using the combination of pre-heater and reactor instead of a single reactor with longer tubing is that the heating is more uniform and a temperature monitoring point can be inserted between the pre-heater and the reactor. Before each run, the apparatus was hydrostatically pressure tested when cold, and was then heated with a flow of pure water (1.5 mL min–1) at a given pressure. Once the operating temperature had been reached, the feedstock was switched from pure water to the ACN aqueous solution. Thirty minutes were allowed for the apparatus to equilibrate, and samples were collected every 15 min for a period of 3 min. Typically, an experiment was performed over approximately 2 h.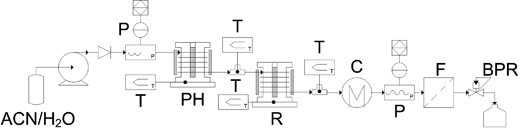 | ||
| Fig. 1 Scheme of the supercritical water continuous-flow reactor for the reaction of ACN to CPL (P: pressure monitor; T: thermocouple; PH: pre-heater; R: reactor; C: cooler; F: filter; BPR: back pressure regulator). | ||
ACN was supplied by INVISTA Performance Technologies. All the other chemicals were used as received (Aldrich) and H2O was HPLC grade triply distilled. Residence time was calculated from the total reactor volume (i.e., the volume of the pre-heater plus that of the reactor) divided by the volumetric flow rate at operating temperature and pressure. The volumetric flow rate of the reaction mixture was calculated using the physical properties of H2O at the high-temperature high-pressure reaction conditions, as published by the US National Institute of Science and Technology (NIST). Samples were analysed by gas chromatography (GC), using a temperature programmable PERKIN ELMER AutoSystem instrument equipped with a 30 m × 0.32 mm × 1.0 mm non-polar capillary column, a flame ionization detector, and He as the carrier gas. The temperature gradient was 20 °C min–1 from 100 °C to 300 °C. The retention times were 4.6 min (ACN), 5.6 min (CPL) and 6.7 min (ACA). The calculation of yields and selectivity was based on the molar amount of each compound.
Since the thermo-physical properties of the reaction mixture are unknown, pure H2O is taken as the model fluid. Density and viscosity data are from NIST; Static dielectric constant and ionic product data were calculated by using the empirical equations from Franck et al.25,26
Results and discussion
Stability of CPL in HTW
To check the feasibility of the synthetic route from ACN to CPL (see Scheme 1), the stability of CPL was initially examined. A solution of CPL in H2O (2.4 mol l–1) was pumped through the system at the flow rates, pressures and temperatures shown in Table 1. Greater decomposition of CPL was observed at lower flow rates. However, the stability cannot be attributed only to the effect of residence time as suggested by entries 2 and 5 because the real properties of this mixture and its flow type in the tubular reactor are uncertain. Even at the longest residence time (44 s), the recovery of CPL was still over 80%. Therefore CPL is sufficiently stable for the reaction to be carried out in HTW.| Entry a | T/°C | p/bar | Flow rate/mL min–1 | Recovery of CPL (%) b | Residence time/sc |
|---|---|---|---|---|---|
| a The starting material was an aqueous solution of CPL (2.4 mol l–1). b The recovery of CPL was calculated by GC, reproducible to ±2%. c For calculation of residence time, see Experimental. | |||||
| 1 | 25 | 200 | 1.5 | 100 | —— |
| 2 | 400 | 200 | 1.5 | 85 | 9 |
| 3 | 400 | 350 | 1.5 | 84 | 44 |
| 4 | 400 | 200 | 5 | 96 | 3 |
| 5 | 400 | 350 | 5 | 97 | 13 |
Effect of temperature
Temperature is one of the important parameters in HTW, because it affects the yield not only by changing the solvent properties of H2O, but also by modifying the rate of the reaction. Vogel et al.14 reported that the conversion of ACN at high temperature and 250 bar displayed pseudo-first-order kinetics with a low temperature dependency.We have studied the effect of temperature (250–450 °C) on the synthesis of CPL from ACN in HTW at 200 bar (a pressure in the near-critical region where the concentrations of H+ and OH– can change greatly with temperature), with a flow rate of 1.5 mL min–1. The concentration of feedstock was 30% (v/v). At the highest temperature (450 °C), oil-like and even solid by-products (insoluble in methanol) were formed, and were not characterised by GC. For all the other runs, CPL was formed selectively as the sole, stable final product from the intermediate ACA. From Fig. 2, it can be seen that the conversion of ACN rose from 14% to 34% with increasing temperature. The selectivities to ACA and CPL vary with temperature; the yield of ACA first decreased to its minimum (less than 5%) and then started to increase (>10%), while the yield of CPL first increased to its maximum (up to 24%) and then started to decrease.
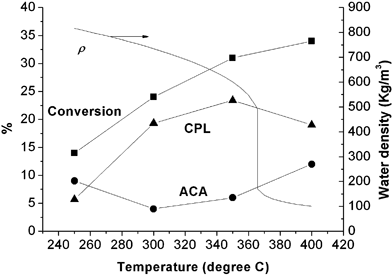 | ||
| Fig. 2 The effect of temperature on the reaction of 30% (v/v) ACN in HTW with a flow rate of 1.5 mL min–1 and a pressure of 200 bar. When the temperature increased, the conversion of ACN increased up to 34%; the yield of CPL first increased to the maximum of 23% and then decreased; and the yield of ACA first decreased to the minimum of 4% and then increased. The density of H2O decreased with temperature from 816 kg m–3 to 101 kg m–3, causing a decrease of calculated residence time from 75 s to 6 s (-■-: conversion of ACN; -●-: yield of ACA; -▲-: yield of CPL). | ||
Vogel et al. also noticed that the selectivity to CPL decreased considerably above 380 °C. However, this could also be the effect of fluid density. In a continuous-flow system, a compressible fluid will expand when the system temperature is increased at constant pressure; this results in a decrease in density and consequently in the residence time of the substrate. In these runs, the calculated residence time decreased from 75 s at 250 °C to 6 s at 400 °C. Thus, the effect of temperature on the reaction can be rationalised as follows: high temperatures improve the conversion of ACN and ACA, but the conversion of the intermediate ACA to CPL is limited by the reduction in residence time. However, it is also possible that the rate coefficients of the two reaction steps respond differently to changes in temperature. Furthermore, the hydrolysis of nitriles is known to follow a proton-catalysed mechanism, involving several proton transfers.27 At higher temperature, therefore, the factors of faster molecular motion, lower solvent viscosity (from 1.1 × 10–4 Pa s at 250 °C to 2.6 × 10–5 Pa s at 400 °C) and breakdown of H-bond network28 enhance the efficiency of the reaction even though the ionic product is lower.
Effect of pressure
Since the conversion of ACN was found to be favoured at 400 °C, a series of experiments was carried out to study the effect of pressure at this temperature at a constant flow rate of 1.5 mL min–1. The same concentration of ACN aqueous solution, 30% (v/v), was used at pressures between 150 and 400 bar. For all runs, CPL was formed selectively as the stable final product from the intermediate ACA. It can be seen from Fig. 3 that the conversion of ACN and the yield of CPL were favoured at higher pressure (increasing to 66% and 58% respectively), while the yield of ACA was reduced at higher pressure (lowered to less than 3%).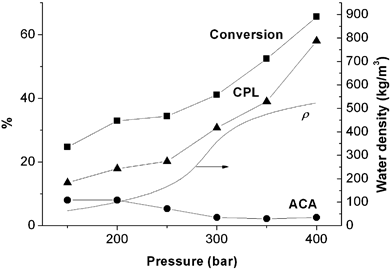 | ||
| Fig. 3 Observed effect of pressure on the reaction of 30% (v/v) ACN in HTW with a flow rate of 1.5 mL min–1 and a temperature of 400 °C. The conversion of ACN (up to 66%) and yield of CPL (up to 58%) increased with pressure. The yield of ACA decreased to less than 5% at higher pressures. The corresponding increase in the density of H2O is from 64 kg m–3 to 523 kg m–3, as indicated on the right hand axis. | ||
One of the advantages of HTW compared to traditional solvents, is that pressure can be used to adjust the solvent power of the fluid, and also to alter the concentration of H+ and OH– in a reaction environment. In the present reaction, pressure can tune the solvent properties of HTW to favour formation of CPL; the ionic product of pure H2O increases with pressure at 400 °C, up to 10–12.5 at 400 bar (compared to 10–14 for H2O at ambient temperatures). Also, according to isothermal molecular dynamics simulations by Laria et al.,29 the overall proton transport increases with pressure. The catalytic properties of H2O are therefore enhanced at elevated pressure. At the same time, higher pressure favours the addition of H2O to the nitrile group of ACN, since the activation volume ΔV‡ is negative.
Furthermore, when this compressible fluid (HTW) is under plug flow conditions in a continuous-flow system, increasing pressure at constant temperature (isothermally) will increase the density of the fluid, and hence increase the residence time of the substrate. For the study of pressure at 400 °C with a constant flow rate of 1.5 mL min–1, the calculated residence time increased from 6 s at 150 bar, to 48 s at 400 bar. Since the reaction in HTW is very selective, longer residence time will lead to a higher yield of the final product and less ACA will remain.
Reproducibility of the synthesis
Although there are no deactivation issues because H2O is the only catalyst, the reproducibility of this process is important from an industrial point of view. Therefore, reaction was carried out for 12 h by pumping a 30% (v/v) aqueous solution of ACN continuously into the unit at 1.5 mL min–1 and at 400 °C and 400 bar, giving a calculated residence time of 48 s, with a sample being collected every hour (for a period of 3 min) and analysed by GC. The results shown in Fig. 4 suggest that CPL was formed reproducibly over this extended period with yields of CPL between 53–57%, and with ACN conversions remaining constant ca. 65%.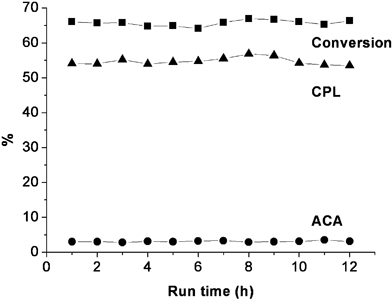 | ||
| Fig. 4 Study of the reproducibility on the reaction of 30% (v/v) ACN in HTW with a flow rate of 1.5 mL min–1 under a temperature of 400 °C and a pressure of 400 bar. CPL yields between 53–57% were achieved (with an average of 54.7% and standard deviation of 1.1%), while ACN conversions remained constant ca. 65% (65.7 ± 0.8%). The yield of ACA was kept below 5%. | ||
Effect of feedstock concentration
In this system, both the starting material (ACN) and the product (CPL) have a high solubility in H2O at 20 °C; CPL is 82% (w/w),1 and ACN is 50% (v/v). From an economic point of view, there is an obvious interest in producing CPL in HTW at the highest feedstock concentration. The reaction was therefore investigated with feedstock concentrations between 5% and 40% (v/v) at 400 °C and 400 bar using a total flow rate of 1.5 mL min–1 (see Fig. 5). In all the runs, CPL was formed selectively as the stable final product via the intermediate 6-aminocaproic acid amide (ACA), which was obtained in less than 5% yield. Although no exotherm was observed, a higher concentration of starting material led to higher conversion (up to 73%) and higher yield of the product (up to 61%) with the same calculated residence time of 48 s. The feedstock from 5% to 40% changed the yield from 47% to 61%. This suggests the high productivity is a function of the solubility of the starting material in H2O. For practical reasons, a 30% concentration was favoured because particles of ACN could possibly precipitate and cause a blockage, at higher concentration of feedstock.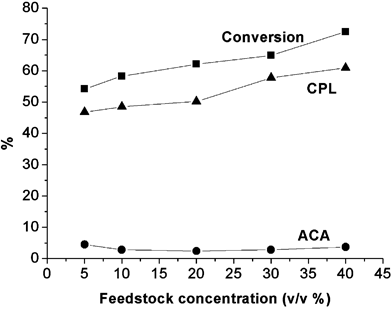 | ||
| Fig. 5 Effect of feedstock concentration on the reaction of ACN in HTW at 400 °C and 400 bar, with a flow rate of 1.5 mL min–1 (calculated residence time = 48 s). The conversion of ACN and yield of CPL increase with increasing feedstock concentration (-■-: conversion of ACN; -●-: yield of ACA; -▲-: yield of CPL). | ||
Vogel et al. reported that the conversion of 5% (w/w) ACN in HTW could be described by pseudo-first order kinetics.14 However, we found that the conversion of ACN increased with increasing feedstock concentration, so that this reaction is unlikely to be first order but could possibly be second order. Alternatively, increasing feedstock concentration might change the properties of the reaction mixture and hence change the reaction rate.
Effect of the residence time
Our final objective was establishing whether the conversion of ACN could be improved at higher residence time. A simple way of increasing the residence time is to collect the solution emerging from the reactor and to pump it through the reactor a second or even a third time under the same conditions of temperature and pressure. Our experiments were carried out as before at 400 °C and 400 bar at a flow rate of 1.5 mL min–1 and a calculated residence time of 48 s. To compare with the results of Vogel et al.,14 two concentrations of ACN, 5% (v/v) and 30% (v/v), were used, see Table 2.| Entry | Feedstock concentration (v/v%) | Conversion of ACN (%)a | Yield of ACA (%)a | Yield of CPL (%)a | Calculated residence timeb/s |
|---|---|---|---|---|---|
| a The conversion and yields were calculated by GC, reproducible to ±2%. b For calculation of residence time, see Experimental. c The results of Vogel et al.14 | |||||
| 1c | 5 | 70 | 4 | 45 | 240 |
| 2 | 5 | 53 | 5 | 47 | 48 |
| 3 | 5 | 68 | — | 67 | 2 × 48 |
| 4 | 5 | 85 | — | 80 | 3 × 48 |
| 5 | 30 | 65 | 2 | 58 | 48 |
| 6 | 30 | 94 | 1 | 90 | 2 × 48 |
It can be seen from Table 2 that 3 runs were needed for the 5% (v/v) feedstock to achieve an ACN conversion of 85% and a CPL yield of 80%, while only 2 runs were necessary for the 30% (v/v) feedstock to obtain an ACN conversion of 94% and a CPL yield of 90%. According to the best result of Vogel et al.,14 an ACN conversion of ca. 70% and a selectivity of CPL of ca. 66% gave an actual CPL yield of <50% at 350 °C and 250 bar with a residence time of 240 s. Thus, it is evident that our process, using a higher temperature, pressure and concentration of ACN, is an improvement on the previously published method. However, it should be noted that our process of repeated runs not only increases the residence time, but also, during the depressurization between the runs, releases the NH3 generated from hydrolysis, which appears to inhibit further reaction. The same idea of using two or more reactors in series with provision for mid-process pressure let-down and “refluxing” at atmospheric pressure or less to remove intermediate NH3 was suggested more than 60 years ago in a patent,30 where the reaction was conducted at temperatures below 300 °C. However, we found that this “refluxing” method worked better with 30% (v/v) feedstock than that with 5% (v/v), suggesting that it could be a combined effect of releasing NH3 and phase behaviour of the reaction mixture.
Conclusion
The results presented here demonstrate that CPL can be formed selectively and continuously from ACN in HTW. A yield of 90% CPL and a conversion of 94% ACN have been achieved within a calculated residence time of 96 s at 400 °C and 400 bar. Also increasing the concentration of ACN (up to 40% v/v aqueous solution) improves the efficiency of the reaction.A study of the effect of temperature, pressure and residence time performed here shows that HTW is an easily adjustable environment which allows simple optimisation of the reaction.
Compared to previous studies of this reaction, our process in HTW is the first to achieve a high conversion and high yield without using either an additional catalyst or an organic solvent. Also the reaction time has been shortened from hours to seconds, compared to those patented processes discussed in this paper. In addition, the higher temperatures used permit more efficient recovery of reaction exotherm. This highly efficient and eco-friendly system has promise for industrial applications.
Acknowledgements
We thank S. D. Housley, W. B. Thomas, G. R. Aird and M. L. Thomas for their advice. The authors would also like to thank M. Guyler, R. Wilson and P. A. Fields for their help and INVISTA Performance Technologies and the University of Nottingham for financial support.References
- F. Ullmann, Ullmann's encyclopaedia of industrial chemistry [electronic resource], John Wiley & Sons, Inc., New York, 2002 Search PubMed.
- K. Weissermel and H.-J. Arpe, Industrial Organic Chemistry, VCH, New York, 1997 Search PubMed.
- G. Dahlhoff, J. P. M. Niederer and W. F. Hoelderich, Catal. Rev. Sci. Eng., 2001, 43(4), 381–441 CrossRef CAS.
- H. Ichihashi and H. Sato, Appl. Catal., A, 2001, 221(1–2), 359–366 CrossRef CAS.
- C. O'Driscoll, Eur. Chem. News, 2004, 81(2111), 16 Search PubMed.
- H. Sato, K. Hirose, N. Ishii and Y. Umada, Sumitomo Chemical Co., Ltd., Japan, JP 62123167, 1987.
- H. Sato, K. Hirose, M. Kitamura, Y. Umada, N. Ishii and H. Tojima, Sumitomo Chemical Co., Ltd., Japan, EP 242960, 1987.
- P. Roffia, M. Padovan, E. Moretti and G. De Alberti, Montedipe S.p.A., Italy, EP 208311, 1987.
- U. Schuchardt, W. A. Carvalho and E. V. Spinace, Synlett, 1993(10), 713–718 Search PubMed.
- U. Schuchardt, D. Cardoso, R. Sercheli, R. Pereira, R. S. da Cruz, M. C. Guerreiro, D. Mandelli, E. V. Spinace and E. L. Pires, Appl. Catal., A, 2001, 211(1), 1–17 CrossRef CAS.
- L. Gilbert, N. Laurain, P. Leconte and C. Nedez, Rhone-Poulenc Fiber & Resin Intermediates, Fr.; Rhodia Polyamide Intermediates, EP 748797, 1996.
- F. Ohlbach, A. Ansmann, P. Bassler, R.-H. Fischer, H. Luyken, S. Maixner and J.-P. Melder, BASF Aktiengesellschaft, Germany, WO 2001083441, 2001.
- G. Achhammer and E. Fuchs, BASF A.-G., Germany, US 5495016, 1996.
- A. Kramer, S. Mittelstadt and H. Vogel, Chem. Eng. Technol., 1999, 22(6), 494–500 CrossRef CAS.
- D. Broll, C. Kaul, A. Kramer, P. Krammer, T. Richter, M. Jung, H. Vogel and P. Zehner, Angew. Chem., Int. Ed., 1999, 38(20), 2999–3014 CAS.
- CRC Handbook of Chemistry and Physics : a ready-reference book of chemical and physical data, ed. D. R. Lide, CRC Press, Boca Raton, FL, 2002 Search PubMed.
- P. A. Aleman, C. Boix and M. Poliakoff, Green Chem., 1999, 1(2), 65–68 RSC.
- E. Venardou, E. Garcia-Verdugo, S. J. Barlow, Y. E. Gorbaty and M. Poliakoff, Vib. Spectrosc., 2004, 35(1–2), 103–109 CrossRef CAS.
- C. Boix, J. M. de la Fuente and M. Poliakoff, New J. Chem., 1999, 23(6), 641–643 RSC.
- P. A. Hamley, T. Ilkenhans, J. M. Webster, E. Garcia-Verdugo, E. Venardou, M. J. Clarke, R. Auerbach, W. B. Thomas, K. Whiston and M. Poliakoff, Green Chem., 2002, 4(3), 235–238 RSC.
- E. Garcia-Verdugo, E. Venardou, W. B. Thomas, K. Whiston, W. Partenheimer, P. A. Hamley and M. Poliakoff, Adv. Synth. Catal., 2004, 346(2–3), 307–316 CrossRef CAS.
- E. Garcia-Verdugo, J. Fraga-Dubreuil, P. A. Hamley, W. B. Thomas, K. Whiston and M. Poliakoff, Green Chem., 2005, 7(5), 294–300 RSC.
- C. Boix and M. Poliakoff, Tetrahedron Lett., 1999, 40(23), 4433–4436 CrossRef CAS.
- L. M. Dudd, E. Venardou, E. Garcia-Verdugo, P. Licence, A. J. Blake, C. Wilson and M. Poliakoff, Green Chem., 2003, 5(2), 187–192 RSC.
- M. Uematsu and E. U. Franck, J. Phys. Chem. Ref. Data, 1981, 9(4), 1291–1306.
- W. L. Marshall and E. U. Franck, J. Phys. Chem. Ref. Data, 1981, 10(2), 295–304 CAS.
- J. Clayden, N. Greeves, S. Warren and P. Wothers, Organic Chemistry, Oxford University Press, Oxford, 2001 Search PubMed.
- M. Boero, T. Ikeshoji, C. C. Liew, K. Terakura and M. Parrinello, J. Am. Chem. Soc., 2004, 126(20), 6280–6286 CrossRef CAS.
- D. Laria, J. Marti and E. Guardia, J. Am. Chem. Soc., 2004, 126(7), 2125–2134 CrossRef CAS.
- E. L. Martin, E. I. du Pont de Nemours & Co., US 2301964, 1942.
| This journal is © The Royal Society of Chemistry 2008 |