The bulk oxypropylation of chitin and chitosan and the characterization of the ensuing polyols
Susana
Fernandes
,
Carmen Sofia Rocha
Freire
*,
Carlos Pascoal
Neto
and
Alessandro
Gandini
CICECO and Department of Chemistry, University of Aveiro, Campus de Santiago, 3810-193 Aveiro, Portugal. E-mail: cfreire@ua.pt; Fax: +351 234 370 084; Tel: +351 234 401 507
First published on 14th November 2007
Abstract
Chitin and chitosan were converted into viscous polyols through a simple oxypropylation reaction, with the aim of valorising the less noble fractions or by-products of these valuable renewable resources. This process bears “green” connotations, given that it requires no solvent, leaves no by-products and no specific operations (separation, purification, etc.) are needed to isolate the entire reaction product. Chitin or chitosan samples were preactivated with KOH and then reacted with an excess of propylene oxide (PO) in an autoclave. In all instances, the reaction product was a viscous liquid made up of oxypropylated chitin or chitosan and PO homopolymer. The two fractions were separated and thoroughly characterized by FTIR and NMR spectroscopy, DSC, TGA and viscosity.
Introduction
The exploitation of renewable resources for both energy and materials has been attracting considerable attention recently, mainly in response to the economical and environmental problems associated with the use of fossil counterparts. In particular, the vast quantities of by-products arising from agricultural, marine and forestry activities represent a very promising first generation of natural resources, available for specific chemical modifications aimed at generating novel materials. Two aspects are relevant here, viz., the fact of (i) selecting only the less noble parts of these commodities for the modifications, leaving the more valuable ones for well-established uses (food, timber, papermaking, pharmaceuticals, etc.) and (ii) valorising all the components of a given resource through the biorefinery working hypothesis, instead of selecting only that or those which appear to be more valuable. Indeed, a growing number of studies show that the so-called by-products can in fact be the precursors to materials with remarkable properties and high added value.Among the numerous approaches investigated within this context,1–4 only the bulk oxypropylation of natural polymeric substrates is discussed here. Lignin, sugar beet pulp and cork powder are secondary products of major industrial activities, mainly used today as sources of energy by combustion. In previous studies, they have been efficiently converted by a single-step oxypropylation reaction into liquid viscous polyols,5–11 which are interesting macromonomers for the synthesis of polyurethanes and polyesters.6,12,13 These systems are particularly straightforward in terms of their implementation, since they only involve the mixing of the activated solid substrate with propylene oxide (PO) in an autoclave and the subsequent heating of the ensuing suspension until the total consumption of the added PO. The recovery of the final polyol mixture is extremely simple, because it does not require the removal of any solvent or other component, nor any separation or purification procedure. The green connotations of the entire operation are therefore quite relevant. However, it is important to emphasize that PO is a hazardous chemical in terms of toxicity and flammability. In our systems, both in the past and in the present context, the fact that PO was always totally used up in the oxypropylation represents an element of safety, but, of course, particular care must be taken during these reactions to avoid any loss of control, which might lead to PO contamination.
Chitin is the second most abundant natural polymer and its partly deacetylated derivative, chitosan, represents one of the most actively investigated materials because of its unique properties and applications.14
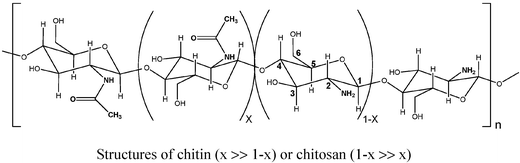
To the best of our knowledge, this specific approach had never been applied to chitin, and only once very succinctly to chitosan.9 The studies related to the preparation of hydroxypropyl chitin and chitosan bearing very short grafts15–22 and destined to biomedical applications, all involved the use of a solvent and therefore required a laborious workup, isolation, and purification of the final product. This state of affairs prompted us to extend to chitin and chitosan the successful approach already applied to other natural polymers,5–13 with the aim of valorising the less noble fractions and by-products arising from the industrial process consisting of the extraction of chitin and its conversion into chitosan.
This paper describes a preliminary study of the bulk oxypropylation of chitin and chitosan and the characterization of the ensuing polyols.
Experimental
Materials
The chitin (Mw ∼ 6 × 105) and chitosan (Mw = 5.4 × 105) samples used in this work were generously provided by Mahtani Chitosan Pvt. Ltd. (India). The deacetylation degree (DDA) of the chitosan was 92%. Propylene oxide (PO) 99% was supplied by Sigma–Aldrich and employed as received.Oxypropylation
The oxypropylation reactions were carried out in a 300 cm3 stainless steel autoclave equipped with stirring, a heating resistance and temperature and pressure sensors. The chitin and chitosan samples (10 g) were preactivated in the reaction vessel with an ethanol–KOH solution for 1 h at room temperature under a nitrogen atmosphere. The dried activated-substrate was then mixed with PO (40 and 20 mL, respectively for chitin and chitosan) and allowed to react, with constant mechanical stirring at 1000 rpm, at 140 °C (set 1) and 120 °C (set 2). The higher amount of PO in the experiments with chitin was employed to guarantee a thorough impregnation of the substrate, which was particularly fluffy. The onset of the oxypropylation reaction was revealed by a rapid increase in temperature (max. 220 °C) and pressure (max. 15 bar) and its completion by the return of the pressure to its atmospheric value. The reaction time varied between 1 and 2 h. In all these experiments, no unreacted PO was detected after opening the autoclave.The viscous products were diluted with dichloromethane and filtered through a highly porous cotton fabric. The solid residues (SR) were washed several times with dichloromethane, dried and weighed in order to determine the percentage of unreacted, or poorly reacted, substrate. The solvent and other volatile components of the filtrates were removed in a rotary evaporator leaving a viscous polyol, which was then extracted with n-hexane in order to separate the oxypropylated chitin or chitosan (hexane insoluble material, PL) from the PO homopolymer fraction (HP). The separation procedures were only applied in order to characterize all the products of the reaction. In practice, however, the polyol mixtures can be used, as such, without any separation or purification.
Some of the solid products were submitted to a second oxypropylation in the same conditions in order to verify whether they were intractable structures or simply residual polysaccharides which had not been sufficiently modified during the first treatment. The fact that in all instances, liquid polyols were obtained, proved that the latter reason had been the cause of these incomplete oxypropylations.
Characterization
The FTIR spectra were taken with a Mattson 7000 spectrophotometer equipped with a single horizontal Golden Gate ATR cell. The thermogravimetric tests were carried out with a Shimadzu TGA 50 analyser equipped with a platinum cell. Samples were heated at a constant rate of 10 °C min–1 from room temperature to 800 °C, under a nitrogen flow of 20 mL min–1. The thermal decomposition temperature was taken as the onset of a significant (≥0.5%) weight loss from the heated sample, after the initial moisture loss. The elemental analyses were conducted with a Leco CNHS Elemental Analyser. The DSC thermograms were traced with a Setaram analyzer scanning at 10 °C min–1 in a stream of helium. Scanning at 2 °C min–1 gave essentially the same results. The 1H–NMR (in CDCl3 for the HP e PL fractions and in CD3CO2D (1%)/D2O for chitosan) spectra were recorded with a Bruker Avance (Wissembourg, France) spectrometer working at 300.13 MHz. Chemical shifts are expressed in δ (ppm) values relative to tetramethylsilane (TMS) as the internal reference. The viscosities of the chitin- and chitosan-based polyols (PL) were measured with a controlled-stress AR 1000 TA rheometer, fitted with a cone-plate geometry (40 mm diameter and 4° angle).Results and discussion
The purpose of this investigation was to establish the feasibility of converting chitin and chitosan into viscous polyolsvia a simple oxypropylation reaction, without any attempt to optimize the processes through a systematic study of the role of each parameter. The mechanism of these bulk oxypropylations calls upon the activation of some of the substrate OH groups by a Brønsted or Lewis base to produce the corresponding oxianions, from which PO oligomers are grafted by its ring-opening anionic polymerization. Because transfer reactions inevitably occur in this process, the formation of some PO homopolymer (PPO) always accompanies the actual oxypropylation.With the conditions described above, the extent of oxypropylation of both chitin and chitosan was relatively high, but not complete, as measured by the relatively modest amounts of unreacted or poorly oxypropylated solid residues (5–15% and ∼25%, respectively for chitin and chitosan). The lower reactivity of chitosan was interpreted on the basis of its higher cohesive energy23 arising from the very strong intermolecular hydrogen bonds involving both its OH and NH2groups, because the reoxypropylation of the corresponding solid residue gave the same result (∼25% of unreacted material), suggesting that the added amount of PO had not been the limiting factor in the first treatment. It is important to emphasize here that there is no doubt in our minds that the systematic study of both these systems will provide the appropriate conditions insuring total conversion of the substrates into liquid products.
As mentioned above, the oxypropylation of OH-containing substrates, such as polysaccharides, inevitably gives two products, namely the oxypropylated macromolecules and some PPO.5–8,11 Their relative proportion, which depends on the reaction conditions, obviously influences the physical properties, as well as the reactivity of these polyol mixtures. It has been shown that these two polymers can be efficiently separated by extracting the reaction mixture with n-hexane,8 since the HP fraction is selectively removed by this solvent. In the present study, the proportion of HP formed was systematically around ∼40%, in close agreement with that obtained with other natural substrates oxypropylated under similar reaction conditions.5,6,8
FTIR analysis
Fig. 1 shows typical FTIR spectra of chitosan and the two liquid polyol fractions (HP and PL) resulting from its oxypropylation. As expected, the spectrum of HP displayed the same bands as those of a commercial sample of PPO, viz. around 3380 cm–1, assigned to the OH stretching modes; in the range 2870–2970 cm–1 for the C–H stretching modes of the aliphatic CH3 CH2 and CH groups; and around 1080 cm–1 for the C–O–C moieties.24 The spectrum of the chitosan PL also showed the latter features, plus an additional peak around 1590 cm–1, assigned to the N–H deformation mode of primary amines,24 arising from the chitosan monomer units. These results corroborated the occurrence of the oxypropylation reaction through the grafting of PPO chains onto the polysaccharide backbone and the efficiency of the use of n-hexane as a discriminating solvent.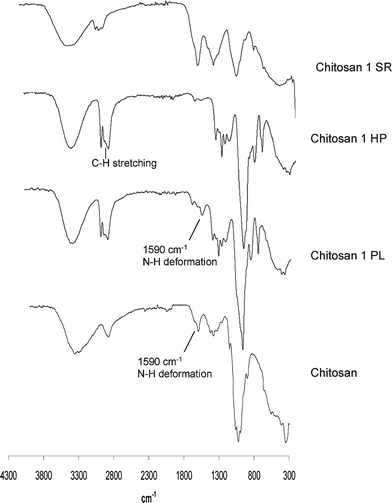 | ||
| Fig. 1 Typical FTIR spectra of chitosan, the two liquid polyol fractions (HP and PL) and the solid residue (SR) resulting from its oxypropylation at 140 °C. | ||
The two polyol fractions obtained in the oxypropylation of chitin also presented different FTIR spectra, but with the carbonyl amide band at 1680–1630 cm–1, resulting from the chitin monomer units, as the main distinguishing feature.
The solid residues isolated in both chitin and chitosan modification reactions showed evidence of a modest oxypropylation, since their infrared spectra differed from those of the respective initial substrates, mostly because of the presence of the characteristic PO-unit CH3 peak around 2960 cm–1.
1H–NMR analysis
Fig. 2 shows typical 1H–NMR spectra of native chitosan and of the two chitosan-related products, following the extraction with n-hexane. The 1H spectrum of chitosan, obtained in an acidic solution at 50 °C, is in close agreement with the literature.25,26 On the other hand, the soluble material gave a spectrum very similar to that of commercial PPO, except for a slightly higher integration in the 3–5 ppm region, characteristic of CH–O and CH2–O protons, compared with that of the methyl groups around 1 ppm, suggesting that small amounts of the oxypropylated chitosan had also been extracted, since for PPO alone those integrations are identical. The spectrum of the n-hexane-insoluble polyol showed, as expected, a higher contribution of the ether-type peaks, reflecting the presence of the chitosan backbones. In the case of the corresponding products relative to the oxypropylation of chitin, the extracted material gave a spectrum virtually identical to that of PPO, whereas that of the residue showed a less pronounced relative integration of the ether protons, compared with the chitosan-based counterpart. These results corroborated the previous indication related to the lower reactivity of chitosan in these oxypropylation conditions.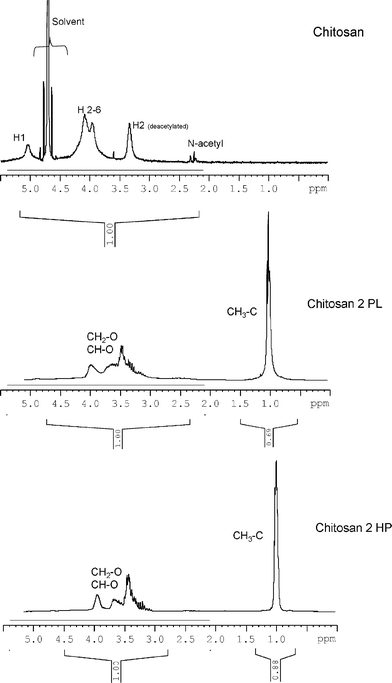 | ||
| Fig. 2 1H–NMR spectra of chitosan and of the two fractions (HP and PL) obtained after its oxypropylation at 120 °C. | ||
Elemental analysis
Table 1 gives a selection of results related to the elemental analysis of the different fractions isolated following the oxypropylation of both substrates. The low, but non-zero nitrogen content of the HP fractions confirmed the conclusions drawn from the 1H–NMR spectra concerning the fact that n-hexane actually extracted the PPO and a very small amount of oxypropylated substrate, more so in the case of chitosan. The nitrogen content of the oxypropylated chitin and chitosan fractions was however much higher than that of their corresponding homopolymeric fractions, as expected for these truly grafted samples.| Sample | C | N | H |
|---|---|---|---|
| Chitin | 41.87 | 6.03 | 6.41 |
| Chitosan | 37.22 | 7.16 | 6.86 |
| Chitin1 HP | 57.79 | 0.42 | 9.90 |
| Chitin2 HP | 57.42 | 0.47 | 10.22 |
| Chitin1 PL | 56.25 | 1.82 | 8.79 |
| Chitin2 PL | 55.24 | 1.84 | 8.25 |
| SR chitin | 52.54 | 2.57 | 8.10 |
| ROx chitin HP | 57.72 | 0.47 | 9.70 |
| ROx chitin PL | 53.19 | 1.59 | 8.46 |
| Chitosan1 HP | 53.09 | 1.21 | 10.16 |
| Chitosan2 HP | 54.85 | 0.65 | 9.86 |
| Chitosan1 PL | 54.29 | 2.23 | 8.45 |
| Chitosan2 PL | 53.75 | 2.67 | 8.20 |
| SR chitosan | 42.36 | 4.54 | 6.55 |
| ROx chitosan HP | 56.16 | 0.41 | 9.79 |
| ROx chitosan PL | 47.52 | 2.04 | 7.67 |
In addition, the nitrogen contents of the solid residues were lower than those of the initial substrates, but higher than those of the corresponding n-hexane insoluble products, confirming that the residues were composed mainly of weakly oxypropylated chitin or chitosan, as already suggested on the basis of the FTIR analysis and of the reoxypropylation experiments. Interestingly, these second experiments gave, not only a similar percentage of solid residue, but also elemental analyses which replicated those related to the corresponding first run, as shown in Table 1.
Thermogravimetric analysis
The two fractions resulting from the oxypropylation of these natural substrates showed in all cases markedly different TGA profiles (Fig. 3). Thus, the HP fractions displayed a typical single weight loss and a maximum decomposition temperature at 240–290 °C, characteristic of POP. On the other hand, the oxypropylated counterparts gave profiles which were a combination of those of the corresponding natural polymer and of PPO, with two main losses at 250–270 and 350–370 °C, indicating that the grafted architecture of these materials did not alter their thermal degradation in relation to their separate components.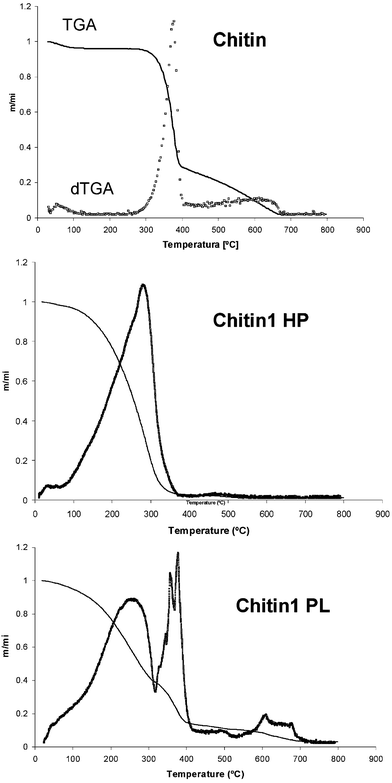 | ||
| Fig. 3 TGA thermograms of the two fractions (HP and PL) obtained from the oxypropylation of chitin at 140 °C. | ||
Differential scanning calorimetry
The Tg of the HP products were consistently around –75 °C, i.e. the typical value for low molecular-weight PPO (Fig. 4). The n-hexane-insoluble products gave Tg values of about –55 °C for both oxypropylated polysaccharides (Fig. 4). This increase in Tg reflects the stiffening role of the natural polymer backbone, but the modest increment suggests that the PPO grafts played a predominant plasticizing role in these structures. These results are in good agreement with those previously published for similar products obtained in the oxypropylation of other natural substrates.6,7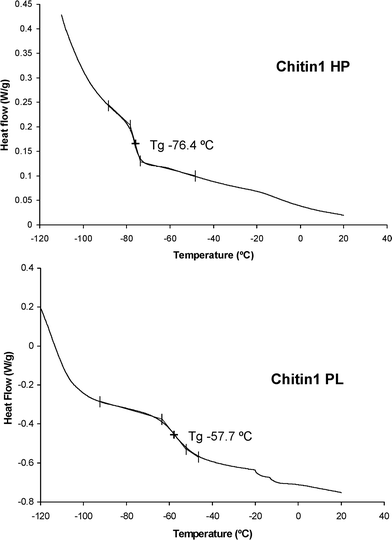 | ||
| Fig. 4 DSC thermograms of the two fractions (HP and PL) obtained from the oxypropylation of chitin at 140 °C. | ||
Viscosity
The viscosities of chitosan's PLs (50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 Pa s) were some 5 times higher than those of chitin's homologues. This further confirmed the lower reactivity of the former polysaccharide. Of course, all the polyol mixtures before extraction were some 100 times less viscous, given the low viscosity of the accompanying PPO oligomers.
000 Pa s) were some 5 times higher than those of chitin's homologues. This further confirmed the lower reactivity of the former polysaccharide. Of course, all the polyol mixtures before extraction were some 100 times less viscous, given the low viscosity of the accompanying PPO oligomers.
Hence, these mixtures, as recovered after the oxypropylation reaction, without any separation or purification, are the actual polyols which constitute the interesting macromonomers to be exploited in polycondensations based on the use of their OH groups. The oxypropylated polymer has a high OH functionality, indeed the same as that of the substrate, since the grafting reaction only brings the OH group out of its initial core structure. The PPO has an OH functionality of two and will therefore act as a chain extender during the polycondensation reactions in which the grafted polymer is responsible for branching and ultimately cross-linking.
Conclusions
This short study provides irrefutable qualitative evidence about the possibility of transforming chitin and chitosan into viscous polyol mixtures, by an extremely simple process which only involves the activated substrate and propylene oxide. It is extremely important to underline that the various separation procedures described above were only applied in order to characterise the different products of these reactions. In practice, the polyol mixtures can be simply removed as such from the reaction vessel, without the need of any other operation, since they constitute viable macromonomers for the synthesis of polyurethanes, polyethers or polyesters. In other words, these systems are a good example of green chemistry, in that they do not require any solvent, leave no residue and call upon the exploitation of renewable resources.Acknowledgements
The authors thank Mahtani Chitosan Pvt. Ltd. (India) for their generous gifts of chitosan and chitin. Susana Fernandes thanks the Fundação para a Ciência e a Tecnologia (Portugal) for a Scientific Research grant (SFRH/BI/33050/2007).References
- D. S. Argyropoulos, ACS Symp. Ser., 2007, vol. 954 Search PubMed.
- Renewable Bioresources, ed. C. V. Stevens, R. Verhé, John Wiley,New York, 2004 Search PubMed.
- A. J. Ragauskas, C. K. Williams, B. H. Davison, G. Britovsek, J. Cairney, C. A. Eckert, W. J. Frederick, J. P. Hallett, D. J. Leak, C. L. Liotta, J. R. Mielenz, R. Murphy, R. Templer and T. Tschaplinski, Science, 2006, 311(5760), 484–489 CrossRef.
- L. Yu, K. Dean and L. Li, Prog. Polym. Sci., 2006, 31(6), 576–602 CrossRef CAS.
- M. Evtiouguina, A. M. Barros, J. J. Cruz-Pinto, C. P. Neto, M. N. Belgacem, C. Pavier and A. Gandini, Bioresour. Technol., 2000, 73(2), 187–189 CrossRef CAS.
- M. Evtiouguina, A. Barros-Timmons, J. J. Cruz-Pinto, C. P. Neto, M. N. Belgacem and A. Gandini, Biomacromolecules, 2002, 3(1), 57–62 CrossRef CAS.
- C. Pavier and A. Gandini, Ind. Crops Prod., 2000, 12(1), 1–8 CrossRef CAS.
- C. Pavier and A. Gandini, Carbohydr. Polym., 2000, 42(1), 13–17 CrossRef CAS.
- P. Velazquez-Morales, J. F. Le Nest and A. Gandini, Electrochim. Acta, 1998, 43(10–11), 1275–1279 CrossRef.
- A. Gandini, M. N. Belgacem, Z. X. Guo and S. Montanari, in Chemical Modification, Properties, and Usage of Lignin, ed. T. Q. Hu, Kluwer Academy/Plenum Publishers, 2002, pp. 57–80 Search PubMed.
- H. Nadji, C. Bruzzese, M. N. Belgacem, A. Benaboura and A. Gandini, Macromol. Mater. Eng., 2005, 290(10), 1009–1016 CrossRef CAS.
- M. Evtiouguina, A. Gandini, C. P. Neto and M. N. Belgacem, Polym. Int., 2001, 50(10), 1150–1155 CrossRef CAS.
- C. Pavier and A. Gandini, Eur. Polym. J., 2000, 36(8), 1653–1658 CrossRef CAS.
- M. Rinaudo, Prog. Polym. Sci., 2006, 31(7), 603–632 CrossRef CAS.
- D. Asahina, T. Matsubara, Y. Miyashita and Y. Nishio, Sen-I Gakkaishi, 2000, 56(9), 435–442 Search PubMed.
- S. J. Kim, S. S. Kim and Y. M. Lee, Macromol. Chem. Phys., 1994, 195(5), 1687–1693 CrossRef CAS.
- S. S. Kim, S. J. Kim, Y. D. Moon and Y. M. Lee, Polymer, 1994, 35(15), 3212–3216 CrossRef CAS.
- Y. Liu, G. Chen and K. A. Hu, J. Mater. Sci. Lett., 2003, 22(19), 1303–1305 CrossRef CAS.
- I. K. Park and Y. H. Park, J. Appl. Polym. Sci., 2001, 80(13), 2624–2632 CrossRef CAS.
- Y.F. Peng, B. Q. Han, W. S. Liu and X. J. Xu, Carbohydr. Res., 2005, 340(11), 1846–1851 CrossRef CAS.
- Y. Wan, K. A. M. Creber, B. Peppley and T. V. Bui, J. Polym. Sci., Part B: Polym. Phys., 2004, 42(8), 1379–1397 CrossRef CAS.
- W. M. Xie, P. X. Xu, W. Wang and Q. Liu, Carbohydr. Polym., 2002, 50(1), 35–40 CrossRef CAS.
- M. N. Belgacem, A. Blayo and A. Gandini, J. Colloid Interface Sci., 1996, 182(2), 431–436 CrossRef CAS.
- L. J. Bellamy, in The Infrared Spectra of Complex Molecules, Chapman and Hall, London, 3rd edn, 1975, p. 433 Search PubMed.
- A. Hirai, H. Odani and A. Nakajima, Polym. Bull., 1991, 26, 87–94 CrossRef CAS.
- K. M. Varum, M. W. Anthonsen, H. Grasdalen and O. Smitsrod, Carbohydr. Res., 1991, 211, 17–23 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2008 |