X-ray crystallography identifies two chloride binding sites in the oxygen evolving centre of Photosystem II
James W.
Murray
a,
Karim
Maghlaoui
a,
Joanna
Kargul
a,
Naoko
Ishida
b,
Thanh-Lan
Lai
b,
A. William
Rutherford
b,
Miwa
Sugiura
c,
Alain
Boussac
b and
James
Barber
*a
aDivision of Molecular Biosciences, Imperial College London, London, UK SW7 2AZ. E-mail: j.barber@imperial.ac.uk; Tel: +44 20 759 45266; Fax: +44 20 759 45267
biBiTec-S, URA CNRS 2096, CEA Saclay, 91191, Gif sur Yvette, France
cCell-Free Science and Technology Research Center, Ehime University, Bunkyo-cho, Matsuyama Ehime 790-8577, Japan
First published on 2nd July 2008
Abstract
Bromide anomalous X-ray diffraction analyses have been used to locate chloride binding sites in the vicinity of the water splitting/oxygen evolving centre (OEC) of Photosystem II. Three-dimensional crystals of PSII from Thermosynechococcus elongatus were grown from (i) isolated PSII crystals infiltrated with bromide or (ii) PSII obtained from cells cultured in a medium in which the chloride content was totally replaced by bromide. In either case, the anomalous diffraction yielded the same result, the existence of two bromide binding sites in the vicinity of the OEC. Neither are in the first coordination sphere of the Mn and Ca ions which form the catalytic centre of the OEC, being about 6 to 7 Å from the metal-cluster. Site 1 is located close to the side chain nitrogen of D2-K317 and the backbone nitrogen of D1-Glu333 while Site 2 is adjacent to backbone nitrogens of CP43-Glu354 and D1-Asn338. Their positioning close to postulated hydrophilic channels may suggest a role in proton removal from, or substrate access to, the OEC.
Introduction
In the region of 3 billion years ago biology developed the capacity to efficiently capture solar energy and use it to power the synthesis of organic molecules. This photosynthetic process set into motion an unprecedented explosion in biological activity allowing life to prosper on an enormous scale as witnessed by the fossil records and by the extent and diversity of living organisms on our planet today. Indeed, it was the process of photosynthesis over eons of time which has provided us with the oil, gas and coal needed to power our technologies, heat our homes and produce the wide range of chemicals and materials that support everyday life.Today, it is estimated that photosynthesis produces more than 100 billion tons of dry biomass annually, which would be equivalent to a hundred times the weight of the total human population on our planet at the present time and equal to a mean storage rate of about 100 TW. The success of this energy generating and storage system stems from the fact that the raw materials and energy needed to drive the synthesis of biomass are available in almost unlimited amounts; sunlight, water and carbon dioxide. At the heart of the reaction is the splitting of water by sunlight into dioxygen and reducing equivalents. Prior to the evolution of the water splitting reaction, photosynthetic organisms had relied on hydrogen/electron donors such as H2S, NH3, organic acids and Fe2+, which were in limited supply compared with the vast quantities of water available on our planet.
The appearance of the water splitting reaction of photosynthesis had dramatic consequences, converting the atmosphere of Earth from anoxygenic to oxygenic and at the same time allowing the ozone layer to be established. With oxygen available, the efficiency of metabolism increased significantly since aerobic respiration provides in the region of twenty times more cellular energy than anaerobic respiration. It was probably this substantial improvement in cellular efficiency, due to aerobic metabolism, which drove the subsequent evolution of eukaryotic cells and multicellular organisms. The establishment of the ozone layer provided a shield against harmful UV radiation allowing organisms to explore new habitats and especially to exploit the terrestrial environment.
The photosynthetic enzyme that uses light energy to split water is known as Photosystem II (PSII). It is a multiprotein complex contained within the thylakoid membranes of all types of plants, algae and cyanobacteria.1 In contrast to chemical and electrochemical water splitting, which are thermodynamically highly demanding, the PSII-catalyzed biological water-splitting mechanism is truly remarkable since it proceeds with very little over-potential.2–4 The processes underpinning this reaction are initiated by the absorption of visible light by chlorophyll (Chl) and other pigments that act as an antenna system for the reaction centre (RC) where energy is initially stored by charge transfer. The primary electron donor consisting of Chla is located within the RC, composed of the D1 and D2 proteins, and is called P680. Excited P680 (P680*) donates an electron to the primary electron acceptor, pheophytin (Pheo) a. The formation of a radical pair P680˙+Pheo˙− takes place in a few picoseconds and occurs across the membrane. Stabilisation of the charge transfer state is accomplished by electron transfer from Pheo˙- to a plastoquinone acceptor QB in the microsecond to millisecond time domain according to the redox state of QB. This electron transfer is aided by an intermediate plastoquinone molecule QA. Unlike QB, QA plastoquinone is tightly bound within the RC, functions as a single electron acceptor and does not undergo protonation. The QBplastoquinone, however, accepts two electrons and is fully protonated prior to its departure from the RC via the hydrophobic lipid phase of the membrane. In this way reducing equivalents leave PSII and with the aid of a second light reaction occurring in Photosystem I (PSI) are used to reduce NADP+ and ultimately CO2.
On the oxidising side of the PSII RC, which is localised towards the lumenal surface of the thylakoid membrane, P680˙+ is used to split water. Four oxidising equivalents are needed to form dioxygen. Since each water splitting reaction occurs at a single catalytic centre, the formation of dioxygen requires four photochemical turnovers of the PSII RC and the storage of four oxidising equivalents. This storage of oxidising potential is driven by electron transfer to P680˙+via a redox active tyrosine YZ and occurs in a catalytic centre containing four Mn ions and a Ca2+. The S-state cycle of Joliot and Kok5,6 provides a framework for describing the chemical intermediates of the catalytic cycle, where the energy of each photon absorbed by PSII drives the conversion from S0 to S1, S1 to S2, S2 to S3 and S3 to S4, where the S4 to S0 transition is a dark reaction giving rise to the formation of dioxygen. In this cycle the S1-state is stable in the dark so that maximum oxygen emission occurs from dark-adapted cells on the third, single turnover, flash followed by a subsequent period of four. The details of the S-state cycle and the chemistry of the water splitting reaction have been emerging over many years through the application of a wide range of techniques (see various articles in ref. 7 and 8) being particularly spurred by the recent structural analyses of PSII by X-ray absorption spectroscopy9–12 and X-ray crystallography.13–17 These studies, coupled with quantum mechanical analyses have provided detailed schemes for the water splitting chemistry leading to O–O bond formation18–20 and refinement of the structure of the oxygen evolving centre (OEC).21,22 However, one factor which has been difficult to consider is the apparent involvement of chloride in the chemistry of the water splitting reaction.
The recognition that chloride is required as a cofactor for photosynthetic oxygen evolution stems from the pioneering studies of Arnon and Whatley,23 Bove et al.24 and Izawa et al.25 Since then this requirement has been investigated in great depth as detailed in the recent review of van Gorkom and Yocum.26 However the role and function of Cl− within the S-state cycle is not understood and remains the subject of a good deal of debate.26–28 In plant PSII, spectroscopic and enzymological studies have led to proposed models with either a single Cl−-site with two different binding affinities29 or two independent binding sites (ref 30–32, see also ref. 33 for a recent discussion).
The location of the Cl−−binding site (or sites) is also uncertain, with some authors favouring a Cl− directly liganded to one of the Mn ions of the cluster34 or to Ca2+,35,36 and others preferring the Cl− to be outside the first coordination sphere of the metal cluster.37–39 To date the resolutions of the crystal structures available are insufficient to assign electron density to this anion. Bromide is known to substitute for chloride in the OEC and give almost complete enzyme function.40 Indeed, a recent X-ray absorption study, using Br−-containing PSII,39 has provided what the authors describe as “tentative evidence” for a Br− (and hence Cl−) at a distance of ≈5 Å from a metal (presumably Ca or Mn): too far to be a direct ligand.
X-Ray anomalous dispersion, a specific and sensitive method to detect heavy atoms within crystals,41 has been successfully used, even at low resolution, to confirm the localisation of the Sr ion in the Mn4Sr cluster of the OEC after a biosynthetic Ca2+/Sr2+ exchange.42Chloride has no accessible absorption edges, but bromide does at 13.47 keV, a near-optimal wavelength for many synchrotron beam lines and therefore ideal for X-ray anomalous diffraction studies.
In the present work we have prepared two kinds of 3-dimensional crystals of PSII isolated from Thermosynechococcus elongatus: (i) standard PSII crystals infiltrated with bromide and (ii) PSII crystals obtained from cells grown in a culture medium in which the chloride content was replaced by bromide. In both cases, anomalous X-ray diffraction analyses were conducted to detect bromide binding sites at or close to the OEC. We find similar results in both types of sample: the presence of two bromide binding sites close to the Mn4Ca cluster but too far to be ligands to the metals.
Material and methods
Culturing of cells
For the bromide-infiltration experiments, PSII was isolated from Thermosynechococcus elongatus wild type (WT) cells, while for the experiments involving cells grown in bromide-containing media33 a strain was used which had a His6-tag on the CP43 subunit43 with only the psbA3gene (WT*).44For the bromide-infiltration experiments, small cultures of WT Thermosynechococcus elongatus (100 mL and 1 L) were grown in Cl−-containing DTN-medium. Cell suspensions were incrementally sub-cultured from 100 mL to 1 L before inoculating a 20 L culture. These 20 L cultures were grown at 57 °C while bubbling continuously with 5% CO2-enriched air and illuminating with white light (Growlux, Sylvania) of increasing intensity (50–250 μmoles photons m−2 s−1) according to cell density. Cells were harvested in the log phase after 8 days at a final OD685 ∼ 1.8. For the Cl/Br physiologically exchange experiments, the WT* cells were grown as previously reported.33
Isolation of thylakoids and PSII
Br − containing PSII from WT* cells was purified exactly as reported previously.33Chloride was substituted with bromide in all buffers. PSII from WT cells were isolated as described previously,45 with the following modifications. WT cells were disrupted with two passes in a French press at 6000 psi and thylakoids resuspended at a Chl concentration of 1.6 mg mL−1 in 20 mM MES pH 6.5, 25% (v/v) glycerol, 20 mM CaCl2, 10mM MgCl2 prior to solubilisation with 0.5% (w/w) β-DDM (Biomol, Germany). Core complexes were isolated after 2 successive anion exchange chromatography steps as described by Kern et al.,46 yielding only dimeric PSII. The sample was washed with 40 mM MES–NaOH, pH 6.0, 20 mM CaBr2, 0.02% (w/w) β-DDM, 5% (w/w) glycerol to remove traces of chloride and concentrated in a Millipore ultra-free centrifugation device (100 KD MWCO) at 6000g.Crystallisation
For the bromide-infiltration experiments (Br-infiltrated), PSII core complexes purified from WT cells grown in the presence of chloride were incubated on ice for 30 minutes in 40 mM MES–NaOH, pH 6.0, 100 mM CaBr2, 0.02% (w/w) β-DDM, 5% (w/w) glycerol and spun for 30 minutes at 18![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000g in a microfuge at 4 °C. The Chl concentration was adjusted to 1.75 mg mL−1 and no chloride was present in the crystallisation solutions. PSII core complexes were mixed 1 : 1 with a solution twice as concentrated as the reservoir solution, consisting of 100 mM Hepes pH 7.5, 100 mM (NH4)2SO4 20% glycerol, 10–16% PEG-4000 (Fluka), 1 mM of methyl mercurial chloride (Hampton Research), 0.11 mM polyoxyethylene lauryl ether (C12E8) (Hampton Research), as described in Ferreira et al.15 Crystals were grown at 17 °C using the hanging drop vapour diffusion method.
000g in a microfuge at 4 °C. The Chl concentration was adjusted to 1.75 mg mL−1 and no chloride was present in the crystallisation solutions. PSII core complexes were mixed 1 : 1 with a solution twice as concentrated as the reservoir solution, consisting of 100 mM Hepes pH 7.5, 100 mM (NH4)2SO4 20% glycerol, 10–16% PEG-4000 (Fluka), 1 mM of methyl mercurial chloride (Hampton Research), 0.11 mM polyoxyethylene lauryl ether (C12E8) (Hampton Research), as described in Ferreira et al.15 Crystals were grown at 17 °C using the hanging drop vapour diffusion method.
PSII core complexes purified from WT* cells grown in the presence of bromide (Br-physiologically substituted) were washed with 40 mM MES–NaOH, pH 6.0, 20 mM CaBr2, 0.02% (w/w) β-DDM, 5% (w/w) glycerol and concentrated in a Millipore ultra-free centrifugation device (100 KD MWCO) at 6000g. The sample was spun at 18000g in a microfuge at 4 °C and crystallisation trials performed as described above.
Crystals obtained in both experiments reached maximum dimensions of 400–500 microns in the longest dimension and were harvested after 3–5 days. Crystals were cryo-protected by stepwise addition of mother liquor with glycerol replacing water to a final concentration of 25% (v/v) and flash frozen in liquid nitrogen. Crystals were viewed with SZX-12 Olympus Stereomicroscope coupled to a DP12 digital camera.
X-Ray data collection and analysis
X-ray data sets were collected using the undulator MAD beamline X06SA at the Swiss Light Source from crystals derived from PSII soaked with 100mM CaBr2 (Br-infiltrated) as well as from crystals obtained from WT* in which chloride had been physiologically replaced by bromide (Br-physiological substituted). Datasets were integrated and scaled using XDS and XSCALE,47 and further processed using programs of the CCP4 suite (CCP4, 1994).48 Molecular replacement was performed using the PSII model 2AXT16 with PHASER,49 which was also used to calculate phases. Phased anomalous difference maps were calculated in COOT50 and subjected to 2-fold non-crystallographic averaging (see Table 1 for the crystallographic data). These maps were searched for peaks which could be identified as bromide sites. Figures were prepared using the PyMol molecular graphics system.51| Br-infiltrated [ca. 450 xtal] | Br-physiologically substituted [806 xtal] | |
|---|---|---|
| Detector | MarCCD | PILATUS 6M |
| wavelength/Å (energy/keV) | 0.90500 (13.6999) | 0.9150 (13.55018) |
| Cell | a = 131.73 Å b = 226.68 Å c = 306.71Å | a = 133.42 Å b = 226.82 Å c = 309.52 Å |
| Resolution/Å | 10–4.45 (4.8–4.45) | 10–4.21 (4.3–4.2) |
| Observations | 223263 (27217) |
914340 (64387) |
| Unique observations | 100620 (15188) |
132169 (9076) |
| Multiplicity | 2.21 (1.79) | 6.92 (7.09) |
| Completeness (%) | 92.3 (68.2) | 99.9 (99.8) |
| <I/sigI> | 5.68 (2.41) | 13.45 (2.12) |
| Rmeas | 0.116 (0.702) | 0.096 (1.08) |
Results and discussion
For the Br-infiltrated crystals, two peaks in the phased anomalous difference map not attributable to sulfur atoms or other known PSII anomalous ions such as Ca and Fe, were identified within a radius of 10 Å of the OEC; Site 1 at 13.6σ and Site 2 at 12.1σ which are very close to the OEC (see Fig. 1A). These peaks are interpreted as bromide sites as they are not present in equivalent anomalous diffraction maps calculated from crystals of PSII containing no bromide. The 9.7σ peak at the OEC is due to the anomalous Mn signal. Site 1 is close to the terminal nitrogen of D2-Lys 317 and the backbone nitrogen of D1-Glu 333, while Site 2 is in the vicinity of the backbone nitrogens of D1-Asn 338 and of CP43-Glu 354, where CP43 is a light harvesting Chl-binding protein of PSII. However, their distance from the closest Mn ion, no matter which model for the cluster is used,10,15,16 indicates that under the conditions of the experiment, the identified halide binding sites are not in ligating distance to Mn or to the Ca2+ within the OEC.The results presented in Fig. 1A show potential chloride binding sites in PSII which are readily exchangeable with bromide and are close to the OEC. However, there is a possibility that a high affinity chloride-binding site exists within the OEC which does not readily exchange during the 100 mM CaBr2 treatment of isolated PSII sample prior to crystallisation. For this reason we cultured T. elongatus in a medium in which its chloride content was replaced entirely by bromide following the procedure of Ishida et al.33 The resulting anomalous difference map derived from Br-physiologically substituted PSII is shown in Fig. 1B where it can be seen that the three main peaks in the anomalous difference map are similar to those shown in Fig. 1A and that there are no other visible strong peaks, indicative of additional bromide-binding sites within 10 Å of the OEC. In this case the sigma values for Site 1 and Site 2 were 1.6 and 2.5, respectively, while the anomalous Mn signal at the OEC was at 5.15σ. Site 1 in the Br-physiologically substituted crystal was 4.3 Å from the side chain nitrogen of D2-K317 and 3.4 Å from the backbone nitrogen of D1-Glu333. Site 2 is 3.5 Å from both the backbone nitrogens of CP43-Glu354 and D1-Asn338 which agree, within the resolution of the data, with the results obtained with the Br-infiltrated PSII crystals.
Agreement between the two sets of data also applies for the distances between the OEC and the two bromide sites. Note that the contouring levels between the two data sets shown in Fig. 1 differ. This is due to different levels of signal, with the physiologically substituted being better because the quality of the crystals used for data collection were poorer for Br-infiltrated PSII.
Coordinates for the two sites, either from the Br-infiltrated or Br-physiologically substituted are given in Table 2.
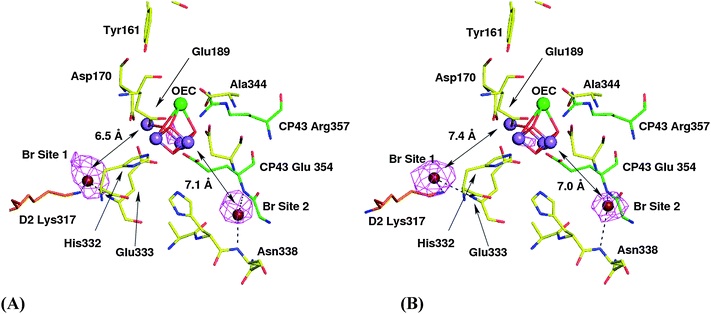 | ||
| Fig. 1 The bromide sites identified at the OEC in context of the Ferreira et al.15 structure (1S5L). Amino acids belonging to the D1 protein are in yellow, those of D2 protein are in orange and those of CP43 are in green, with selected residues labelled. 2-Fold averaged phased anomalous difference maps are shown. Panel (A) shows data from a crystal from Br-infiltrated PSII contoured at 3σ. (B) Shows data from a crystal grown with Br-physiologically replacing chloride contoured at 9σ. The distances shown are between the peak of anomalous Br diffraction for site 1 and site 2 and the closest Mn of the Ferreira et al.15 model. | ||
| PDB | Br-infiltrated | Br-physiologically substituted |
|---|---|---|
| 1S5L (site 1) | 24.882, 34.256, 64.135 | 23.572, 34.771, 63.300 |
| 2AXT (site 1) | 24.707, 32.614, 63.526 | 23.419, 33.141, 62.669 |
| 1S5L (site 2) | 28.402, 35.568, 75.946 | 29.306, 35.990, 75.957 |
| 2AXT (site 2) | 27.994, 33.862, 75.410 | 28.901, 34.273, 75.445 |
Under the experimental conditions used, our data show that neither of the two Br−-binding sites is close enough to form direct ligands to Mn or Ca2+. Indeed, several studies suggest that Cl− is not a direct ligand to the OEC metal-cluster.38,39,52 For example, in the recent extended X-ray absorption fine structure (EXAFS) work on PSII (isolated from spinach) which had been biochemically infiltrated with bromide, Haumann et al.39 concluded that a bromide ion is bound at ≈5 Å from Mn or Ca. This distance differs from those reported here, but this difference is probably within the limitation imposed by the resolution of the X-ray diffraction. It is worth pointing out that Haumann et al.39 only saw the ≈5 Å back-scattering peak at low Br− concentrations. This peak was lost as the Br− concentration was increased with an apparent concentration dependence that matched the Br−-induced recovery of enzyme activity. In the present work, X-ray data was collected from crystals exposed to variable levels of Br− (Br-infiltrated, Br− ≈ 50 mM; Br-physiologically substituted, Br− ≈ 10 mM or less) without any significant change in the data.
The demonstration here of the existence of two halide-binding sites does not eliminate the earlier one-site two-state model,29 since no correlation has been made with activity. However, the presence of the two halides in the crystallographic data is clearly more consistent with a two-site model (ref. 30–32; see also ref. 38).
Site 1 is close to D2-Lys317, which is believed to form part of a proton channel leading from the OEC to the lumen.15,53 The bound Cl− may help to maintain a proton-relay in this proposed channel, a suggestion previously made in the absence of structural information.37,38 Site 2 is close to the backbone nitrogen of D1-Asn 338 and CP43-Glu 354 where the latter is a residue contained within a conserved 310-helix of the large extrinsic lumenal loop of CP43 and almost certainly acts as a ligand for a Mn ion within the OEC.15 It has also been suggested that CP43-Glu354 is a component of a second water/proton channel linking the OEC with the lumen,53 and again the Cl− bound at site 2 could facilitate proton transfer. The proximity of the Cl− to a direct ligand of a Mn ion could be consistent with perturbations of the Mn EPR signal seen when Cl− is removed.32,54
Since our analysis relates to the S1-state or a modification of it, it does not rule out Cl−-binding to the Mn or Ca2+ in other S-states, where it may have a more direct role in water oxidation catalysis.33,55 Indeed, there is evidence from studies with synthetic binuclear Mn-compounds that Cl− will form direct ligands when Mn is in its high oxidative state, apparently playing a role in charge neutralisation and redox potential control.56,57 However, since both binding sites are located in regions postulated to be hydrophobic channels, then the suggestion that chloride facilitates substrate access35,58 and proton release37,38 to and from the OEC would be consistent with the proposal that one of the two halides is not absolutely required for the water splitting reaction and that removal of Cl− slows the kinetics of the S-state cycle but does not inhibit O2-evolution completely.29,32
Although it is possible that during the collection of the X-ray data the oxidation state of the Mn ions within the OEC may have changed due to the generation of free electrons in the crystal,59,60 it is unlikely that any associated conformational changes would be sufficiently large to affect the interpretation of the diffraction data presented here given its intermediate resolution. It is possible, however, that X-ray induced reduction of the Mn-cluster by the X-ray beam could bring about a change in the chloride binding. Given this proviso, our results indicate two bromide (and by analogy chloride) binding sites within the vicinity of the OEC but not sufficiently close to the metal-cluster to form direct ligands to it.
Abbreviations
Chl, chrorophyll; RC, reaction centre; β-DDM, dodecyl-β-D-maltoside; C12E8, polyoxyethylene lauryl ether; EXAFS, extended X-ray absorption fine structure; OEC, oxygen evolving complex; MAD, multiple-wavelength anomalous diffraction; MES, 2(N-morpholino)ethanesulphonic acid; P680, primary electron donor of photosystem II; Pheo, pheophytin; PSII, photosystem II; PSI, photosystem I; QA, QB, fixed and mobile plastoquinones of PSII; WT, wild type;Acknowledgements
We acknowledge financial support for this work from the Biotechnology and Biological Science Research Council (BBSRC) and from The Royal Society UK–Japan exchange programme. This work was also supported in part by the JSPS and CNRS under the Japan–France Research Cooperative Program and the SolarH and SolarH2 programs from the European Community (contract 212508). NI was supported in part by the Bio-Hydrogen program of CEA. We also wish to acknowledge our access to the facilities and staff of the Swiss Light Source, particularly Clemens Schulze-Briese.References
- J. Barber, Photosystem II: The engine of life, Biophys. Quart. Rev., 2003, 36, 71–89 Search PubMed.
- B. A. Diner and G. T. Babcock, in Oxygenic Photosynthesis:The Light Reactions, ed. D. R. Ort and C. F. Yocum, Kluwer Academic Publishers, Dordrecht, The Netherlands, 1996, pp. 213–247 Search PubMed.
- R. D. Britt, in Oxygenic Photosynthesis:The Light Reactions, ed. D. R. Ortand C. F. Yocum, Kluwer Academic Publishers, Dordrecht, The Netherlands, 1996, pp .137–159 Search PubMed.
- B. A. Diner and F. Rappaport, Structure, dynamics and energetics of the primary photochemistry of photosystem II of oxygenic photosynthesis, Annu. Rev. Plant Biol., 2002, 53, 551–580 Search PubMed.
- P. Joliot, G. Barbieri and R. Chabaud, Un nouveau modele des centres photochimiques du system II., Photochem. Photobiol., 1969, 10, 309–329 CrossRef CAS.
- B. Kok, B. Forbush and M. McGloin, Cooperation of charges in photosynthetic O2 evolution-I. A linear four step mechanism, Photochem. Photobiol., 1970, 11, 457–475 CrossRef CAS.
- T. J. Wydrzynski and K. Satoh, Photosystem II. The Light-Driven Water:Plastoquinone Oxidoreductase, Advances in Photosynthesis and Respiration, Springer, Dordrecht, The Netherlands, vol. 22, 2005, pp. 1–786 Search PubMed.
- J. Barber and A. W. Rutherford, Revealing how nature uses sunlight to split water, Philos. Trans. R. Soc. London, Ser. B, 2007, 363, 1123–1303.
- V. K. Yachandra, Structure of the Mn complex in Photosystem II: Insights from X-ray spectroscopy, Philos. Trans. R. Soc. London, Ser. B, 2002, 357, 1347–1358 CrossRef CAS.
- J. Yano, J. Kern, K. Sauer, M. J. Latimer, Y. Pushkar, J. Biesiadka, B. Loll, W. Saenger, J. Messinger, A. Zouni and V. K. Yachandra, Where water is oxidised to dioxygen: Structure of the photosynthetic Mn4Ca cluster, Science, 2006, 314, 821–825 CrossRef CAS.
- Y. Pushkar, J. Yano, K. Sauer, A. Boussac and V. Yachandra, Structural Changes in the Mn4Ca Cluster and the Mechanism of Photosynthetic Water Splitting, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 1879–1884 CrossRef CAS.
- J. Yano and V. R. Yachandra, Where water is oxidized to dioxygen: Structure of the photosynthetic Mn4Ca cluster from X-ray spectroscopy, Inorg. Chem., 2008, 47, 1711–1726 CrossRef CAS.
- A. Zouni, H. T. Witt, J. Kern, P. Fromme, N. Krauss, W. Saenger and P. Orth, Crystal structure of photosystem II from Synechococcus elongatus at 3.8 Å resolution, Nature, 2001, 409, 739–743 CrossRef CAS.
- N. Kamiya and J. R. Shen, Crystal structure of oxygen-evolving Photosystem II from Thermosynechococcus vulcanus at 3.7 resolution, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 98–103 CrossRef CAS.
- K. N. Ferreira, T. M. Iverson, K. Maghlaoui, J. Barber and S. Iwata, Architecture of the photosynthetic oxygen-evolving center, Science, 2004, 303, 1831–1838 CrossRef CAS.
- B. Loll, J. Kern, W. Saenger, A. Zouni and J. Biesiadka, Towards complete cofactor arrangement in the 3.0 Å resolution structure of photosystem II, Nature, 2005, 438, 1040–1044 CrossRef CAS.
- J. Barber, Crystal structure of the oxygen-evolving complex of Photosystem II., Inorg. Chem., 2008, 47, 1700–1710 CrossRef CAS.
- P. E. M. Siegbahn and M. Lundberg, The mechanism for dioxygen formation in PSII studied by quantum chemical methods, Photochem. Photobiol. Sci., 2005, 4, 1035–1043 RSC.
- P. E. M. Siegbahn, O–O bond formation in the S4-state of the oxygen evolving complex in Photosystem II., Chem.–Eur. J., 2006, 12, 9217–9237 CrossRef CAS.
- P. E. M. Siegbahn, Theoretical studies of O–O bond formation in Photosystem II., Inorg. Chem., 2008, 47, 1779–1786 CrossRef CAS.
- E. M. Sproviero, J. A. Gascon, J. P. McEvoy, G. W. Brudvig and V. S. Batista, Curr. Opin. Struct. Biol., 2007, 17, 173–180 CrossRef CAS.
- E. M. Sproviero, J. A. Gascón, J. P. McEvoy, G. W. Brudvig and V. S. Batista, A Model of the Oxygen-Evolving Center of Photosystem II Predicted by Structural Refinement Based on EXAFS Simulations, J. Am. Chem. Soc., 2008, 130, 6728–6730 CrossRef CAS.
- D. I. Arnon and F. R. Whatley, Is chloride a coenzyme of photosynthesis?, Science, 1949, 110, 554–556 CrossRef CAS.
- J. M. Bove, C. Bove, F. R. Whatley and D. I. Arnon, Chloride requirement for oxygen evolution in photosynthesis, Z. Naturforsch., 1963, 18b, 683–688 Search PubMed.
- S. Izawa, R. L. Heath and G. Hind, Role of chloride ion in photosynthesis. 3. Effects of artificial electron donors upon electron transport, Biochim. Biophys. Acta, 1969, 180, 388–389 CAS.
- H. J. Van Gorkom, and C. F. Yochum, The calcium and chloride cofactors, in Photosystem II. The Light-Driven Water Plastoquinone Oxido-reductase, ed. T. Wydrzynski, T. J. and K. Satoh, Springer, Dordrecht, The Netherlands, 2005, pp. 307–327 Search PubMed.
- R. J. Debus, The manganese and calcium-ions of photosynthetic oxygen evolution, Biochim. Biophys. Acta, 1992, 1102, 269–352 CAS.
- C. F. Yocum, The calcium and chloride requirements of the O-2 evolving complex, Coord. Chem. Rev., 2008, 252, 296–305 CrossRef CAS.
- K. Lindberg and L.-E. Andreasson, A one-site, two-state model for the binding of anions in Photosystem II, Biochemistry, 1996, 35, 14259–14267 CrossRef CAS.
- P. Homann and Y. Inoue, The anion and calcium requirement of the photosynthetic water splitting complex, in Ion interactions in energy transfer biomembranes, ed. G. Papageorgiou, J. Barber and S. Papa, Plenum Press, New York, 1986, pp. 291–302 Search PubMed.
- A. Boussac, Exchange of chloride by bromide in the manganese photosystem II complex studied by cw- and pulsed-EPR, Chem. Phys., 1995, 194, 409–418 CrossRef CAS.
- P. van Vliet and A. W. Rutherford, Properties of the chloride-depleted oxygen-evolving complex of photosystem II studied by electron paramagnetic resonance, Biochemistry, 1996, 35, 1829–1839 CrossRef CAS.
- N. Ishida, M. Sugiura, F. Rappaport, T.-L. Lai, A. W. Rutherford and A. Boussac, Biosynthetic exchange of bromide for chloride and strontium for calcium in the Photosystem II oxygen-evolving enzyme, J. Biol. Chem., 2008, 283, 13330–13340 CrossRef CAS.
- P. O. Sandusky and C. F. Yocum, The chloride requirement for photosynthetic oxygen evolution; analysis of the effects of chloride and other anions on amine inhibition of the oxygen-evolving complex, Biochim. Biophys. Acta, 1984, 766, 603–611 CrossRef CAS.
- A. W. Rutherford, Photosystem II, the water-splitting enzyme, Trends Biochem. Sci., 1989, 14, 227–232 CrossRef CAS.
- J. P. McEvoy and G. W. Brudvig, Water-splitting chemistry of photosystem II., Chem. Rev., 2006, 106, 4455–4483 CrossRef CAS.
- W. J. Coleman, Chloride binding proteins: Mechanistic implications for the oxygen evolving complex of Photosystem II, Photosynth. Res., 1990, 23, 1–27 CrossRef CAS.
- K. Olesen and L.-E. Andreasson, The function of the chloride ion in photosynthetic oxygenevolution, Biochemistry, 2003, 42, 2025–2035 CrossRef CAS.
- M. Haumann, M. Barra, P. Loja, S. Loscher, R. Krivanck, A. Grundmeier, L.-E. Andreasson and H. Dau, Bromide does not bind to the Mn4Ca complex in its S1 state in Cl-depleted and Br-reconstituted oxygen evolving photosystem II: evidence from X-ray absorption spectroscopy at the Br K-edge, Biochemistry, 2006, 45, 13101–13107 CrossRef CAS.
- G. Hind, H. Y. Nakatani and S. Izawa, The role of Cl− in photosynthesis. The Cl− requirement of electron transport, Biochim. Biophys. Acta, 1969, 172, 277–289 CAS.
- M. E. Than, S. Henrich, G. P. Bourenkov, H. D. Bartunik, R. Huber and W. Bode, The endoproteinase furin contains two essential Ca2+ ions stabilizing its N-terminus and the unique S1 specificity pocket, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2005, 61, 505–512 CrossRef.
- J. Kargul, K. Maghlaoui, J. W. Murray, Z. Deak, A. Boussac, A. W. Rutherford, I. Vass and J. Barber, Purification, crystallization and X-ray diffraction analyses of the T. elongatus PSII core dimer with strontium replacing calcium in the oxygen-evolving complex, Biochim. Biophys. Acta, 2007, 1767, 404–413 CAS.
- M. Sugiura and Y. Inoue, Highly purified thermo-stable oxygen-evolving photosystem II core complex from the thermophilic cyanobacterium Thermosynechococcus elongatus having His-tagged CP43, Plant Cell Physiol., 1999, 40, 1219–1231 CAS.
- M. Sugiura, A. Boussac, T. Noguchi and F. Rappaport, Influence of Histidine-198 of the D1 subunit on the properties of the primary electron donor, P680, of photosystem II in Thermosynechococcus elongatus, Biochim. Biophys. Acta, 2008, 1777, 331–342 CAS.
- A. Boussac, F. Rappaport, P. Carrier, J.-M. Verbavatz, R. Gobin, D. Kirilovsky, A. W. Rutherford and M. Sugiura, Biosynthetic Ca2+/Sr2+ exchange in the photosystem II oxygen evolving enzyme of Thermosynechococcus elongatus, J. Biol. Chem., 2004, 279, 22809–22819 CrossRef CAS.
- J. Kern, B. Loll, C. Lüneberg, D. DiFiore, J. Biesiadka, K. D. Irrgang and A. Zouni, Purification, characterisation and crystallisation of photosystem II from Thermosynechococcus elongatus cultivated in a new type of photobioreactor, Biochim. Biophys. Acta, 2005, 1706, 147–157 CAS.
- W. J. Kabsch, Automatic processing of rotation diffraction data from crystals of initially unknown symmetry and cell constants, J. Appl. Crystallogr., 1993, 26, 795–800 CrossRef CAS.
- Collaborative Computational Project No 4 (1994), The CCP4 suite: programs for protein crystallography, Acta Crystallogr., Sect. D: Biol. Crystallogr., 1994, 50, 760–763 CrossRef.
- A. J. McCoy, R. W. Grosse-Kunstleve, P. D. Adams, M. D. Winn, L. C. Storoni and R. J. Read, Phaser crystallographic software, J. Appl. Crystallogr., 2005, 40, 658–674.
- P. Emsley and K. Cowtan, Coot: model-building tools for molecular graphics, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2004, 60, 2126–2132 CrossRef.
- W. L. DeLano, The PyMOL Molecular Graphics System. DeLano Scientific, San Carlos, CA, USA, 2002. http://www.pymol.org Search PubMed.
- A. Rashid and P. H. Homann, Properties of iodide activated photosynthetic water oxidizing complexes, Biochim. Biophys. Acta, 1992, 1101, 303–310 CAS.
- J. W. Murray and J. Barber, Structural characteristics of channels and pathways in photosystem II including the identification of an oxygen channel, J. Struct. Biol., 2007, 159, 228–237 CrossRef CAS.
- T.-A. Ono, J.-L. Zimmermann and A. W. Rutherford, EPR evidence for a modified S-state transition in chloride depleted Photosystem II., Biochim. Biophys. Acta, 1986, 851, 193–201 CrossRef CAS.
- H. Wincencjusz, H. J. van Gorkom and C. F. Yocum, The photosynthetic oxygen evolving complex requires chloride for its redox state S-2->S-3 and S-3->S-0 transitions but not for S-0->S-1 or S-1->S-2 transitions, Biochemistry, 1997, 36, 3663–3670 CrossRef CAS.
- J. E. Sheats, R. S. Czernuszewicz, G. C. Dismukes, A. L. Rheingold, V. Petrouleas, J. Stubbe, W. H. Armstrong, R. H. Beer and S. J. Lippard, Binuclear manganese(III) complexes of potential biological significance, J. Am. Chem. Soc., 1987, 109, 1435–1444 CrossRef CAS.
- T. K. Lal and R. Mukherjee, Modeling the Oxygen-Evolving Complex of Photosystem II. Synthesis, Redox Properties, and Core Interconversion Studies of Dimanganese Complexes Having {MnIII2(μ–O)(μ–OAc)2}2+, {MnIIIMnIV(μ–O)2(μ–OAc)}2+, and {MnIV2(μ–O)2(μ–OAc)}3+ Cores with MeL as a Terminal Ligand: A New Asymmetric Mixed-Valence Core, Inorg. Chem., 1998, 37, 2373–2382 CrossRef CAS.
- K. Beckmann, N. Ishida, A. Boussac and J. Messinger, Effects of the Calcium/Strontium and Chloride/Bromide substitution on substrate water exchange rates in Photosystem II, Photosynth. Res., 2007, 91 Search PubMed vol. 2–3.
- J. Yano, J. Kern, K. D. Irrgang, M. J. Latimer, U. Bergmann, P. Glatzel, Y. Pushkar, J. Biesiadka, B. Loll, K. Sauer, J. Messinger, A. Zouni and V. K. Yachandra, X-ray damage to the Mn4Ca complex in single crystals of photosystem II: a case study for metalloprotein crystallography, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 12047–12052 CrossRef CAS.
- M. Grabolle, M. Haumann, C. Muller, P. Liebisch and H. Dau, Rapid loss of structural motifs in the manganese complex of oxygenic photosynthesis by X-ray irradiation at 10–300 K, J. Biol. Chem., 2006, 281, 4580–4588 CAS.
- K. Diederichs and P. A. Karplus, Improved R-factors for diffraction data analysis in macromolecular crystallography, Nat. Struct. Biol., 1997, 4, 269–275 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2008 |