Hydrogen production using cobalt-based molecular catalysts containing a proton relay in the second coordination sphere†
George M.
Jacobsen
b,
Jenny Y.
Yang
a,
Brendan
Twamley
c,
Aaron D.
Wilson
b,
R. Morris
Bullock
a,
M.
Rakowski DuBois
a and
Daniel L.
DuBois
*a
aChemical and Materials Sciences Division, Pacific Northwest National Laboratory, Richland, WA 99352, USA
bDepartment of Chemistry and Biochemistry, University of Colorado, Boulder, CO 80309, USA
cDepartment of Chemistry, University of Idaho, Moscow, ID 83844, USA
First published on 11th June 2008
Abstract
The cobalt analogue of a highly active nickel electrocatalyst for hydrogen production has been synthesized and characterized as [Co(PPh2NPh2)2(CH3CN)](BF4)2. In the presence of triflic acid in acetonitrile solution, the complex loses one cyclic diphosphine ligand to form [Co(PPh2NPh2)(CH3CN)3](BF4)2, which has been synthesized independently and stucturally characterized. The latter complex serves as an electrocatalyst for hydrogen formation with a turnover frequency of 90 s−1 and an overpotential of 285 mV using bromoanilinium tetrafluoroborate as the acid. A similar cobalt complex with a related diphosphine ligand that does not contain a pendant base is not catalytically active, confirming an important role for the pendant amine in the catalytic reaction.
Introduction
The development of highly efficient catalysts for the interconversion of electrical energy and fuels could have a significant impact on the feasibility of future energy distribution systems. The simplest fuel is hydrogen, and its production from electricity requires the reduction of two protons by two electrons to form H2. Both platinum metal and hydrogenase enzymes efficiently catalyze this reaction. Because the hydrogenase enzymes use either iron or a combination of iron and nickel in their active sites, these enzymes demonstrate that expensive precious metals such as platinum are not required for this reaction. X-ray diffraction studies have provided information on the structures of the active sites of [FeFe] and [NiFe] hydrogenase enzymes,1–5 and this has led to efforts to develop simple structural analogs of these active sites with the goal of developing synthetic catalysts and probing mechanistic features.6–14 Other molecular electrocatalysts for hydrogen production based on first row transition metal complexes have also been reported.15For the [FeFe] hydrogenase enzyme it has been suggested that a pendant base in the second coordination sphere facilitates the heterolytic cleavage of H2 and the transfer of the resulting proton to a proton-conduction channel leading to the exterior of the protein.2 These considerations led us to synthesize nickel and iron complexes containing diphosphine ligands with pendant nitrogen bases that could play a similar role in simple functional models of the active site. These pendant bases can facilitate H2 binding, heterolytic cleavage of H2, proton exchange with bases in solution, and proton coupled electron transfer events. These results have been summarized in a recent review.16
The facile reversibility of the heterolytic cleavage of hydrogen in the proposed active site implies that the hydride acceptor ability of the iron center and the proton acceptor ability of the base are carefully balanced so that the free energy of this reaction is close to 0 kcal mol−1. Studies of those features controlling the hydride acceptor abilities of metal complexes containing diphosphine ligands have also been carried out in our laboratories.17,18 Efforts to match the hydride acceptor ability of the metal center, controlled largely by features of the first coordination sphere, with the proton acceptor ability of a pendant base in the second coordination sphere, led to the development of nickel-based catalysts for H2 production and oxidation having the general features shown in structure 1. When R = R′ = Ph, the complex [Ni(PPh2NPh2)2](BF4)2 serves as a catalyst for the electrochemical reduction of protons to produce H2. When R = cyclohexyl and R′ = benzyl, an electrocatalyst for H2 oxidation is obtained.19 Further studies of these catalysts have shown that they are not inhibited by CO concentrations of several percent.20 In contrast, derivatives of iron(II) that contain two cyclic diphosphine ligands are six coordinate octahedral complexes that do not show catalytic activity.21
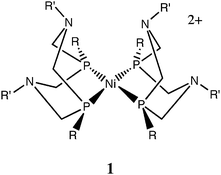
In an effort to extend this work to other first row transition metals, we report the preparation of new cobalt complexes containing either the cyclic PPh2NPh2 ligand or other related acyclic diphosphines. Only the derivatives with diphosphine ligands containing pendant amines demonstrated the ability to catalyze the electrochemical reduction of protons to H2. In contrast to nickel complexes, for which optimal catalytic activity is obtained when two cyclic ligands with pendant bases are coordinated to nickel, the catalytic activity for proton reduction observed for [Co(PPh2NPh2)2(CH3CN)](BF4)2 was found to result from loss of a diphosphine ligand to form [Co(PPh2NPh2)(CH3CN)3](BF4)2 in the presence of strong acid. The latter complex has been isolated and characterized and shown to be the true electrocatalyst for hydrogen production.
Results
Synthesis and characterization of cobalt complexes
The addition of two equivalents of the cyclic diphosphine ligand 1,3,5,7-tetraphenyl-1,5-diaza-3,7-diphosphacyclooctane, PPh2NPh2, to an acetonitrile solution of [Co(CH3CN)6](BF4)2 followed by workup provides a convenient synthesis of [Co(PPh2NPh2)2(CH3CN)](BF4)2. Elemental analysis of the product is consistent with this formulation, and a dichloromethane solution of this complex exhibited a broad EPR signal at room temperature with a g value of 2.11. This value is similar to the values observed previously for [Co(PP3)(CH3CN)](BF4)2 (g = 2.11, where PP3 = (Ph2PCH2CH2)3P) and [Co(dppe)2(CH3CN)](BF4)2 (g = 2.20, where dppe = Ph2PCH2CH2PPh2) in dichloromethane.22Recrystallization of the new complex from dichloromethane/ethanol solutions at room temperature under a slow stream of nitrogen over a period of two days resulted in the formation of the chloride adduct, [Co(PPh2NPh2)2(Cl)](BF4). A mass spectrum obtained using electrospray ionization from acetonitrile solutions showed evidence for a parent cation and a fragment corresponding to the loss of chloride. An X-ray diffraction study was performed on the crystals obtained by slow room-temperature recrystallization from methylene chloride to confirm the nature of this complex. The crystal consists of discrete cations and BF4− anions. A drawing of the cation is shown in Fig. 1, and a list of selected bond lengths and angles are given in Table 1. The cation is best described as a trigonal bipyramid with the chloride ligand occupying an equatorial position. The relatively small bite angles observed for the two diphosphine ligands (81.02° and 82.15°) of [Co(PPh2NPh2)2(Cl)]+ result in a structure that is distorted somewhat from an ideal trigonal bipyramid, but the P1–Co–P3 bond angle of 177.34° formed by the two axial phosphine ligands is close to the ideal 180°.
| Bond distances | |||
| Co(1)–P(1) | 2.2133(8) | Co(1)–P(4) | 2.2303(8) |
| Co(1)–P(2) | 2.2368(8) | Co(1)–Cl(1) | 2.2870(8) |
| Co(1)–P(3) | 2.2014(9) | ||
| Bond angles | |||
| P(1)–Co(1)–P(2) | 82.15(3) | P(2)–Co(1)–P(4) | 125.04(3) |
| P(3)–Co(1)–P(4) | 81.02(3) | P(1)–Co(1)–Cl(1) | 90.30(3) |
| P(1)–Co(1)–P(4) | 100.09(3) | P(2)–Co(1)–Cl(1) | 124.55(3) |
| P(2)–Co(1)–P(3) | 95.23(3) | P(3)–Co(1)–Cl(1) | 91.57(3) |
| P(1)–Co(1)–P(3) | 177.34(4) | P(4)–Co(1)–Cl(1) | 110.40(3) |
![Drawing of the [Co(PPh2NPh2)2(Cl)]+ cation showing atom numbering scheme. Only the ipso-carbon atoms of the phenyl rings are shown and the hydrogen atoms are omitted for clarity. Thermal ellipsoids are shown at 30% probability.](/image/article/2008/EE/b805309j/b805309j-f1.gif) | ||
| Fig. 1 Drawing of the [Co(PPh2NPh2)2(Cl)]+ cation showing atom numbering scheme. Only the ipso-carbon atoms of the phenyl rings are shown and the hydrogen atoms are omitted for clarity. Thermal ellipsoids are shown at 30% probability. | ||
The addition of one equivalent of either PPh2NPh2 or 1,3-(diphenylphosphino)propane (dppp) to an acetonitrile solution of [Co(CH3CN)6](BF4)2 followed by workup provides a convenient synthesis of [Co(PPh2NPh2)(CH3CN)3](BF4)2 and [Co(dppp)(CH3CN)3](BF4)2, respectively. Structural studies of crystals isolated from these preparations are consistent with their formulations. A perspective drawing and numbering scheme for the structure of [Co(PPh2NPh2)(CH3CN)3](BF4)2 are shown in Fig. 2, and selected bond distances and angles are given in Table 2. The geometry of the cation is best described as a square pyramid with two acetonitrile ligands and the two phosphorus atoms of the diphosphine ligand forming the basal plane, and the third acetonitrile ligand occupying the axial position.
| Bond distances | |||
| Co(1)–P(1) | 2.1918(5) | Co(1)–N(34) | 2.0776(15) |
| Co(1)–P(2) | 2.1978(5) | Co(1)–N(37) | 1.9427(15) |
| Co(1)–N(31) | 1.9516(15) | ||
| Bond angles | |||
| P(1)–Co(1)–P(2) | 82.450(17) | N(34)–Co(1)–P(1) | 98.58(4) |
| N(31)–Co(1)–N(37) | 88.69(6) | N(37)–Co(1)–P(2) | 91.91(4) |
| N(34)–Co(1)–N(37) | 96.53(6) | N(31)–Co(1)–P(2) | 172.61(4) |
| N(31)–Co(1)–N(34) | 90.15(6) | N(34)–Co(1)–P(2) | 97.09(4) |
| N(37)–Co(1)–P(1) | 164.41(5) | N(31)–Co(1)–P(1) | 95.08(4) |
2. Hydrogen atoms are omitted for clarity, and thermal ellipsoids are drawn at 30% probability.](/image/article/2008/EE/b805309j/b805309j-f2.gif) | ||
| Fig. 2 Perspective drawing and numbering scheme for [Co(PPh2NPh2)(CH3CN)3](BF4)2. Hydrogen atoms are omitted for clarity, and thermal ellipsoids are drawn at 30% probability. | ||
The Co–N bond length for the axial acetonitrile is 0.13 Å longer than those of the equatorial acetonitrile ligands. The six-membered ring adjacent to the axial acetonitile ligand adopts a chair conformation to minimize steric interactions. The second six-membered ring of the diphosphine ligand has a boat conformation, and the phenyl ring is folded over the cobalt atom resulting in Co(1)–C(24) and Co(1)–C(25) distances of 3.03 and 3.09 Å, respectively. A similar structure of lower quality has been determined previously for a complex without a pendant amine in the diphosphine ligand, [Co(dppp)(CH3CN)3](BF4)2.23 The greatest structural differences are the larger P–Co–P angle of 92.99° for the dppp ligand compared to 82.45° for the PPh2NPh2 ligand and the absence of the N-phenyl ring in the ligand backbone of the dppp ligand.
A similar reaction of [Co(CH3CN)6](BF4)2 with one equivalent of bis(diethylphospinomethyl)methyl amine (Et2PCH2N(Me)CH2PEt2, PNP) was carried out to form [Co(PNP)(CH3CN)3](BF4)2. The product was not structurally characterized, but is presumed to have a structure similar to those of the other mono(diphosphine) derivatives. The PNP ligand, which forms a 6-membered chelate ring upon coordination, is assumed to be in the more stable chair conformation, as observed for the nickel and iron complexes with this ligand.24,25
Electrochemical studies
The cyclic voltammogram of [Co(PPh2NPh2)2(CH3CN)](BF4)2 consists of a single reversible wave at E1/2 = −0.58 V (ΔEp = 71 mV) followed by a quasi-reversible reduction wave at E1/2 = −2.04 V (ΔEp = 100 mV), as seen in Fig. 3 (blue trace). The reversible reduction at −0.58 V is a diffusion-controlled one-electron wave assigned to the Co(II/I) couple. Based on the peak height (1.8 times that of the wave at −0.58), the quasi-reversible reduction wave at −2.04 V appears to involve two electrons corresponding to an overall Co(I/−I) reduction process. A second oxidation wave with Ep = −1.81 V is associated with the reduction wave at E1/2 = −2.04 V. The Co(−I) species is a highly strained tetrahedral complex that could relax by forming bimetallic complexes with bridging diphosphine ligands (tentatively assigned to the peak at −1.81 V) as observed for nickel and palladium complexes containing bis(diphenylphosphino)methane, a diphosphine ligand with a small bite angle.262 (blue trace) and [Co(PPh2NPh2)(CH3CN)3](BF4)2 (red trace). Conditions: scan rate = 200 mV s−1, 0.3 M NBu4BF4 (supporting electrolyte) acetonitrile solution, glassy carbon working electrode.](/image/article/2008/EE/b805309j/b805309j-f3.gif) | ||
| Fig. 3 Cyclic voltammogram of 3 × 10−3 M solutions of [Co(PPh2NPh2)2(CH3CN)](BF4)2 (blue trace) and [Co(PPh2NPh2)(CH3CN)3](BF4)2 (red trace). Conditions: scan rate = 200 mV s−1, 0.3 M NBu4BF4 (supporting electrolyte) acetonitrile solution, glassy carbon working electrode. | ||
The cyclic voltammograms of the complexes containing a single diphosphine ligand have also been studied, and these also exhibit a well defined, reversible or quasi-reversible, one-electron reduction in acetonitrile solution assigned to the Co(II/I) couple. Parameters associated with this couple are included in Table 3. All of the complexes exhibit other poorly defined and irreversible waves at more negative potentials, but the Co(II/I) couple is the most relevant couple for the catalytic reactions described below. The red trace in Fig. 3 is a cyclic voltammogram of [Co(PPh2NPh2)(CH3CN)3](BF4)2. The potential for this derivative (−0.99 V) is significantly more negative than that observed for the bis(diphosphine) analogue discussed above (−0.58 V). The CVs of [Co(PPh2NPh2)(CH3CN)3](BF4)2 obtained from different syntheses consistently show that a small amount (ca. 15%) of the bis(diphosphine) complex is present. This may be due to a ligand redistribution equilibrium in this solvent, but analogous contaminants are not observed in the CVs of the other mono(diphosphine) complexes. The Co(II/I) potentials for the other [Co(diphosphine)(CH3CN)3](BF4)2 complexes in this series are also more negative than −0.9 V, as shown in Table 3, and the range in these potentials reflects the electronic differences in the diphosphine ligands in the series. The most negative Co(II/I) potential in the complexes studied was observed for [Co(CH3CN)6](BF4)2 (E1/2 = −1.21 V, ΔEp = 95 mV).
| Complex | E 1/2/V | ΔEp/mV | i a/ic |
|---|---|---|---|
| a Volts versus the FeCp2+/FeCp2 couple in 0.2 M NEt4BF4/CH3CN solutions. b Separation of cathodic and anodic peak potentials at a scan rate of 100 mV s−1. Under these conditions ferrocene exhibited ΔEp values of 70 ± 5 mV. c Ratio of anodic and cathodic peak currents at 100 mV s−1. | |||
| [Co(PPh2NPh2)2(CH3CN)](BF4)2 | −0.58 | 71 | 1.0 |
| [Co(PPh2NPh2)(CH3CN)3](BF4)2 | −0.99 | 70 | 1.0 |
| [Co(PNP)(CH3CN)3](BF4)2 | −1.15 | 150 | 0.9 |
| [Co(dppp)(CH3CN)3](BF4)2 | −0.91 | 72 | 0.9 |
| [Co(CH3CN)6](BF4)2 | −1.21 | 95 | 0.9 |
Studies of catalytic hydrogen production
Fig. 4 shows successive cyclic voltammograms of [Co(PPh2NPh2)2(CH3CN)](BF4)2 recorded in acetonitrile with increasing concentrations of triflic acid. In the presence of triflic acid, the cyclic voltammogram of [Co(PPh2NPh2)2(CH3CN)](BF4)2 exhibits a catalytic wave for the reduction of protons at a half peak potential of −1.00 ± 0.05 V. This potential does not correspond to the Co(II/I) or Co(I/−I) couples of the parent complex. The height of the Co(II/I) wave associated with [Co(PPh2NPh2)2(CH3CN)](BF4)2 at −0.58 V decreases with increasing acid concentration while the height of the catalytic wave at −0.99 V increases several fold. The potential of the catalytic wave matches that of the Co(II/I) couple of [Co(PPh2NPh2)(CH3CN)3](BF4)2, which is proposed to be the active catalyst. The left graph in Fig. 5 shows plots of icat/i (where icat is the peak current in the presence of acid and i is the peak current in the absence of acid) versus triflic acid concentration over several different scan rates for this catalytic formation of hydrogen. Linear plots are observed over the range of acid concentration (0–0.025 M) and scan rates (50–800 mV s−1), which indicates a second-order dependence on [H+] for the catalytic reaction.27 A first order dependence of the catalytic current on the concentration of catalyst is demonstrated by the linear dependence of the catalytic current on catalyst precursor concentration at constant acid concentration (0.022 M, right graph in Fig. 5). This gives an overall rate law for the production of hydrogen of rate = k [H+]2[catalyst].2 in acetonitrile at increasing concentrations of triflic acid. Conditions: scan rate = 200 mV s−1, 0.3 M NBu4BF4 (supporting electrolyte), glassy carbon working electrode. Potentials are referenced to the ferrocenium/ferrocene couple shown at 0.0 V.](/image/article/2008/EE/b805309j/b805309j-f4.gif) | ||
| Fig. 4 Successive cyclic voltammograms of a 1.2 × 10−3 M solution of [Co(PPh2NPh2)2(CH3CN)](BF4)2 in acetonitrile at increasing concentrations of triflic acid. Conditions: scan rate = 200 mV s−1, 0.3 M NBu4BF4 (supporting electrolyte), glassy carbon working electrode. Potentials are referenced to the ferrocenium/ferrocene couple shown at 0.0 V. | ||
2 at several different scan rates (left graph). (b) Plot of catalytic current vs. concentrations of [Co(PPh2NPh2)2(CH3CN)](BF4)2 demonstrating first order kinetic behavior (right graph). Conditions: 0.3 M NBu4BF4 (supporting electrolyte) acetonitrile solution, glassy carbon working electrode.](/image/article/2008/EE/b805309j/b805309j-f5.gif) | ||
| Fig. 5 Plots of icat/ivs. triflic acid concentration for a 1.2 × 10−3 M solution of [Co(PPh2NPh2)2(CH3CN)](BF4)2 at several different scan rates (left graph). (b) Plot of catalytic current vs. concentrations of [Co(PPh2NPh2)2(CH3CN)](BF4)2 demonstrating first order kinetic behavior (right graph). Conditions: 0.3 M NBu4BF4 (supporting electrolyte) acetonitrile solution, glassy carbon working electrode. | ||
The catalytic activity of the proposed catalyst, [Co(PPh2NPh2)(CH3CN)3](BF4)2, was studied using p-bromoanilinium tetrafluoroborate ([BrC6H4NH3][BF4]) as the acid. In the presence of this acid (or tetrafluoroboric acid), the cyclic voltammogram of [Co(PPh2NPh2)(CH3CN)3](BF4)2, exhibits a catalytic wave for the reduction of protons with a half peak potential (−0.99 V) that corresponds to the Co(II/I) couple of this complex. A plot of icat/iversus acid concentration is shown in Fig. 6 (blue diamonds). The linear region of this plot is again consistent with a second order dependence of the overall catalytic rate on acid at low acid concentrations. At higher concentrations of acid, no acid dependence is observed, and this is interpreted in terms of a rate-limiting elimination of H2 under these conditions. Using the limiting current in this acid-independent region, a turnover frequency of 90 s−1 can be calculated for this catalyst.27 A controlled potential electrolysis experiment was performed on the complex at −1.1 V in the presence of tetrafluoroboric acid, and the evolution of H2 was confirmed by gas chromatography (current efficiency = 101 ± 5%).
2 (blue diamonds), [Co(dppp)(CH3CN)3](BF4)2 (solid red squares) and [Co(CH3CN)6](BF4)2 (green triangles). The scan rate was 0.05 V s−1 for all data. The E1/2 values for the respective catalysts are given in Table 3.](/image/article/2008/EE/b805309j/b805309j-f6.gif) | ||
| Fig. 6 Plots of the icat/i ratio as a function of the concentration (M) of p-bromoanilinium tetrafluoroborate for [Co(PPh2NPh2)(CH3CN)3](BF4)2 (blue diamonds), [Co(dppp)(CH3CN)3](BF4)2 (solid red squares) and [Co(CH3CN)6](BF4)2 (green triangles). The scan rate was 0.05 V s−1 for all data. The E1/2 values for the respective catalysts are given in Table 3. | ||
The catalytic activities of related cobalt diphosphine complexes with bromoanilinium tetrafluoroborate have also been investigated by cyclic voltammetry. During the stepwise addition of this acid to [Co(dppp)(CH3CN)3](BF4)2 only a small current enhancement is observed for the Co(II/I) couple at −0.91 V, which is consistent with a reversible electron transfer followed by an irreversible protonation reaction. However, the current enhancement is not sufficiently large to indicate catalysis. Fig. 6 shows a plot of the ratio of the current observed in the presence of acid to the peak current observed in the absence of acid for different acid concentrations of this complex (solid red squares).
The addition of bromoanilinium tetrafluoroborate to acetonitrile solutions of [Co(PNP)(CH3CN)3](BF4)2 results in the loss of the diphosphine ligand as indicated by a bleaching of the solution. However, in the presence of the weaker acid anisidinium tetrafluoroborate, no bleaching is observed and ratios of icat/i exceeding 15 (−1.15 V and a scan rate of 0.05 V s−1) indicate an active catalyst for H2 production. As a final control, the addition of bromoanilinium tetrafluoroborate to acetonitrile solutions of [Co(CH3CN)6](BF4)2 results in icat/i ratios of 3–5 at −1.21 V (green triangles in Fig. 6) that are clearly much smaller than those observed for [Co(PPh2NPh2)(CH3CN)3](BF4)2.
Discussion
The complex [Co(PPh2NPh2)2(CH3CN)](BF4)2 provides for a direct comparison of structural features, redox properties and catalytic activity with the highly active nickel catalyst for hydrogen production, [Ni(PPh2NPh2)2(CH3CN)](BF4)2. In an effort to grow crystals of the cobalt complex in dichloromethane, crystals of [Co(PPh2NPh2)2(Cl)]+ were obtained instead. The source of the chloride ligand is presumed to be a trace HCl contaminant in the solvent. The structure of this cation confirmed the presence of two coordinated cyclic diphosphine ligands with the fifth ligand in an equatorial position of a trigonal bipyramidal geometry. Two of the chelate rings of the diphosphine ligands are in boat conformations with Co⋯N distances of 3.50 and 3.52 Å, while the two chelate rings adjacent to the coordinated fifth ligand assume chair conformations to minimize steric interactions. The structure is very similar both in geometry and bond parameters to that observed previously for the nickel acetonitrile analogue.19The cyclic voltammogram of [Co(PPh2NPh2)2(CH3CN)]2+ can be compared with those observed for other M(diphosphine)2 complexes of the first row metal ions. Because both the seventeen-electron Co(II) cation, of [Co(PPh2NPh2)2(CH3CN)]2+, and the corresponding eighteen-electron Co(I) cation are five-coordinate, the observed reversibility of the Co(II/I) couple at −0.58 V is expected. Only one additional, quasi-reversible reduction wave is observed at −2.04 V. In contrast, a related diphosphine complex [Co(dppe)2(CH3CN)](BF4)2 has three reversible one-electron reduction waves at −0.70, −1.56, and −2.03 V.18 It is known from studies of nickel(II) and palladium(II) bis(diphosphine) complexes that smaller chelate bite sizes result in large negative shifts in the potential of the d8/d9 couples and a decrease in the separation between the d8/d9 and d9/d10 couples.28 The small chelate bite of the heterocyclic diphosphine ligand PPh2NPh2 apparently results in similar shifts in potentials for the cobalt complex to give a two-electron reduction for the Co(I/−I) couple. The current amplitude of the wave at −2.04 V is consistent with this assignment.
The onset potential of approximately −1.0 V for the production of hydrogen in the presence of triflic acid in acetonitrile solutions of [Co(PPh2NPh2)2(CH3CN)](BF4)2 does not correspond to the Co(II/I) potential, nor does it correspond to the more negative potentials associated with the Co(I/−I) couple of this complex. The decrease in the peak height of the Co(II/I) wave of [Co(PPh2NPh2)2(CH3CN)](BF4)2 as the acid concentration increased indicated that this complex was decomposing in the presence of triflic acid and suggested that one or more of the diphosphine ligands was being removed by protonation of the ligand at high acid concentrations. This type of acid-induced ligand loss was not observed in the previously studied nickel complex [Ni(PPh2NPh2)2(CH3CN)](BF4)2, and this reveals a significant difference in the chemistry of the two five-coordinate metal complexes. The trend is consistent with the generally observed greater lability of Co(II) vs. Ni(II) derivatives.
The complex [Co(PPh2NPh2)(CH3CN)3](BF4)2 is readily synthesized by addition of one equivalent of the cyclic ligand to [Co(CH3CN)6](BF4)2. In contrast, the addition of one equivalent of ligand to [Ni(CH3CN)6](BF4)2 under similar conditions led only to the formation of a mixture of [Ni(PPh2NPh2)2(CH3CN)](BF4)2 and the starting solvated complex. An X-ray diffraction study of the cobalt product confirmed that the square pyramidal dication contains only one cyclic ligand with one chelate ring in the chair form and the second in a boat. The Co⋯N distance to the proximal amine is 3.24 Å, and in addition, the phenyl ring of that amine is oriented over the top of the cobalt ion with Co–C(24) and Co–C(25) distances of 3.03 and 3.09 Å, respectively. The structural data alone are not definitive of a bonding interaction between the Co and a C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond of the Ph ring. At these long distances, any interaction would be quite weak, though a weak bonding interaction was found at similar distances (W–C distances 2.901(13) and 3.072(13) Å) in CpW(CO)2(IMes)+ (IMes = 1,3-bis(2,4,6-trimethylphenyl)-imidazol-2-ylidene) where one CC of a mesityl ring had an interaction with the metal.29
C bond of the Ph ring. At these long distances, any interaction would be quite weak, though a weak bonding interaction was found at similar distances (W–C distances 2.901(13) and 3.072(13) Å) in CpW(CO)2(IMes)+ (IMes = 1,3-bis(2,4,6-trimethylphenyl)-imidazol-2-ylidene) where one CC of a mesityl ring had an interaction with the metal.29
The E1/2 of the Co(II/I) couple of [Co(PPh2NPh2)(CH3CN)3](BF4)2, −0.99 V, corresponds to the potential observed for the catalytic wave of [Co(PPh2NPh2)2(CH3CN)](BF4)2 in acetonitrile in the presence of triflic acid. This confirms that the actual catalytic species for hydrogen formation is [Co(PPh2NPh2)(CH3CN)3](BF4)2. The negative shift in the half-wave potential of the Co(II/I) couple from −0.58 V for the bis(diphosphine) complex to −0.99 V for the mono(diphosphine) derivative suggests that the hard nitrogen donors of the acetonitrile ligands are more electron-donating than the softer diphosphine ligand in this system. Because a strong acid such as triflic acid is no longer required to remove one of the diphosphine ligands, catalytic activity can now be observed using weaker acids, and this allows the catalysis to proceed with a lower overpotential. For example, a catalytic wave is observed at −0.99 V in the presence of p-bromoanilinium tetrafluoroborate (pKa = 9.4 in acetonitrile30) and anisidinium tetrafluoroborate (pKa = 11.8 in acetonitrile30). Because of the lower acidity of anisidinium tetrafluoroborate, the catalytic currents are much smaller than for p-bromoanilinium tetrafluoroborate. At high concentrations of the p-bromoanilinium salt, the catalytic current observed for the production of H2 is independent of acid concentrations and a limiting turnover frequency of 90 s−1 at 22 °C was determined. From the operating potential of this catalyst and the pKa value of p-bromoanilinium tetrafluoroborate (9.4), the overpotential for hydrogen production can be calculated to be 285 mV using the standard NHE potential in acetonitrile suggested by Evans et al.31
The cobalt derivative [Co(PPh2NPh2)(CH3CN)3](BF4)2 shows a slower rate for proton reduction than that reported for [Ni(PPh2NPh2)2(CH3CN)](BF4)2 (350 s−1), while the overpotential is somewhat reduced compared to that estimated for the nickel system (ca. 350 mV). Mechanistic studies have established the same rate law for both systems: rate = k′[H+]2[catalyst] at low acid concentrations and rate = k[catalyst] at high acid concentrations. Theoretical and experimental studies of the nickel catalysts have suggested that the positioning of two amines near the nickel ion is critical for high catalytic activity because interaction with both amines stabilizes the formation of a dihydrogen ligand on the Ni(II) ion.19 A similar effect cannot be achieved for the cobalt catalyst, which contains only one positioned amine base. However, the complex [Co(dppp)(CH3CN)3](BF4)2, which contains no pendant base in the ligand backbone of the diphosphine ligand, exhibits no significant catalytic activity for hydrogen production under the same conditions that [Co(PPh2NPh2)(CH3CN)3](BF4)2 exhibits relatively high catalytic rates. This suggests that the positioned amine in the cyclic ligand plays an important role in the catalytic activity of [Co(PPh2NPh2)(CH3CN)3](BF4)2. Previous NMR studies of diamagnetic iron diphosphine complexes containing a single pendant amine have shown that the base facilitates both intermolecular proton exchange between the protonated complex and protons in solution as well as intramolecular Fe–H⋯H–N exchange.32 Similar exchange processes appear likely for this cobalt system. In addition, a mechanistic step may be proposed in which an intramolecular interaction of a cobalt-hydride with the adjacent protonated amine promotes formation of a H–H bond. Similar M–H⋯H–N interactions have been proposed previously in our studies of the electrocatalysis of hydrogen oxidation promoted by [Ni(PNP)2]2+.24
The complex [Co(PNP)(CH3CN)3](BF4)2 was prepared in an attempt to further probe the role of the pendant base in the catalytic activity of this system. The PNP complex incorporates a more basic amine as a result of more electron donating substituents than those in [Co(PPh2NPh2)(CH3CN)3](BF4)2 (methyl and ethyl versus phenyl). As a result of the increased basicity, the weaker acid, anisidinium tetrafluoroborate (pKa = 11.8 in acetonitrile), can be used, and catalytic hydrogen production is observed. These observations further support the postulate that a base in the second coordination sphere plays an important role in hydrogen formation. Additional studies on the cobalt systems with PNP ligands will be reported in a subsequent publication.
It is interesting that catalytic waves for hydrogen production are observed by cyclic voltammetry when tetrafluoroboric acid is added to the simple solvated complex [Co(CH3CN)6](BF4)2. The onset potential for the catalytic wave corresponds to the potential of the Co(II/I) couple for this complex at −1.21 V. However as shown in Fig. 6, the complex is nearly inactive when the weaker p-bromoanilinium acid is used. The negative potential and strong acid required for this catalytic reaction indicate that it proceeds with a large overpotential and does not significantly contribute to the observed catalytic activity of [Co(PPh2NPh2)(CH3CN)3](BF4)2 or [Co(PNP)(CH3CN)3](BF4)2.
Summary and conclusions
This work has extended studies on the electrocatalytic production of H2 using first row transition metal complexes containing diphosphine ligands with pendant nitrogen bases. [Co(PPh2NPh2)(CH3CN)3](BF4)2 has a catalytic turnover frequency of 90 s−1 with an estimated overpotential of 285 mV. In contrast to previously studied nickel complexes, two cyclic diphosphine ligands with positioned nitrogen bases are not required for high activity. A comparison of the catalytic behavior of [Co(PPh2NPh2)(CH3CN)3](BF4)2 with that of [Co(dppp)(CH3CN)3](BF4)2, for which no catalytic activity is observable by cyclic voltammetry, indicates that the pendant amine is playing an important role in the catalysis of H2 production. This is supported by the observation that the complex containing the flexible acyclic diphosphine incorporating an amine base, [Co(PNP)(CH3CN)3](BF4)2, is also a catalyst in the presence of a weak acid.An important conclusion from this work is that cobalt complexes containing diphosphine ligands with pendant bases can exhibit catalytic rates for hydrogen production comparable to those of previously reported nickel complexes. The amine base in the second coordination sphere plays a crucial role in these catalytic processes, but for cobalt complexes this high catalytic activity is obtained with only one positioned pendant base rather than two as required in the nickel complexes.
Experimental
Materials and instrumentation
The syntheses of [Co(CH3CN)6](BF4)2,33 PPh2NPh2,19 and PNP24 have been reported elsewhere. Synthetic reactions were carried out under nitrogen. Electrochemical measurements were performed at room temperature under a nitrogen atmosphere in acetontirile solutions containing either 0.30 M tetrabutylammonium tetrafluoroborate (Bu4NBF4) or 0.20 M tetraethylammonium tetrafluoroborate. Cyclic voltammetry data were collected using a Cypress Systems model CS-1200 computer aided potentiostat with a three-electrode system. The working electrode was a 2 mm diameter glassy carbon disk and the counter electrode was an uncoated glassy carbon rod with a 3 mm diameter. The reference electrode was a silver wire coated with silver chloride and immersed in a 0.20 M electrolyte solution of tetraethylammonium tetrafluoroborate in CH3CN and separated from the sample solution by a Vycor tip. However, all potentials are referenced to the ferrocenium/ferrocene couple (FeCp2+/FeCp2), using ferrocene as an internal standard.Synthesis of [Co(PPh2NPh2)2(CH3CN)](BF4)2
A mixture of PPh2NPh2 (0.306 g, 0.672 mmol) and [Co(CH3CN)6](BF4)2 (0.160 g, 0.336 mmol) in acetonitrile was stirred at room temperature for 6 h. The brownish solution was reduced to approximately 1 mL, and 5 mL of methylene chloride was added. Fifteen mL of methanol was added with stirring, and the volume of the solution reduced until a precipitate began to appear. Additional precipitate formed upon cooling to −15 °C overnight, which was isolated by filtration. This process was repeated a second time to yield an orange–brown microcrystalline product. Yield: 0.171 g, 43%. Elemental analyses calcd. for C58H59B2CoF8N5P4: C, 58.91; H, 5.03; N, 5.92. Found: C, 58.28; H, 4.79; N, 5.69. ESI+ MS, m/z (CH3CN): 1002 {[Co(PPh2NPh2)2]Cl}+, 483 {[Co(PPh2NPh2)2]}2+.[Co(PPh2NPh2)(CH3CN)3](BF4)2
[Co(CH3CN)6](BF4)2 (0.20 g, 0.42 mmol) was dissolved in 5 mL acetonitrile and PPh2NPh2 (0.19 g, 0.42 mmol) was added. The brown solution was stirred at room temperature overnight, and then solvent was removed in vacuo. The resulting brown solid was recrystallized from acetonitrile/ether and identified by an X-ray diffraction study. Yield: 70%. Elemental analyses calcd. for C34H37B2CoF8N5P2: C, 50.40; H, 4.60; N, 8.65. Found: C, 50.16; H, 4.87; N, 8.65. Electrochemical data are reported in Table 3. The procedure described here was followed for all the [Co(diphosphine)(CH3CN)3](BF4)2 derivatives reported.X-Ray diffraction studies
A suitable crystal of [Co(PPh2NPh2)2Cl]BF4 or [Co(PPh2NPh2)(CH3CN)3](BF4)2 was selected, attached to a glass fiber and data were collected at 90(2) K using a Bruker/Siemens SMART APEX instrument (Mo Kα radiation, λ = 0.71073 Å) equipped with a Cryocool NeverIce low temperature device. For each structure, data were measured using omega scans 0.3° per frame for 20 s, and a full sphere of data was collected. A total of 2400 frames were collected in each case with a final resolution of 0.83 Å for [Co(PPh2NPh2)2Cl]BF4 and 0. 77 Å for [Co(PPh2NPh2)(CH3CN)3](BF4)2. Cell parameters were retrieved using SMART34 software and refined using SAINTPlus35 on all observed reflections. Data reduction and correction for Lorentzian polarization and decay were performed using the SAINTPlus software. Absorption corrections were applied using SADABS.36 Each structure was solved by direct methods and refined by least squares method on F2 using the SHELXTL program package.37 No decomposition was observed during data collection.The structure of [Co(PPh2NPh2)2Cl]BF4 was solved in the space group C2/c (#15) by analysis of systematic absences. Aryl rings C9–C14 and C48–C53 were disordered and modeled in two positions with occupancies 65 : 35%. One of the half occupied BF4 groups was also disordered over an inversion center. The diffraction contribution from disordered solvent molecules was removed using the subroutine SQUEEZE in the PLATON software suite.38 All other non-hydrogen atoms were refined anisotropically. The structure of [Co(PPh2NPh2)(CH3CN)3](BF4)2 was solved in the space group P2(1)/n (#14) by analysis of systematic absences. All non-hydrogen atoms were refined anisotropically. Details of the data collections and refinements are given in Table 4. Complete structural data are provided in the ESI.†
| Formula | C56H56BClCoF4N4P4 | C34H37B2CoF8N5P2 |
|---|---|---|
| Formula weight | 1090.12 | 810.18 |
| Temperature | 90(2) K | 90(2) K |
| Crystal system | Monoclinic | Monoclinic |
| Space group | C2/c | P2(1)/n |
| Unit cell dimensions | a = 34.2939(13) Å | a = 10.0584(4) Å |
| α = 90° | α = 90° | |
| b = 12.9425(5)Å | b = 20.1715(7) Å | |
| β = 121.872(1)° | β = 93.1020(10)° | |
| c = 28.6361(11) Å | c = 17.6050(6) Å | |
| γ = 90° | γ = 90° | |
| Volume | 10793.8(7) Å3 | 3566.7(2) Å3 |
| Z | 8 | 4 |
| Density(calc) | 1.342 Mg m−3 | 1.509 Mg m−3 |
| Absorption coefficient | 0.541 mm−1 | 0.647 mm−1 |
| F(000) | 4520 | 1660 |
| Crystal size, mm | 0.47 × 0.20 × 0.09 | 0.29 × 0.16 × 0.10 |
| Theta range for data collection | 1.40 to 25.25° | 1.54 to 27.50° |
| Reflections collected | 62971 | 52775 |
| Independent reflections | 9775 [R(int) = 0.0446] | 8183 [R(int) = 0.0384] |
| Absorption correction | Semi-empirical | Semi-empirical |
| Goodness-of-fit on F2 | 1.065 | 1.034 |
| Final R indices [I > 2σ(I)] | R1 = 0.0514, wR2 = 0.1372 | R1 = 0.0345, wR2 = 0.0838 |
| R indices (all data) | R1 = 0.0634, wR2 = 0.1446 | R1 = 0.0449, wR2 = 0.0899 |
| Largest diff. peak and hole | 1.597 and −0.816 e Å−3 | 0.466 and −0.266 e Å−3 |
Acknowledgements
This work was supported by the Chemical Sciences program of the Office of Basic Energy Sciences of the Department of Energy. The Pacific Northwest National Laboratory is operated by Battelle for the US Department of Energy.The Bruker (Siemens) SMART APEX diffraction facility was established at the University of Idaho with the assistance of the NSF-EPSCoR program and the M. J. Murdock Charitable Trust, Vancouver, WA, USA.
References
- A. Volbeda, E. Garcin, C. Piras, A. L. de Lacey, V. M. Fernandez, E. C. Hatchikian, M. Frey and J. C. Fontecilla-Camps, J. Am. Chem. Soc., 1996, 118, 12989–12996 CrossRef CAS; E. Garcin, X. Vernede, E. C. Hatchikian, A. Volbeda, M. Frey and J. C. Fontecilla-Camps, Structure, 1999, 7, 557–565 CrossRef CAS.
- Y. Nicolet, A. L. de Lacey, X. Vernède, V. M. Fernandez, E. C. Hatchikian and J. Fontecilla-Camps, J. Am. Chem. Soc., 2001, 123, 1596–1601 CrossRef CAS; A. Volbeda and J. C. Fontecilla-Camps, J. Chem. Soc., Dalton Trans., 2003, 4030–4038 RSC.
- J. W. Peters, W. N. Lanzilotta, B. J. Lemon and L. C. Seefeldt, Science, 1998, 282, 1853–1858 CrossRef CAS; J. W. Peters, Curr. Opin. Struct. Biol., 1999, 9, 670–676 CrossRef CAS.
- A. S. Pereira, P. Tavares, I. Moura, J. J. G. Moura and B. H. Huynh, J. Am. Chem. Soc., 2001, 123, 2771–2782 CrossRef CAS.
- Y. Higuchi, H. Ogata, K. Miki, N. Yasuoka and T. Yagi, Structure, 1999, 7, 549–556 CrossRef CAS.
- E. J. Lyon, I. P. Georgakaki, J. H. Reibenspies and M. Y. Darensbourg, J. Am. Chem. Soc., 2001, 123, 3268–3278 CrossRef CAS; X. Zhao, I. P. Georgakaki, M. L. Miller, J. C. Yarbrough and M. Y. Darensbourg, J. Am. Chem. Soc., 2001, 123, 9710–9711 CrossRef CAS; R. Mejia-Rodriguez, D. Chong, J. H. Reibenspies, M. P. Soriaga and M. Y. Darensbourg, J. Am. Chem. Soc., 2004, 126, 12004–12014 CrossRef CAS; J. W. Tye, J. Lee, H.-W. Wang, R. Mejia-Rodriguez, J. H. Reibenspies, M. B. Hall and M. Y. Darensbourg, Inorg. Chem., 2005, 44, 5550–5552 CrossRef CAS; J. W. Tye, M. Y. Darensbourg and M. B. Hall, Inorg. Chem., 2006, 45, 1552–1559 CrossRef CAS; T. Liu and M. Y. Darensbourg, J. Am. Chem. Soc., 2007, 129, 7008–7009 CrossRef CAS.
- J. D. Lawrence, H. Li, T. B. Rauchfuss, M. Bénard and M.-M. Rohmer, Angew. Chem., Int. Ed., 2001, 40, 1768–1771 CrossRef CAS; F. Gloaguen, J. D. Lawrence and T. B. Rauchfuss, J. Am. Chem. Soc., 2001, 123, 9476–9477 CrossRef CAS; A. K. Justice, R. C. Linck, T. B. Rauchfuss and S. R. Wilson, J. Am. Chem. Soc., 2004, 126, 13214–13215 CrossRef CAS; C. A. Boyke, J. I. Van der Vlugt, T. B. Rauchfuss, S. R. Wilson, G. Zampella and L. De Gioia, J. Am. Chem. Soc., 2005, 127, 11010–11018 CrossRef CAS; J. I. Van der Vlugt, T. B. Rauchfuss, C. M. Whaley and S. R. Wilson, J. Am. Chem. Soc., 2005, 127, 16012–16013 CrossRef; A. K. Justice, R. C. Linck and T. B. Rauchfuss, Inorg. Chem., 2006, 45, 1552–1559 CrossRef CAS; J. L. Stanley, Z. M. Heiden, T. B. Rauchfuss, S. R. Wilson, L. DeGioia and G. Zampella, Organometallics, 2008, 27, 119–125 CrossRef CAS.
- D. J. Evans and C. J. Pickett, Chem. Soc. Rev., 2003, 32, 268–275 RSC; S. J. Borg, T. Behrsing, S. P. Best, M. Razavet, X. Liu and C. J. Pickett, J. Am. Chem. Soc., 2004, 126, 16988–16999 CrossRef CAS; C. Tard, X. Liu, S. K. Ibrahim, M. Bruschi, L. De Gioia, S. C. Davies, X. Yang, L.-S. Wang, G. Sawers and C. J. Pickett, Nature, 2005, 433, 610–613 CrossRef CAS; M. H. Cheah, C. Tard, S. J. Borg, X. Liu, S. K. Ibrahim, C. J. Pickett and S. P. Best, J. Am. Chem. Soc., 2007, 129, 11085–11092 CrossRef CAS.
- S. Ott, M. Kritikos, B. Åkermark, L. Sun and R. Lomoth, Angew. Chem., Int. Ed., 2004, 43, 1006–1009 CrossRef CAS; L. Schwartz, G. Ellers, L. Eriksson, A. Gogoll, R. Lomoth and S. Ott, Chem. Commun., 2006, 520–521 RSC.
- P. Das, J.-F. Capon, F. Gloaguen, F. Y. Pétillon, P. Schollhammer, J. Talarmin and K. W. Muir, Inorg. Chem., 2004, 43, 8203–8205 CrossRef CAS; J.-F. Capon, S. El Hassnaoui, F. Gloaguen, P. Schollhammer and J. Talarman, Organometallics, 2005, 24, 2020–2022 CrossRef CAS; S. Ezzaher, J.-F. Capon, F. Gloaguen, F. Y. Pétillon, P. Schollhammer, J. Talarmin, R. Pichon and N. Kervarec, Inorg. Chem., 2007, 46, 3426–3428 CrossRef CAS.
- L.-C. Song, J. Gao, H.–T. Wang, Y.-J. Hua, H.-T. Fan, X.-G. Zhang and Q.-M. Hu, Organometallics, 2006, 25, 5724–5729 CrossRef CAS; L.-C. Song, B.-S. Yin, Y.-L. Li, L.-Q. Zhao, J.-H. Ge, Z.-Y. Yang and Q.-M. Hu, Organometallics, 2007, 26, 4921–4929 CrossRef CAS.
- G. A. N. Felton, A. K. Vannucci, J. Chen, L. T. Lockett, N. Okumura, B. J. Petro, U. I. Zakai, D. H. Evans, R. S. Glass and D. L. Lichtenberger, J. Am. Chem. Soc., 2007, 129, 12521–12530 CrossRef CAS.
- W. Gao, J. Liu, B. Åkermark and L. Sun, Inorg. Chem., 2006, 45, 9169–9171 CrossRef CAS; W. Gao, J. Ekstrom, J. Liu, C. Chen, L. Eriksson, L. Wang, B. Åkermark and L. Sun, Inorg. Chem., 2007, 46, 1981–1991 CrossRef CAS.
- I. A. De Carcer, A. DiPasquali, A. L. Rheingold and D. M. Heinekey, Inorg. Chem., 2006, 45, 8000–8002 CrossRef CAS.
- M. Razavet, V. Artero and M. Fontecave, Inorg. Chem., 2005, 44, 4786–4795 CrossRef CAS; V. Artero and M. Fontecave, Coord. Chem. Rev., 2005, 29, 1518–1535 CrossRef; C. Baffent, V. Artero and M. Fontecave, Inorg. Chem., 2007, 46, 1817–1824 CrossRef; X. Hu, B. Cossairt, B. Brunschwig, N. Lewis and J. C. Peters, Chem. Commun., 2005, 4723–4725 RSC; X. Hu, B. Brunschwig and J. C. Peters, J. Am. Chem. Soc., 2007, 129, 8988–8998 CrossRef CAS.
- M. Rakowski DuBois and D. L. DuBois, C. R. Chim., 2008 DOI:10.016/j.crci.2008.01.019.
- D. Berning, B. Noll and D. DuBois, J. Am. Chem. Soc., 1999, 121, 11432–11447 CrossRef CAS; D. Berning, A. Miedaner, C. J. Curtis, B. C. Noll, M. Rakowski DuBois and D. L. DuBois, Organometallics, 2001, 20, 1832–1839 CrossRef CAS; C. J. Curtis, A. Miedaner, W. W. Ellis and D. L. DuBois, J. Am. Chem. Soc., 2002, 124, 1918–1925 CrossRef CAS; A. Price, R. Ciancanelli, B. C. Noll, C. J. Curtis, D. L. DuBois and M. Rakowski DuBois, Organometallics, 2002, 21, 4833–4839 CrossRef CAS.
- R. Ciancanelli, B. C. Noll, D. L. DuBois and M. Rakowski DuBois, J. Am. Chem. Soc., 2002, 124, 1926–1832 CrossRef CAS.
- A. D. Wilson, R. H. Newell, M. J. McNevin, J. T. Muckerman, M. Rakowski DuBois and D. L. DuBois, J. Am. Chem. Soc., 2006, 128, 358 CrossRef CAS; A. D. Wilson, R. Shoemaker, A. Miedaner, D. L. DuBois and M. Rakowski DuBois, Proc. Nat. Acad. Sci. U. S. A., 2007, 104, 6951–6956 CrossRef CAS.
- A. D. Wilson, K. Fraze, S. Miller, B. Twamley, D. L. DuBois and M. Rakowski DuBois, J. Am. Chem. Soc., 2008, 130, 1061–1068 CrossRef CAS.
- G. M. Jacobsen, R. K. Shoemaker, M. MacNevin, M. Rakowski DuBois and D. L. DuBois, Organometallics, 2007, 26, 5003–5009 CrossRef CAS.
- D. L. DuBois and A. Miedaner, Inorg. Chem., 1986, 25, 4642–4650 CrossRef CAS.
- R. Ciancanelli, PhD thesis, University of Colorado, Boulder, CO, 2002.
- C. J. Curtis, A. Miedaner, R. Ciancanelli, W. W. Ellis, B. C. Noll, M. Rakowski DuBois and D. L. DuBois, Inorg. Chem., 2003, 42, 216–227 CrossRef CAS.
- G. M. Jacobsen, R. K. Shoemaker, M. Rakowski DuBois and D. L. DuBois, Organometallics, 2007, 26, 4964–4971 CrossRef CAS.
- C. T. Hunt and A. L. Balch, Inorg. Chem., 1981, 20, 2267–2270 CrossRef; A. Miedaner, R. C. Haltiwanger and D. L. DuBois, Inorg. Chem., 1991, 30, 417–427 CrossRef CAS; A. Miedaner and D. L. DuBois, Inorg. Chem., 1988, 27, 2479–2484 CrossRef CAS.
- P. Delehay and G. L. Stiehl, J. Am. Chem. Soc., 1952, 74, 3500–3505 CrossRef; R. S. Nicholson and I. Shain, Anal. Chem., 1964, 36, 706–723 CrossRef CAS; J. M. Saveant and E. Vianello, Electrochim. Acta, 1965, 10, 905–920 CrossRef CAS; J. M. Saveant and E. Vianello, Electrochim. Acta, 1967, 12, 629–646 CrossRef CAS.
- J. W. Raebiger, A. Miedaner, C. J. Curtis, S. M. Miller and D. L. DuBois, J. Am. Chem. Soc., 2004, 126, 5502–5514 CrossRef CAS.
- F. Wu, V. K. Dioumaev, D. J. Szalda, J. Hanson and R. M. Bullock, Organometallics, 2007, 26, 5079–5090 CrossRef CAS.
- I. Kaljurand, A. Kutt, L. Soovali, T. Rodima, V. Maemets, I. Leito and I. A. Koppel, J. Org. Chem., 2005, 70, 1019–1028 CrossRef CAS.
- G. A. N. Felton, R. S. Glass, D. L. Lichtenberger and D. H. Evans, Inorg. Chem., 2006, 45, 9181–9184 CrossRef CAS.
- R. M. Henry, R. K. Shoemaker, R. H. Newell, G. M. Jacobsen, D. L. DuBois and M. Rakowski DuBois, Organometallics, 2005, 24, 2481 CrossRef CAS; R. M. Henry, R. K. Shoemaker, D. L. DuBois and M. Rakowski DuBois, J. Am. Chem. Soc., 2006, 128, 3002–3010 CrossRef CAS.
- R. A. Heintz, J. A. Smith, P. S. Szalay, A. Weisgerber, K. R. Dunbar, K. Beck and D. Coucouvanis, Inorg. Synth., 2002, 33, 75–83 CAS; B. J. Hathaway, D. E. Holah and A. E. Underhill, J. Chem. Soc., 1962, 2444–2448 RSC.
- SMART: v. 5.632, Bruker AXS, Madison, WI, 2005 Search PubMed.
- SAINTPlus: v. 7.23a, Data Reduction and Correction Program, Bruker AXS, Madison, WI, 2004 Search PubMed.
- SADABS: v.2007/4, an empirical absorption correction program, Bruker AXS Inc., Madison, WI, 2007 Search PubMed.
- SHELXTL: v. 6.14, Structure Determination Software Suite, G. M. Sheldrick, Bruker AXS Inc., Madison, WI, 2004 Search PubMed.
- A. L. Spek, PLATON: A Multipurpose Crystallographic Tool, Utrecht University, The Netherlands, 2006 Search PubMed; A. L. Spek, Acta Crystallogr., Sect A, 1990, 46, C34 Search PubMed.
Footnote |
| † CCDC reference numbers 683301 and 683302. For crystallographic data in CIF or other electronic format see DOI: 10.1039/b805309j |
| This journal is © The Royal Society of Chemistry 2008 |