Synthesis of ammonia borane for hydrogen storage applications†
David J.
Heldebrant
,
Abhi
Karkamkar
,
John C.
Linehan
and
Tom
Autrey
*
Pacific Northwest National Laboratory, Richland, WA 99352, USA
First published on 20th June 2008
Abstract
A new synthetic procedure to make the condensed phase hydrogen storage material, ammonia borane (NH3BH3, abbreviated as AB), is described and compared with previous literature procedures. Ammonia borane with a gravimetric density ca. 194 gm H2 kg−1 and a volumetric density ca. 146 H2 litre−1, is a promising chemical hydrogen storage material for fuel cell powered applications. The work shows that ammonium borohydride, NH4BH4, formed in situ by the metathesis of NH4X and MBH4 salts (M = Na, Li; X = Cl, F) in liquid NH3, can be induced to decompose in an organic ether to yield AB in near quantitative yield. The purity of the AB prepared by this one-pot synthetic strategy is sufficient to meet the thermal stability requirements for on-board hydrogen storage.
Introduction
For many years, the synthesis, isolation, and characterization of the simple Lewis acid–base complex AB was an elusive endeavour.1,2 It was well established that Lewis acids such as CO and Me3N reacted with diborane to give the monomeric donor–acceptor adducts, (CO)–BH3 and Me3N–BH3, respectively; however, the reaction of ammonia with diborane appeared to proceed by a very different pathway.3 Alfred Stock had shown that the stoichiometry was unusual; two moles of ammonia react with one mole of diborane to form a salt-like compound represented by the empirical formula B2H6·2NH3.4 Subsequent molecular weight determinations by others confirmed the dimeric nature of the diammoniate of diborane (DADB);5,6 however, there was considerable ambiguity in the identity of the molecular nature of the structure ranging from the ammonium cation, I, the borohydride anion, II, to a structure containing both an ammonium cation and a borohydride anion separated by an aminoborane, III.7–9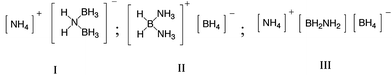
In a classic series of papers from Parry's laboratory in the 1950s, the structure of DADB was finally proven to be the species containing the boronium cation and borohydride ion pair, which is species II in the scheme shown above.10–16 It was during these experimental studies that serendipity played a role in providing AB. Sheldon Shore had added NH4Cl to the DADB in an attempt to prepare the corresponding chloride salt [NH3BH2NH3]+ [Cl]−, eqn (1), in analogy with the previous work of Schultz who showed that the addition of NH4Br to DADB in liquid ammonia yielded the corresponding bromide salt [NH3BH2NH3]+ [Br]−, eqn (2).1 However, Shore discovered an unexpected side reaction that formed AB.17 As it turned out, the choice of the solvent changed the course of the reaction, and he subsequently demonstrated that AB could be formed directly from a mixture of NH4Cl and LiBH4 in diethyl ether when a trace of NH3 was present, eqn (3). Consequently, the metathesis reaction in organic solvents has been one of the most used synthetic strategies to prepare ammonia borane.18,19
| NH4Cl + II → NH3BH3 + [NH3BH2NH3]+ [Cl]− + H2 | (1) |
| NH4Br + II → NH4BH4 + [NH3BH2NH3]+ [Br]− | (2a) |
| NH4BH4 → II + H2 | (2b) |
| NH4Cl + LiBH4 → NH3BH3 (THF with trace NH3) | (3) |
A few years later, Shore and Böddeker described reaction conditions that enabled the preparatory scale synthesis of DADB and mixtures of DADB with AB.20 They showed that passing diborane gas into liquid ammonia gave the asymmetric cleavage product DADB, eqn (4), in quantitative yield at temperatures below −78 °C.21 On the other hand, when they distilled liquid NH3 onto a solution of BH3–THF complex at −78 °C, they observed both DADB and the symmetric cleavage product AB in equimolar amounts, eqn (5).
Asymmetric cleavage

| (4a) |

| (4b) |
Symmetric cleavage:

| (5a) |
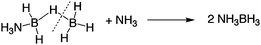
| (5b) |
Subsequently, Mayer reported that the basicity of the solvent had an influence on the competing symmetric and asymmetric reaction pathways.22 He found that AB could be prepared with 68–76% yields in diglyme if gaseous ammonia was added to diborane solutions of ethers. However, when diborane was added to ammonia dissolved in the same solvents the yields were lower and more erratic (i.e., 32–60%). In a paper published the following year, Mayer reported that DADB could be converted to AB, eqn (6), with no apparent H2 evolution in diglyme containing a trace amount of diborane (yield 80–91%, 40 h at 25 °C).23,24 He noted that this was in sharp contrast to the fate of DADB in ether solvents containing traces of ammonia. In ether, with a trace of ammonia, Shore observed significant hydrogen evolution with a polymeric product, (NH2BH2)n, and a limited quantity of AB, eqn (7), (ca. <20%).2
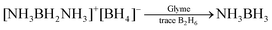 | (6) |
 | (7) |
In this paper, we describe the details of a modified synthetic strategy for preparing AB for hydrogen storage applications, and we report the surprising result that AB can be prepared in near quantitative yields in a one-pot synthetic strategy. The high yields of isolated AB were surprising for the following two reasons: (1) we found that it was not necessary to remove all traces of ammonia prior to addition of the organic solvent, and (2) we found that it was not necessary to add trace quantities of diborane to get quantitative yields of AB. A synthetic approach to prepare AB in quantitative yields in a single pot will provide researchers with a simple procedure to prepare AB. Furthermore, we envision that this simpler procedure can be scaled up and the solvents can be recycled.25 Efficient routes to the synthesis of material are an important aspect for R&D focused on discovering materials that could be used to store high densities of hydrogen for fuel cell powered applications. Ammonia borane, with a 144 g H2 L−1 volumetric density and a 194 g H2 kg−1 gravimetric density, is under investigation by many research groups26–29 that are looking for hydrogen storage materials to meet system-based hydrogen storage targets (i.e., >82 g H2 L−1 volumetric density and >90 g H2 kg−1 gravimetric density) that have been established by the US Department of Energy (DOE).30–32
Experimental
Sequential addition
Anhydrous NH3 (25 mL) was condensed in an oven-dried, 100 mL, three-neck, round-bottom flask fitted with a stir bar. The flask was cooled in a dry-ice/isopropanol bath (−78 °C) that was open to a nitrogen atmosphere. Both NH4Cl (1 g, 18.6 mmol) and NaBH4 (0.71 g, 18.6 mmol) were added to the liquid ammonia using a solids addition funnel. The mixture was stirred for 2 h under a nitrogen atmosphere at −78 °C. Ammonia then was removed by vacuum, leaving a white polycrystalline powder that was assumed to be NH4BH4 and NaCl. Anhydrous THF (100 mL) was cannulated under a nitrogen atmosphere over the residual white solid at −78 °C open to a nitrogen bubbler. After THF addition, the white solid began to foam and release hydrogen. The slurry was stirred at −78 °C for 30 min and then slowly warmed to room temperature. At room temperature, the slurry was stirred for an additional 60 min at which time gas evolution had ceased. The NaCl was filtered and the THF was removed by rotary evaporation to yield 0.57 g of a microcrystalline powder, mp = 110 °C. Analysis of this powder using 1H NMR in d-glyme showed a triplet at 3.8 ppm (NH3, JN–H = 45 Hz) and a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1:1:1 quartet centered at 1.5 ppm (BH3, JB–H = 94 Hz), and analysis using 11B NMR showed a quartet at −23 ppm (BH3, JB–H = 93 Hz), thus providing ca. 99% of the theoretical yield of AB.
:1:1:1 quartet centered at 1.5 ppm (BH3, JB–H = 94 Hz), and analysis using 11B NMR showed a quartet at −23 ppm (BH3, JB–H = 93 Hz), thus providing ca. 99% of the theoretical yield of AB.
Parallel addition I
Anhydrous NH3 (10 mL) was condensed in an oven-dried, 100 mL, three-neck, round-bottom flask fitted with a stir bar. The flask was cooled in a dry-ice/isopropanol bath (−78 °C) that was open to a nitrogen atmosphere. As before, the NH4Cl (1.06 g, 18.6 mmol) and NaBH4 (0.71 g, 18.6 mmol) were added by a solids addition funnel to the three-neck flask, and the reaction was stirred for 2 h under nitrogen at −78 °C. Anhydrous THF (100 mL) was slowly added drop-wise to the flask via an addition funnel, and NH3 and H2 were allowed to evaporate while thawing the reaction to room temperature. At room temperature, the flask was stirred for another hour. The precipitated NaCl was removed by filtration, and the THF was removed by rotary evaporation, and the remaining microcrystalline powder then was dried overnight under vacuum. The amount of AB recovered as a microcrystalline powder (mp = 107 °C) was 0.58 g (18.4 mmol, 99% yield). Analysis using 1H NMR in d-glyme showed a small triplet at 3.8 ppm (NH3, JN–H = 45 Hz), and a 1:1:1:1 quartet centered at 1.5 ppm (BH3, JB–H = 94 Hz). Analysis using 11B NMR showed a quartet at −23 ppm (BH3, JB–H = 93 Hz).
Parallel addition II
Anhydrous NH3 (25 mL) was condensed in an oven-dried, 100 mL, three-neck, round-bottom flask fitted with a stir bar. The flask was cooled in a dry-ice/isopropanol bath (−78 °C) that was open to a nitrogen atmosphere. A 9.3 mL volume of 2 M LiBH4:THF (18.6 mmol) was added to 50 mL anhydrous THF, and then loaded in an addition funnel and slowly added dropwise to the liquid NH3. NH4F (0.408 g, 11.0 mmol) was massed in a glove box into a solids addition funnel. The solids addition funnel was connected to the flask, and the NH4F was slowly added to the THF–ammonia solution and stirred for 1.25 h under nitrogen at −78 °C. After gas evolution had ceased, the flask was warmed to room temperature, and the precipitated NaF was separated from the remaining liquid by filtering. The THF was removed by rotary evaporation and the remaining material then was dried under vacuum overnight to yield 0.420 g of a microcrystalline powder that was composed of a mixture of LiBH4 and AB. Analysis using 11B NMR in d-glyme showed a quartet at −23 ppm (BH3, JB–H = 94 Hz, 87%) and a pentet at −41 ppm (BH4, JB–H = 82 Hz).
Results and discussion
The following three general procedures for preparing AB are described in the literature:1. Condensation of B2H6 in organic ethers followed by treatment with gaseous NH3. This procedure provides AB in yields varying from 45–60%.20,22
2. Metathesis of NH4X and MBH4 in an organic solvent. This procedure provides AB in yields ranging from 30–85%.2,12,18,19,33
3. Dissolving DADB in glyme solvent containing a trace of diborane.23
The first approach requires making diborane in stoichiometric quantities and results in mixtures of DADB and AB that depend on the reaction temperature and solvent used in the preparation. The second approach does not require diborane; however, to get reasonable yields of AB from the metathesis approach, it has been suggested that the synthesis must be performed under dilute solvent reaction conditions. The third approach, dissolving DADB in glyme containing a trace of diborane, makes one consider if it may be possible to make AB from DADB prepared in situ from decomposition of the NH4BH4. There does not appear to be any need to isolate DADB. The judicious observations reported by Parry, Schultz, and Shore in their series of eight sequential papers in J. Am. Chem. Soc. combined with the reports from Mayer's22,23 and Geanangels'33 research groups led us to consider alternative approaches to prepare AB for hydrogen storage studies. Some of the important observations are summarized below:
• NH4BH4 is stable at −40 °C and in liquid NH3.
• NH4BH4 in ether slurries, with a trace of NH3, decomposes to AB.
• NH4BH4 decomposes to DADB in the solid state.
• DADB in ether, with a trace of NH3, decomposes and gives low yields of AB.
• DADB in diglyme, with a trace of B2H6, gives high yields of AB.
The differences in reactivity of both NH4BH4 and DADB, depending on the reaction environment, were critical to the development of a synthetic strategy to make AB in practical yields. First, DADB decomposes with H2 loss in diethyl ether when there is a trace of NH3 present; however DADB converts AB in diglyme when there is a trace of diborane.23 Two very different pathways for DADB are possible depending on whether there is a trace of NH3 or a trace of B2H6 in the organic ether solvent. Second, the difference between the decomposition of NH4BH4 in the solid state compared to decomposition of NH4BH4 in solution, solid NH4BH4 decomposes to DADB while slurries of NH4BH4 decompose to AB. Two explanations were offered to explain the “role of solvent” and the difference between decomposition of NH4BH4 in the solid state and in ether slurries. Either the ether removes heat from exothermic reaction so there is not sufficient thermal energy to isomerize to DADB, eqn (8), or the ether removes AB from the reaction center so that it does not react with NH4BH4, eqn (9).12
| 2NH4BH4 → 2AB + 2H2 | (8a) |
| 2AB → DADB | (8b) |
| NH4BH4 → AB + H2 | (9a) |
| NH4BH4 + AB → DADB + H2 | (9b) |
Thus, by employing the appropriate reaction conditions, it should be possible to maximize the yield of AB from the metathesis approach. Specifically, reasonable yields of AB can be obtained in a multi-step, one-pot, synthesis that involves making NH4BH4 from MBH4 + NH4X in liquid ammonia, removing all the NH3, and letting the NH4BH4 decompose to DADB. However, the DADB need not be isolated, and when diglyme containing a trace of diborane is added to the solid DADB, it should provide the desired hydrogen storage compound AB at an 80% yield in a single pot. This is a substantial improvement compared to 45% yield when the metathesis is carried out directly in ether (with a trace of ammonia).
Thus we undertook a comparative synthesis study to determine the optimal reaction conditions to prepare AB in a “single pot”.
• Synthesis I—Sequential Reaction Sequence. The NH4BH4 is made in liquid ammonia, and then ammonia is removed before organic ether is added.
• Synthesis II—Parallel Reaction Sequence. The NH4BH4 is made in liquid ammonia in the presence of the organic ether.
Based on work reported in the literature, we expected that we could attain yields as high as 80% using the sequential reaction sequence and as low as 20% using the parallel reaction sequence.
In accordance with procedures reported in the literature, we prepared NH4BH4 from NaBH4 and NH4Cl in liquid ammonia at −78 °C. The reaction mixture was stirred for 1 h under a nitrogen atmosphere before subliming the ammonia solvent under vacuum. The vessel was kept at −78 °C in a slush bath while THF was slowly added by a cannula to the flask, thus resulting in significant gas evolution through the nitrogen bubbler. After stirring the slurry at −78 °C for 30 min, the flask was warmed to room temperature and stirred for an additional 60 min before the insoluble salts were filtered from the THF soluble products. The isolated yield of ammonia borane is nearly quantitative and the purity as measured by 11B NMR and X-ray diffraction34 is better than 99%. Melting point determinations showed decomposition at 110 °C. One critical purity test for hydrogen storage is the stability of the material at 60 °C. In previous work, we have shown that one measure of the relative stability of AB is the induction period prior to hydrogen release at 80 °C.35 We discovered that sources of high-purity AB (i.e., 99% pure as determined by 11B NMR) were sufficiently stable to meet DOE targets. However, sources of AB that are less than 95% pure had to be purified further to enhance stability. Similar isothermal differential scanning calorimetry experiments showed that the AB prepared by the methods described above was sufficiently pure to meet the stability requirements.
In another synthetic trial, THF was present in the liquid ammonia solution containing NaBH4 and NH4Cl with similar results; that is, a quantitative yield of AB was obtained, and the product was 99% pure as determined by 11B NMR when the insoluble salts were filtered and the solvents were removed by vacuum. This result was not expected because of the lower yields of AB formed in the metathesis reactions in organic solvents. Table 1 shows a comparison of results from this work with the results obtained from previously used experimental procedures.
| Entry | Molarity | Solvent | Yield(%) | |
|---|---|---|---|---|
| a Current work. b See reference 2. c See reference 19. d See reference 20. e See reference 33. f See reference 23. g 2 M LiBH4 solution in THF. h Trace amounts of NH3 and B2H6, respectively. | ||||
| 1a | NH4Cl + NaBH4 | 0.74 | NH3–THF | 99 |
| 2a | NH4Cl + NaBH4 | 1.9 | NH3–THF | 99 |
| 3a | NH4F + LiBH4g | 0.74 | NH3–THF | 99 |
| 4b | (NH4)2SO4 + NaBH4 | 0.46 | Et2O | 45 |
| 5c | NH4HCO2 + NaBH4 | 0.165 | THF | 95 |
| 6c | (NH4)2SO4 + NaBH4 | 0.165 | Et2O | 96 |
| 7c | NH4HCO2 + NaBH4 | 1.0 | Dioxane | 95 |
| 8d | NH3 + BH3:THF | 1.0 | THF | 50 |
| 9e | (NH4)2CO3 + NaBH4 | 0.40 | THF | 80 |
| 10b | DADB | 0.173 | Ether(NH3)h | 20 |
| 11f | DADB | 0.12 | Diglyme(B2H6)h | 80 |
The observation of quantitative yields of AB from the metathesis reaction in liquid ammonia solutions suggests the possibility of an alternative pathway to AB relative to the same metathesis reaction in organic solvents. As noted above, Parry and Shore provided two potential mechanisms to explain how NH4BH4 decomposes in ether slurries to yield AB and not DADB as observed in the neat solid.
The first step in both mechanisms is an acid–base reaction to form AB + H2. In the subsequent step, the organic solvent either (1) removes AB from the solid slurry before it could react with another molecule of NH4BH4 to make DADB or (2) it removes the heat from the local environment, thereby slowing isomerization of two AB molecules to DADB. A third explanation may be that DADB is formed but decomposes in the organic solvent to AB as reported by Mayer.23 This is what we expected in the reaction upon adding the THF to the liquid ammonia solution, but this reaction should yield no more than 20% AB given reports of significant decomposition of DADB in ether to yield polymeric products.2,12 Thus, we were surprised with the high yield of AB formed in the ammonia–THF solvent mixture. It appears that nature may have again provided a circuitous pathway to yield AB.1 Additional work is planned to improve our understanding of the decomposition reaction pathways of NH4BH4 in the solid state and in polar solvent mixtures such as ammonia–THF.
Conclusions
Ammonia borane (19 wt% H2), a promising hydrogen-storage material for fuel cell applications, can be prepared in near quantitative yields from NH4BH4. The transformation of NH4BH4 in a solvent containing liquid NH3 is essential to the high yield and purity of AB.Acknowledgements
This work was supported by the Office of Basic Energy Sciences of the Department of Energy, Chemical Sciences program. The Pacific Northwest National Laboratory is operated by Battelle for the US Department of Energy. TA wishes to thank Sheldon Shore for brilliant discussions on the history of AB and DADB.References
- R. W. Parry, J. Chem. Educ., 1997, 74 Search PubMed.
- S. Shore, PhD thesis, University of Michigan, 1956.
- A. B. Burg and H. I. Schlessinger, J. Am. Chem. Soc., 1937, 59, 780 CrossRef CAS; H. I. Schlesinger and A. B. Burg, J. Am. Chem. Soc., 1938, 60, 290–299 CrossRef CAS.
- A. Stock, in Hydrides of Boron and Silicon, Cornell University, Ithaca, NY, USA, 1933, pp. 58 Search PubMed.
- A. Stock and E. Pohland, Ber. Dtsch. Chem. Ges., 1925, 657 CAS.
- G. W. Rathjens and K. S. Pitzer, J. Am. Chem. Soc., 1949, 71, 2783 CrossRef CAS.
- H. I. Schlesinger and A. B. Burg, J. Am. Chem. Soc., 1938, 60, 290 CrossRef CAS.
- G. W. Schaefer, M. D. Adams and F. J. Koenig, J. Am. Chem. Soc., 1956, 78, 725 CrossRef CAS.
- D. R. Schultz, PhD thesis, University of Michigan, 1954.
- R. W. Parry and D. R. Schultz, J. Am. Chem. Soc., 1958, 80, 1 CrossRef CAS.
- D. R. Schultz and R. W. Parry, J. Am. Chem. Soc., 1958, 80, 4 CrossRef CAS.
- S. G. Shore and R. W. Parry, J. Am. Chem. Soc., 1958, 80, 8 CrossRef CAS.
- S. G. Shore and R. W. Parry, J. Am. Chem. Soc., 1958, 80, 12 CrossRef CAS.
- R. W. Parry and S. G. Shore, J. Am. Chem. Soc., 1958, 80, 15 CrossRef CAS.
- S. G. Shore, P. R. Girardot and R. W. Parry, J. Am. Chem. Soc., 1958, 80, 20 CrossRef CAS.
- R. W. Parry, G. Kodama and D. R. Schultz, J. Am. Chem. Soc., 1958, 80, 24 CrossRef CAS.
- S. G. Shore and R. W. Parry, J. Am. Chem. Soc., 1955, 77, 6084 CrossRef CAS.
- (a) M. E. Bowden, G. J. Gainsford and W. T. Robinson, Aust. J. Chem., 2007, 60, 149 CrossRef CAS; (b) J. B. Yang, J. Lamsal, Q. Cai, W. J. James and W. B. Yelon, Appl. Phys. Lett., 2008, 92 Search PubMed , 091916; (c) P. A. Storozhenko, R. A. Svitsyn, V. A. Ketsko, A. K. Buryak and A. V. Ul'yanov, Russ. J. Inorg. Chem., 2005, 50, 980; (d) G. H. Penner, Y. C. P. Chang and J. Hutzal, Inorg. Chem., 1999, 38, 2868 CrossRef CAS; (e) G. Wolf, J. C. van Miltenburgb and U. Wolf, Thermochim. Acta, 1998, 317, 111 CrossRef CAS.
- P. V. Ramachandran and P. D. Gagare, Inorg. Chem., 2007, 9, 1831.
- S. G. Shore and K. W. Boddeker, Inorg. Chem., 1964, 3, 914 CrossRef CAS.
- For a review of symmetric and asymmetric cleavage reactions of diborane see R. W. Parry, J. Organomet. Chem., 2000, 614, 5 Search PubMed.
- E. Mayer, Inorg. Chem., 1972, 11, 866 CrossRef CAS.
- E. Mayer, Inorg. Chem., 1973, 12, 1954 CrossRef CAS.
- Interestingly they report the yield of AB was much lower in monoglyme (22–37% 60 h at 25 °C).
- Our group is designing an apparatus to prepare AB on a 100 g scale. The results will be reported in a subsequent paper.
- G. Wolf, J. Baumann, F. Baitalow and F. P. Hoffmann, Thermochim. Acta, 2000, 343, 19 CrossRef CAS.
- T. B. Marder, Angew. Chem., Int. Ed., 2007, 46, 8116 CrossRef CAS.
- M. E. Bluhm, M. G. Bradley, R. B. Butterick, III, U. Kusari and L. G. Sneddon, J. Am. Chem. Soc., 2006, 128, 7748 CrossRef CAS.
- A. C. Stowe, W. J. Shaw, J. C. Linehan, B. Schmid and T. Autrey, Phys. Chem. Chem. Phys., 2007, 9, 1831 RSC.
- A hydrogen storage system is all the components required to get the hydrogen from the material, e.g., AB, to the fuel cell. For example, the tank, heat exchangers, valves, tubing, pumps, etc., must be included in the system weight.
- System target goals have been developed through the FreedomCAR Partnership between DOE and the US Council for Automotive Research (USCAR), http://www.uscar.org/.
- in “National Research Council and National Academy of Engineering, committee on Alternatives and Strategies for Future Hydrogen Production and Use”, The Hydrogen Economy: Opportunities, Costs, Barriers and R&D Needs, Ed. G. C. M. Dresselhaus and M. Buchanan, National Academic Press, Washington, DC, USA, 2004 Search PubMed.
- M. G. Hu, J. M. van Paasschen and R. Geanangel, J. Inorg. Nucl. Chem., 1977, 39, 2147–2150 CrossRef CAS.
- E. W. Hughes, J. Am. Chem. Soc., 1956, 78, 502 CrossRef CAS; S. G. Shore and R. W. Parry, J. Am. Chem. Soc., 1955, 77, 6084 CrossRef CAS.
- S. D. Rassat, R. S. Smith, T. Autrey, C. L. Aardahl, A. A. Chin, J. W. Magee, G. R. VanSciver and F. J. Lipiecki., Prepr. Symp.–Am. Chem. Soc., Div. Fuel Chem., 2006, 51(2), 517 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: ammonia borane synthesis. See DOI: 10.1039/b808865a |
| This journal is © The Royal Society of Chemistry 2008 |