Size effects and nanostructured materials for energy applications
Palani
Balaya
*
Department of Mechanical Engineering and Engineering Science Programme, National University of Singapore, Singapore 117574. E-mail: mpepb@nus.edu.sg
First published on 15th October 2008
Abstract
Size reduction in nanocrystals leads to a variety of exciting phenomena due to enhanced surface-to-volume ratio and reduced transport length for the mobile charges. We consider some of these anomalous phenomena restricting our discussions to the effects of nano-size on energetics and transport behaviour, with a few examples to illustrate materials challenges for efficient energy conversion and storage. We show that nano-size affects the thermodynamical aspects due to excess surface contributions giving rise to enhanced cell voltage in lithium batteries. An anomalous electrical conduction occurs at nano-size, its relevance is briefly highlighted in the context of energy conversion using fuel cells and excitonic solar cells. The benefit of narrowly spaced interfaces also results in rapid energy storage due to the reduction of the effective diffusion path. Thus, in the context of storage behaviour, nanocrystalline electrodes exhibit high capacity as well as Coulombic efficiency and in some cases high rate performance. An interfacial lithium storage mechanism which is a consequence of nanocrystallinity has been discussed to explain the extra storage when metal is brought in contact with Li2O/LiF at nanosize. Superior storage performance by supercapacitors and efficient waste heat conversion by thermoelectric devices using nanostructured materials are also briefly highlighted. This review thus emphasises fundamental understanding and novel concepts at nano-size for the development of excellent materials which achieve efficient energy conversion and storage, both of which are vital in facing the challenges posed by global warming and the predicted future limited energy resources.
 Palani Balaya Palani Balaya | Dr Palani received a PhD in Solid State Ionics from the University of Hyderabad, India. He has worked at the Indian Institute of Science, Bangalore, the Bhabha Atomic Research Centre, Mumbai, Madras University, Chennai and the Max Planck Institute for Solid State Research, Germany. His research interests include anomalous transport phenomenon at nano-size (mesoscopic phenomenon), energetics of nano-crystals and interfacial lithium storage. Dr. Palani joined the Faculty of Engineering, NUS in January 2007. His current research area is nano-ionics for energy systems specifically in the context of solar energy conversion as well as lithium batteries for energy storage. |
Broader contextApplied energy research has become currently a matter of priority due to increasing demand for energy, rising oil prices, uncertain energy supplies and the fear of global warming. Increased use of renewable energy is recognized to be both an economically and an environmentally sound alternative. However, the performance of renewable energy generation and storage devices under ambient conditions are seriously limited by kinetic issues. Research into novel nanocrystalline materials and new technological solutions will contribute to the development of such energy systems. In nanocrystals, one of ten atoms or even more sit at surfaces/interfaces. Corrrespondingly, the surface or interfacial contributions to overall physical and chemical behaviour are drastically different. For example, the melting point of nano-sized metals are drastically reduced and are attributed to the significant surface contribution to the overall free energies. Recently, various such anomalous phenomena have been observed in energetics and transport behaviour of nanocrystals. The purpose of this review is to highlight these anomalies and discuss their impact in device applications such as fuel cells, solar cells, lithium batteries and supercapacitors. A few advantages of thermoelectric materials at nano-size for energy conversion are also addressed. |
1. Introduction
Significant efforts have been made currently in energy research due to the increasing demand for energy, rising oil prices, uncertain energy supplies and the fear of global warming. Increased use of renewable energy is recognized to be both economically as well as environmentally a sound alternative. Research into novel materials and new technological solutions will contribute to the development of renewable energy sources and storage systems such as solar cells, fuel cells, lithium-ion batteries and supercapacitors. Each of these energy conversion and storage processes has certain limited efficiencies, cost factors and environmental effects. While much of the research underway aims to improve the above, fundamental science and engineering of materials for sustainable energy are important to solve the challenges facing us at the moment.1,2Nanostructured materials in this context have triggered great excitement, due to both fundamental interest as well as potential technological applications especially in energy research.3–5 Size reduction in nanocrystals leads to a variety of exciting phenomena due to enhanced surface-to-volume ratio and reduced scale of transport lengths for both mass and charge transport. While electronic materials have been widely investigated at nano-size,3,4 this is less so in the case of ionic as well as mixed (ionic and electronic) conducting materials, although these have a significant role in a variety of energy applications such as batteries, fuel cells, excitonic solar cells, and supercapacitors. Though classical in nature, size effects on ionic and mixed conducting materials are by no means less striking.5–8 In this review, we emphasize these size effects in materials relevant for energy research and present a few anomalies observed recently in thermodynamic and transport properties of nanostructured materials and their relevance for efficient energy conversion and storage. We also briefly comment on how nanostructuring affects the thermoelectric behaviour of materials relevant for generating electrical energy from waste heat.
2. Energetics of nanocrystalline materials
An often cited example of size effect is the change of thermodynamic stability, such as the suppression of the melting point in the case of nano-sized Au particles by several hundreds of degrees.9 Such anomalies in energetics are due to excess surface free energy of small particles MX (where M is metal and X refers to O, F, S etc.,) according to:µMX(nano) = µ∞MX + (2![[small gamma, Greek, macron]](https://www.rsc.org/images/entities/i_char_e0c4.gif) / /![[r with combining macron]](https://www.rsc.org/images/entities/i_char_0072_0304.gif) )v )v | (1) |
is the average surface tension, is the average radius, and v is the molar volume. Yet another example is the reversal of thermodynamic state from meta-stable to stable or vice versa upon size reduction.10,11 For example, metastable phases such as cubic ZrO2,12 the anatase form of TiO2,10,13 γ-Al2O311 and γ-Fe2O3 (maghemite)14 have been stabilized at nano-size. In the literature, little experimental data is available on oxide surface energies. In particular, very few experiments have been carried out to estimate the excess surface contribution in nanoparticles. Among these, the excess surface enthalpy measured using calorimetry experiments on nanoparticles of alumina and titania by Navrotsky et al. is worth mentioning.10,11 Electrochemical emf measurements are also known to provide thermodynamic data, enthalpy and entropy, if measured as a function of temperature.15–18 Let us look at the energetics of TiO2, being an important material for energy applications, such as the photoanode in dye-sensitized solar cells and an anode material for rechargeable lithium-ion batteries. Recently, we measured the excess surface contribution for rutile and anatase nanoparticles by an electrochemical emf cell using Na-β′′ alumina as the solid electrolyte in the temperature range 520–720 K.
Both the working and reference electrodes of the electrochemical cell consist of a composite of Na2Ti6O13, gold powder and rutile, or anatase separated by Na-ß′′ alumina.18 While the reference electrode contains bulk rutile of 2 µm, the working electrode comprises nanoparticles of either rutile or anatase. Let us briefly comment here on surface contribution to the thermodynamics at nanosize leading to an excess cell potential for the case of 25 nm rutile, and excess Gibbs free energy of formation for different sizes of anatase particles 100 nm, 15 nm and 5 nm.
A stable excess emf of about 62 mV was obtained at 621 K as shown in Fig. 1 for the nano-sized rutile of 25 nm versus 2 µm bulk rutile.19 To a first approximation this emf is related to the Gibbs free energy of coarsening through the Nernst equation by:
| (2/)v = −nFE | (2) |
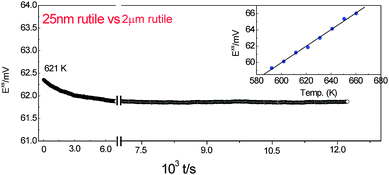 | ||
| Fig. 1 Typical results of emf measurements for a TiO2 binary system comprising of two elements (here titanium and oxygen) at 621 K. After a short transient drift occurring on a temperature change, the excess emf Exs is very stable (∼ 62 mV). The graph top right shows the temperature dependence of emf for 25 nm rutile in the reversible T-range.19 | ||
The advantage of the present electrochemical cell is that emf which is measured directly provides the difference of Gibbs free energies of formation (ΔfG°) of the titania crystals on both sides. It is seen clearly that this stable excess emf is, in the case of a binary system such as the rutile (TiO2) nanoparticle, a consequence of the relatively less mobile Ti4+ and O2− ions with a low self-diffusion coefficient, unlike single component silver nanoparticles that grow with time leading to unstable excess emf values due to an electrochemical Ostwald ripening (because of the high diffusivity of Ag+) in the Ag(nano)/RbAg4I5/Ag (bulk) electrochemical cell.17
On the other hand, Table I presents the Gibbs free energy of formation of anatase particles of 1200 nm, 100 nm, 15 nm and 5 nm at selected temperatures. The higher energetics of nanoparticles are clearly due to the excess surface contribution of anatase.19
| Anatase/nm | Gibbs free energy of formation (ΔfG°)/ kJ mol−1 at selected temperatures | ||
|---|---|---|---|
| T = 572 K | T = 621 K | T = 678 K | |
| 1200 | −837.82 | −828.86 | −819.82 |
| 100 | −829.65 | −819.62 | −810.15 |
| 15 | −816.64 | −807.35 | −798.16 |
| 5 | −789.13 | −780.63 | −771.60 |
Eqn 1 also implicitly or explicitly influences the voltage of various electrochemical cells containing nano-crystalline constituents.17,20,21 Assuming, typical values for and v as 0.1 Jm−2 and 25 cm3 mol−1, the surface contribution for particle sizes of 1 nm is reported to be 10 kJ mol−1 (∼100 mV), that is the typical excess cell voltage of 100 mV that one would expect for the smallest size 1 nm particles.20 Recently, it has been shown that upon further reducing the particle size and making the structure highly disordered, such as with amorphous RuO2 prepared electrochemically or by thin-film deposition, the cell potential for Li insertion can be enhanced by 580 mV compared to bulk RuO2.22
3. Transport anomalies in nanostructured materials
In nanocrystalline materials, owing to a largely enhanced surface area to volume ratio, the overall electrical response may be dominated by the boundary contributions.23,24 Especially when dealing with dense thin films or nanoceramics, such as solid electrolytes in fuel cells or anodes in excitonic solar cells, interfaces play a key role in the transport behaviour due to the formation of space charge layers adjacent to the grain boundaries. The space charges at interfaces might refer to an accumulation or a depletion situation depending on the defect chemistry of the materials investigated. If the spacing of the interfaces (film thickness or the grain size) is perceptibly less than the effective width of the space charge layers, these layers overlap and the sample is charged throughout.6Epitaxial ionic heterolayers of CaF2/BaF2 prepared by molecular beam epitaxy by Sata et al.,25 have shown such an overlap of accumulated space charges when the layer thickness approaches few nanometers (<50 nm) resulting in anomalous ionic transport behaviour. This mesoscopic situation (defined based on the comparable length scales involved – screening length is comparable with the thickness of layers) in which all the electro-neutral bulk has disappeared and the boundary layers overlap – results in an artificial fast ionic conductor.
Recently Guo et al., reported enhanced Ag+ ionic conductivity in polytype AgI-nanoplates by about 4 orders of magnitude compared to that of the macroscopically stable β-AgI phase in the wurtzite structure. This drastic effect is attributed to the size effect where macroscopically unstable 7H and 9R stacking fault arrangements are stabilized due to nano-size and those stacking faults are considered to have hetero-layers composed of β-AgI/γ-AgI whose individual layer thickness is lower than the Debye screening length.26
Acceptor doped CeO2, ZrO2 and SrTiO3 ceramics are well investigated materials for the counterpart, namely depletion situation, where the grain boundaries have been found to be positively charged and accompanied by space charge zones in which electron holes and oxygen vacancies are depleted.27,28 Detailed impedance analysis of dense nanocrystalline SrTiO3 ceramics (80 nm) in comparison to macrocrystalline sample (2.5µm) gives direct and unambiguous evidence of space charge overlap as a characteristic size effect.29,30 While two relaxation times are clearly identified for the macrocrystalline SrTiO3 (2.5µm), in nanocrystalline SrTiO3 only a single relaxation time referring to the low-frequency signal remains. Thus, unlike microcrystalline SrTiO3 which exhibits both bulk and semi-infinite interfacial contributions to conduction, in nanocrystalline SrTiO3 the bulk contribution disappears and space charge effects are observed throughout. The Debye length is deduced to be larger than the grain size, thus confirming the appearance of a mesoscopic phenomenon i.e. that the depleted space charges overlap within the grains.30 This phenomenon is the counterpart of the accumulation layer overlap discussed above,25 but occurs at much larger spacing owing to the larger dielectric constant. Even a change of the conduction mechanism from ionic to electronic has been observed in dense nanocrystalline CeO231 as a consequence of size effect.
3.1 Fuel cells
The enhanced mesoscopic ionic conduction in nanostructured materials discussed earlier25 is relevant in the context of fuel cells application. A fuel cell is an electrochemical conversion device, which produces electricity from various external quantities of fuel (on the anode side) and an oxidant (on the cathode side). These react in the presence of an electrolyte as a separator. A solid oxide fuel cell (SOFC), as the name implies, has a solid ceramic oxide, electrolyte and is intended mainly for stationary applications with an output from 100 W to 2 MW. Presently the SOFC is operated at 900 °C using Y2O3-doped ZrO2 as the solid electrolyte. In the context of SOFC, an increase in electrolyte conductivity may lead to a decrease in the overall operating temperature and opens up new applications for energy conversion. Recently, Ginestra et al.,32 synthesized 30 nm thin dense film of 2.9% Y2O3-doped ZrO2 by atomic layer deposition technique, which exhibits ionic conductivity near 800 °C about 1 order of magnitude higher than values reported for bulk polycrystals with 3% yttria content. Yet another attractive candidate for application as a solid electrolyte is an acceptor doped cerium oxide, due to enhanced ionic conductivity compared to that of zirconia at a given doping concentration and temperature. Merl et al.,33 compared the conductivity of more disordered (less-oriented) CeO2 films with that of well-oriented films, and reported that disordered films exhibit an increased conductivity of about 2–3 orders of magnitude. The enhanced ionic conduction in these thin films is argued to be due to size reduction and the associated effects of grain boundaries and interfaces, that is Dinterface≫Dbulk the reason for this anomalous behaviour is a subject of further investigation.3.2 Solar cells
Solar energy conversion represents an important area of energy research and has a benign effect on the environment and climate, making it an appealing alternative energy source. The solar cell families include thin films, amorphous, polycrystalline and organic materials, each favored due to its own advantages either in cost or in the efficiency of conversion. Although photovoltaics is one of the most versatile means of converting solar energy readily to electricity, the challenge is to raise its conversion efficiency by factors of five or ten so it can compete in cost with other forms of energy from various biofuels and wind. This requires an understanding of the fundamentals of solar conversion phenomena at the nanoscale.Dye-sensitized solar cells (DSCs) are low-cost alternatives to conventional solid-state junction devices, usually made from crystalline or amorphous silicon. In DSCs, a nanoporous inorganic semi-conductor such as near-UV absorbing TiO2 or ZnO, is used to collect the photo-excited electrons.34–38 The visible-light absorption is performed by a monolayer of dye molecules adsorbed on the surface of these inorganic conductors. The pores are filled with either a liquid or a polymer which acts as hole transporting medium (HTM) as shown in Fig. 2. In the process of light harvesting, the interfaces of inorganic/dye, inorganic/HTM and that of dye/HTM play a crucial role in injecting the electrons and holes to the respective charge collectors viz., inorganic and HTM, respectively. While a Grätzel cell has shown an efficiency of 11.4%, these solar cells currently exhibit only moderate conversion efficiency mainly because of charge recombination due to back reactions at the inorganic/HTM interfaces. An ultra thin layer of insulating Al2O3 or ZrO2 is deposited on the surface of the inorganic compounds as shown in Fig. 3 to slow down such back reactions and hence the interfacial charge recombination kinetics.39 While this approach does not show clear benefits on TiO2 photoanodes with liquid electrolytes, it has improved the performance of other photoanodes such as SnO2.40,41 It is worth mentioning that clear benefits have typically been observed in fully-solid-state solar cells, in which the interfacial charge recombination kinetics seem to limit the performance significantly, and requires further investigation for optimization.
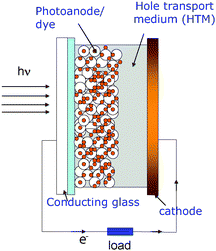 | ||
| Fig. 2 Schematic representation of a dye-sensitized solar cell. White circles refer to a near-UV absorbing photoanode and red filled circles refer to monolayer adsorpted dye molecules for visible light absorption. | ||
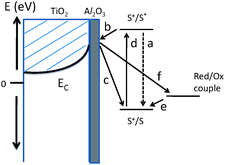 | ||
| Fig. 3 Schematic representation of electron transfer processes in a dye-sensitized solar cell with a blocking-layer on the photoelectrode which facilitates tunneling of photoelectrons to inject into the conduction band of photoanode, but acts as a barrier for the back reaction. Arrows show photogeneration of the dye-excited state (d), electron injection into the conduction band of nanoporous TiO2 (b), regeneration of the dye ground state by electron transfer from the redox couple (liquid electrolyte/conducting polymer) (e), and the wasteful charge recombination pathways of injected electron recombination oxidized dye molecules (c) and with oxidized redox couple (f). Also shown is the dye-excited state decay to ground (a), which competes with electron injection.39 | ||
Concepts related to fully depleted mesoscopic electrical conduction30 cited above in the case of nanocrystalline SrTiO3, find application in improving the efficiency of the dye-sensitized solar cells. Depleted space charge layers at the boundaries of inorganic/hole transporting medium – especially in the all solid-state solar cells – provide a local electrical potential barrier that could effectively be used as a screening medium for this purpose in the place of ultrathin insulators such as alumina, which is used in Fig. 3 to avoid the back reactions causing charge recombination. The space charge potential, or the electrical barrier is found to be sensitive to the size of the nanocrystals. For example, in the case of SrTiO3, the barrier potential is found to be 0.7 eV for 2.5 µm, 0.4 eV for 200 nm and 0.2 eV for 80 nm crystals.30 Thus it can be proven that the space charge potential and the depletion layer width can be well tuned by controlling the size of the particles as well as the dopant concentration that decides the depletion barrier width and depth at the surfaces of nanoparticles. It is thus proposed that the depleted space charges could be used as a local electrical barrier to help avoid back reactions and thereby the charge recombination in dye-sensitized or all solid-state solar cells, hence enhancing their efficiency.
During the transport of electrons within the photoanode, Nakade et al.,42 reported that the diffusion coefficient (D) of electrons increases with the increase of the particle size up to 32 nm and interpreted this due to the decrease of effective surface area, where the trap sites are likely to exist. Nanofibers, where the grains are well organized, offer a higher probability for 1-D conduction for channelized transport of electrons.43 Such a favoured conduction path along with a well-defined electrical potential barrier at the contact of inorganic/HTM discussed above improves the charge collection, suppressing the interfacial charge recombination, and thereby may enhance the efficiency of the solar cells. Charge collection can further be improved by decreasing the boundary impedances at the contacts of grains in nanofibers, by choosing single crystalline nanorods of photoanodes such as ZnO discussed in ref. 44. The challenge with these nanofibers and nanorods is to align them vertically on the transparent conducting oxide substrates for optimal charge collection. Novel synthesis methods are essential to overcome such structural obstacles and to improve the efficiency of dye-sensitized or hybrid solar cells.
Size effects resulting in quantum confinement are well documented in quantum dots of narrow band-gap semiconductors. For example, PbS with its large Bohr radius (20 nm), high dielectric constant (17.3), and narrow band gap of 0.41 eV ensures strong quantum confinements. Most importantly, the band-gap of nano-sized PbS can be tuned to any desired value between 0.41 and 5 eV. In particular, this excellent band-gap tailoring capability can make quantum dots of such narrow band-gap semiconductors highly relevant for solar photovoltaic conversion which demands absorption between 1.4 and 2 eV.45
Yet another effect due to the nano-size is the multiple exciton generation (MEG), that has created much interest recently for efficient generation of multiple excitons per absorbed photon of energy greater than twice the band gap. Evidence of the formation of up to eight excitons from a single photon has been found in quantum dots of direct band gap semiconductors, i.e., PbSe, PbS, PbTe, CdSe, and InAs. Recently, MEG within colloidal Si nanocrystals was reported.46 A challenging issue at the moment is to separate these excitons efficiently and to collect the electrons and holes without significant losses. Such a solar cell in an ideal condition is expected to have an efficiency exceeding the Schockley and Queisser limit.47
The benefit of narrowly spaced interfaces that act as fast pathways for ions and/or electronic charges (holes/electrons) lies not only in the enhanced or reduced effective conductivity influencing the energy conversion as discussed above. Such nano-size also results in the possibility of enhanced energy storage resulting from the reduction of the effective diffusion length. Let us now discuss a few anomalous phenomena related to nanocrystallinity that are relevant to energy storage, specifically in lithium batteries.5,20
4. Anomalous storage behavior in lithium batteries
High-power lithium-ion batteries, the most successful electrochemical devices, offer the promise of higher efficiency, longer life, and easier state-of-charge control at lower weight, volume, and cost. In the classical “lithium-ion battery”, Li+ is transferred from a LixCoO2 (0.5 < x < 1.0) electrode into a graphite electrode during charge while the reverse process occurs via discharging. Thus the Li+ ions which shuttle between these two electrodes during charge–discharge cycling are facilitated by the layer-type crystal structure of the electrodes. The high reversibility of the electrochemical process is caused by the soft insertion/extraction of Li+ ions in these host lattices (the rocking chair concept of lithium intercalation). This lithium-ion battery which is now used commercially has many advantages, high open circuit voltage (∼4 V), excellent cyclic performance (more than 3000 cycles) and high coulombic efficiency (95%), but limited lithium storage capacity. Only 0.5 Li+ could be removed from LiCoO2 resulting in Li0.5CoO2 (140 mAh g−1 using a half cell of LiCoO2 versus Li metal) and 1Li+ could be stored in graphite in the form of LiC6 (370 mAh g−1 using a half cell of graphitevs.Li metal). Since most of today's high-performance portable microelectronic devices demand high energy density, there is great interest in increasing the storage capacity of both the cathode as well as anode electrodes using nanostructured materials operating at high and low potentials, respectively.484.1 Enhanced Li storage at high potential
The proposal of phospo–olivine based materials LiMPO4 (M = Fe, Mn, Ni) by Padhi et al.,49 as an alternative candidate for LiCoO2 has triggered great interest to explore environmentally benign and low-cost cathode materials. Let us limit our discussion to olivine iron phosphate as the cathode material. Despite many other advantages (ease of synthesis, less reactivity towards electrolytes hence highly flat potential during charge/discharge processes50–56), the main disadvantage of LiFePO4 is that the storage capacity (1Li+ which is equivalent to 170 mAh g−1) reduces at high current operation. This is mainly due to the sluggish transport of electrons and Li+ ions in poorly conducting LiFePO4.57–61Recent ab-initio60 calculations and atomistic simulations62 on LiFePO4 have shown that Li+ ions move preferably along the b-axis rather than along a- and c- axes in this crystal with the orthorhombic space groupPnma. Anisotropic measurement of electronic and ionic conduction in LiFePO4 single crystals by Amin et al.57 further claims that Li+ conductivity is essentially two dimensional, nearly four orders of magnitude lower than the electronic conductivity along the b- and c-axes and many orders of magnitude lower along a-axis. Thus, in order to achieve high rate performance it is mandatory to reduce the crystallite size either along the b- or c-axis or if possible along both axes, for rapid insertion/extraction of Li+ and e− simultaneously. Such a criteria of having a morphology with thin b- or c-axis of the LiFePO4 crystallites (rapid extraction of Li+ ions and electrons from the bulk) with exterior decoration using conductive materials such as carbon63,64 or RuO256 (enhancing the electronic wiring among neighboring particles) could result in high rate performances. Saravananet al.,65 have succeeded in synthesizing LiFePO4 nanoplates with a thin b-axis (30–40 nm) and uniform surface carbon coating (2–5 nm) and have demonstrated a storage capacity of 50 mAh g−1 at a rate of 30 °C (here 1 °C refers to drawing a current of 170 mA g−1 in 1 h).
The above example illustrates some of the advantages of nanostructured materials for energy storage using an insertion reaction. Such an investigation suggests that it is possible to store energy in insulating materials i.e. less electronically as well as ionically conducting materials such as LiFePO4, even at high current density, by controlling the morphology of the nano-sized electrode materials within a uniform surface coating of conductive carbon. Several next generation cathode materials such as LiMnPO4 and Li2FeSiO4 are currently being investigated using nanostructured morphologies to improve their energy density as well as power density.
4.2 Enhanced Li storage at low potential
A far reaching reversible Li storage has been achieved recently by Poizot et al.,67 in several transition metal oxides (CoO, NiO, FeOetc.,) by heterogeneous reaction resulting in a reduction to the respective metal. Such a heterogeneous reaction for CoO, could uptake 2Li+ leading to the formation of Co/Li2O composites in which Co metal grains of 2–5 nm are embedded in an amorphous Li2O matrix. This conversion reaction favors a storage capacity in the range 800–1200 mAh g−1 with reasonably good cyclic performances. The process of Li incorporation/ex-corporation, is reversible in a voltage range of 0–3 V with about 75% coulombic efficiency in its first cycle and nearly 100% coulombic efficiency in the following cycles. The reason for the reversibility of Co/Li2O to CoO upon removal of Li, is believed to be related to the formation of extremely small grain sizes of Li2O and Co metals (see Fig. 4).68,69
| CoO + 2Li+ + 2e− ⇆ Co + Li2O | (3) |
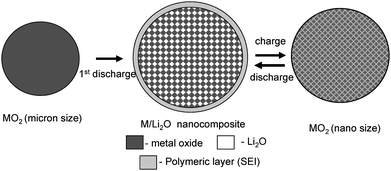 | ||
| Fig. 4 Schematic representation of the conversion reaction for Li storage using a MO2/Li cell where M is a transition metal. | ||
Apart from oxides, this heterogeneous conversion reaction has also been reported in fluorides, such as TiF3, VF3, FeF2, etc.70–72 as well as in oxyfluorides.73 Although enhanced Li storage capacity (in the range 800–1200 mA h g−1) has been achieved through a conversion reaction in the above cited materials, in view of practical applications, the performance is not very satisfactory, as the coulombic efficiency (reversibility) at first cycle is less than 75% for most of the materials, i.e. 25% of the Li is lost for ever.
In order to get more insight into the cause of such poor coulombic efficiency at first charge cycle, a systematic experiment has been performed on NiO and NiF2 using two different electrolytes EC![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :DEC and EC:DEC:DMC (EC: ethylene carbonate, DEC: diethyl carbonate; DMC: dimethyl carbonate) over the voltage window 0.02–3.0 V.74 Irrespective of the electrolytes used, NiO could be fully converted to Ni and Li2O nanocomposites (5–10 nm size) upon incorporation of 2Li+ at a rate of C/20, while NiF2 exhibits poor storage performance for Li incorporation. Only about 30% of NiF2 is found to be converted based on the comparison of capacity at the end of plateau at 1.5 V (no polymeric passivation layer formed at this potential during the conversion reaction), the remaining 70% of NiF2 is not involved in the conversion reaction. Given that the initial particle size is nearly the same, limited incorporation into NiF2 is in good agreement with its highly insulating character when compared to NiO. On the other hand, only about 23% of Li+ ions could be extracted from the Ni/LiF nanocomposite compared to Ni/Li2O nanocompoistes where Li extraction was found to be about 57% using EC:DEC:DMC electrolytes.
:DEC and EC:DEC:DMC (EC: ethylene carbonate, DEC: diethyl carbonate; DMC: dimethyl carbonate) over the voltage window 0.02–3.0 V.74 Irrespective of the electrolytes used, NiO could be fully converted to Ni and Li2O nanocomposites (5–10 nm size) upon incorporation of 2Li+ at a rate of C/20, while NiF2 exhibits poor storage performance for Li incorporation. Only about 30% of NiF2 is found to be converted based on the comparison of capacity at the end of plateau at 1.5 V (no polymeric passivation layer formed at this potential during the conversion reaction), the remaining 70% of NiF2 is not involved in the conversion reaction. Given that the initial particle size is nearly the same, limited incorporation into NiF2 is in good agreement with its highly insulating character when compared to NiO. On the other hand, only about 23% of Li+ ions could be extracted from the Ni/LiF nanocomposite compared to Ni/Li2O nanocompoistes where Li extraction was found to be about 57% using EC:DEC:DMC electrolytes.
Fortunately it was found that this heterogeneous conversion reaction is not necessarily connected to low coulombic efficiency. Such an exception was recently found by Balaya et al.,75 in RuO2, where 98% coulombic efficiency was achieved during first cycle with a reversible capacity of 1110 mAh g−1 corresponding to storage of 5.5 Li+ (in the voltage window of 0.02–4.3 V) as shown in Fig. 5. However, it may be noted that a RuO2/Li half cell could be reversibly operated only for about 4 cycles, beyond which the cell fails due to a large volume expansion (∼100%) during the conversion reaction. Despite this disadvantage and the high cost factor, RuO2 is still considered to be a model material for understanding the physical and chemical reasons for achieving this uniquely favorable combination of high capacity and high coulombic efficiency.
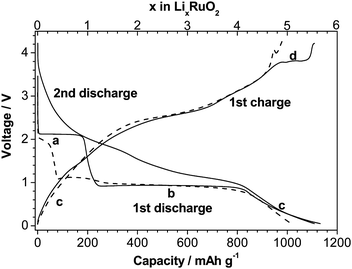 | ||
| Fig. 5 Electrochemical discharge–charge curves for the RuO2/Li cell. Solid lines refer to 100 nm grain size, while the dashed line refers to 10 µm RuO2. Note that the grain size of RuO2 during second discharge (solid line) is ∼5 nm.75 | ||
Based on Raman spectroscopy and high resolution transmission spectroscopy investigations, it is observed that incorporation of 5.6 Li+ (during discharging) transforms the initial RuO2 crystal into a nanocrystalline composite of Ru and Li2O with a grain size of 2–5 nm covered by a 5–10 nm surface solid/electrolyte interphase (SEI) layer. At full Li extraction (5.5 Li+), amorphous RuO2 forms (2–5 nm) again and the SEI layer disappears completely (see Fig. 6)
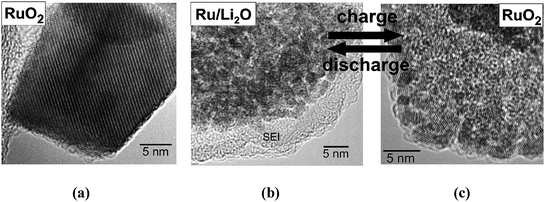 | ||
| Fig. 6 HRTEM images of RuO2electrodes: (a) initial, (b) fully discharged to 0.05 V – uptake of 5.6 Li and (c) charged to 4.3 V – extraction of 5.5 Li.5 | ||
In short, extraction of Li+ and e− from the M/LimX (where M – transition metal ions; X = O or F and m = 1 or 2), is fully achieved only in Ru/Li2O in the first cycle, while in other materials only 70–80% of Li+ can be extracted. It is worth analyzing the cause of this completely reversible reaction of Ru/Li2O into RuO2 in terms of microstructure and transport behavior. It is seen that nano-sized amorphous RuO2 is formed at the interface of Ru and Li2O upon the removal of Li+ and e− from the Ru/Li2O nanocomposites. Unlike NiO and NiF2, rutile RuO2 is an excellant electronic conductor (5 × 104 Scm−1)76 as well as a good mixed conductor for both Li+ and O2− ions. Thus it is believed that short transport length of 2–5 nm of Ru and Li2O grains along with the favorable inherent transport properties (electronic and ionic conduction) are responsible for fully reversible Li storage in RuO2.75
A recent striking observation in M/Li2O nanocomposites (where M is a transition metal such as Co, Cu, Fe, Ni, Mo, etc., that do not alloy with Li) investigated for rechargeable Li batteries, is the occurrence of an extra Li storage at low potential.68,77–79 To make this point clear let us recall the storage behavior of the RuO2electrode material. As mentioned earlier, incorporation of 4Li per RuO2 formula unit leads to the formation of a Ru/Li2O nanocomposite with crystallite sizes of 2–5 nm.73 Further incorporation (up to 5.6Li per RuO2) results in a sloped behavior. On charging, if the voltage is limited between 0.02–1.2 V (as the slope ends at 1.2 V), a reversible Li-storage capacity of 120 mAh g−1 is observed at a slow rate (i.e. discharge in 45 min.) with capacitive behavior.78 A storage capacity of 70 mA h g−1 was achieved at a fast rate (i.e. discharge in 1.3 min.) within this voltage window. This phenomenon is common to most of the nanocomposites M/Li2O and M/LiF investigated. Similar behavior observed in Co/Li2O nanocomposite at low potential has been tentatively explained by a reaction of Li with the conducting-type polymer film formed in situ.77 However, in the case of Ru/Li2O, Ni/Li2O and Ni/LiF nanocomposites, it is clear from the high resolution transmission electron microscope images74,75 that the passivation layer mainly decomposes on charging beyond the sloped region (1.2 V). In addition, similar extra storage has been reported recently by Liao et al.,80 in Fe/LiF composite films operated at a high voltage window, which does not permit formation of SEI layers. Yet another explanation given in the literature81 is the segregation of metal at the grain boundaries/interface and subsequent alloy reaction of metal with the incorporated Li. This possibility is, however, unlikely in the case of the Ru/Li2O nanocomposite, as no alloy reaction has been known between Ru and Li metals. These observations and the fact that the storage can occur even at a fast rate suggest that this extra storage could be due to a process that is different from the formation–decomposition of SEI and other mechanisms such as homogeneous insertion and heterogeneous conversion reactions.
In Ref. 20, a heterogeneous interfacial charge storage mechanism was proposed to explain the origin of this anomaly. As shown in Fig. 7 in this model, Li+ ions are stored on the oxide side of the interface while electrons (e−) are localized on the metallic side resulting in double layer storage. If the spacing of the interface is of the order of the screening length the difference between a capacitor and a battery is blurred, explaining fast rate performance at this low potential.
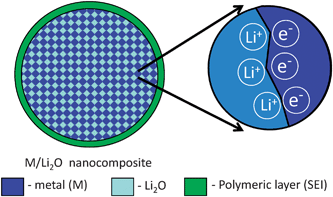 | ||
| Fig. 7 Schematic representation of the interfacial storage within M/Li2O nanocomposite. | ||
In order to check these ideas, ab-initio hybrid DFT-HF B3PW calculations were performed for the Li2O/Ti and LiF/Cu interfaces.78,79 Although it is known that TiO2 cannot be electrochemically reduced to metal, the Li2O/Ti interface has been chosen here as a model interface mainly due to a proper lattice matching and absence of irregular structures and steps at the interface.
Different positions of inserted Li atoms have been considered: positions at the slab surface opposite to the Ti/Li2O or Cu/LiF interfaces, positions at octahedral interstitial sites inside the slab, and finally at the interface between metal and Li2O or LiF slab. Self-consistent calculations in these composite systems show a considerable electron transfer from excess Li atoms towards Ti or Cu atoms if it is close to the interface, providing evidence for the interfacial lithium storage.78,79
We also noticed that M/Li2O interface stores more Li+ at low potential compared to M/LiF nanocomposites. Our theoretical simulations show that this could be due to different composition of interfacial Li atoms in M/Li2O and M/LiF interfaces which possess different crystalline structures. Unlike pure LimX (X = O, F and m = 1 or 2) and transition metal bulk, a M/LiX (saturated with Li) interface can store one (LiF) or two–three (Li2O) monolayers of additional Liper interface with electrons being transferred largely to the metal adlayers, in full accordance with the suggested interfacial storage mechanism.20 While LimX (X = O, F and m = 1 or 2) surface layers or the interfacial core serve as hosts for Li+, transition metals serve as electron sinks, a role which is more pronounced, the thicker the slab owing to the stabilization of the electron. The extent of this anomalous storage effect depends on the electronegativity of transition metal used, for example extra 1 monolayer (ML) of Li placed at the interfaces of Cu/LiF (001) and Ti/Li2O (111) results in more interfacial storage with Cu metal adlayers than with Ti.
The above discussion of enhanced lithium storage in nanostrucured transition metal oxides and transition metal fluorides using heterogeneous reactions such as conversion reaction and interfacial reaction, are examples which highlight revolutionary approaches and concepts due to nanocrystallinity. Further challenges for materials researchers involve optimizing the conditions needed to achieve high cyclic as well as high rate performances in these nanocrystalline materials.
5. Supercapacitance of mesoporous materials
Supercapacitors are electrical storage devices that can deliver a huge amount of energy in a short time. The life time of supercapacitor is quite high (>500000 cycles) as it functions solely on electrostatic surface charge accumulation, unlike a battery that stores energy chemically where the cycle life is limited (<3000 cycles) due to repeated expansion and contraction of the electrodes upon cycling. High performance supercapacitors can be achieved by moving from bulk to nanostructured electrodes.
How such nanomaterials can advance these devices, is clear if we compare the storage capabilities of both battery and conventional capacitors. Lithium batteries store a lot of energy, but discharge power at a relatively low level; traditional capacitors provide a burst of power but do not hold much energy. Supercapacitors are something of a crossbreed: they can both store a cache of energy and release it in pulses of strong power. A commercial supercapacitor comprises current collectors coated with a thin film of highly porous activated carbon filled with an ionic solution – the electrolyte. The nanostructured activated carbon has several orders of magnitude more surface area than an ordinary capacitor. Recent trends in supercapacitors involve the development of high-surface-area mesoporous electrodes to optimize the performance in terms of capacitance and overall conductivity. Schindall's group at MIT82 replaced the activated carbon with a dense microscopic bunch of carbon nanotubes grown on the surface of the current collectors. Another advantage of such nanotubes is that their structure makes them less chemically reactive, so they can operate at higher temperature. It is also believed that certain types of nanotubes, depending on their geometry can be excellent conductors, suggesting that they can supply more power than supercapacitors using activated carbon. Razal et al.83 have produced fibers from carbon nanotubes that when woven into threads can function as a supercapacitor. With the development of novel nano-architecture of finer dimensions and uniform distribution, carbon nanotubes could enable greater energy storage.
Apart from the above non-faradaic electrochemical double layer capacitors, redox supercapacitors which are based on the pseudo-capacitance arising from fast and reversible faradaic redox reactions of electroactive materials are also known to exhibit high storage performances. For example, conducting polymers and transition metal oxides are employed as electrodes.84–86 Hydrous IrO2 has been found to perform in electrochemical capacitor applications with a specific capacitance close to 550 F g−1.87 In addition, hydrous RuO2 is an ideal material suitable for pseudocapacitors because a specific capacitance value of 720 F g−1 was obtained at the scan rate of 2 mV s−1 in H2SO4 electrolyte.88 Non-hydrous nanoporous RuO2 with a surface area of 240 m2 g−1 synthesized by electrochemical lithiation method exhibits a distinctly better storage performance of 350 F g−1 at 2 mV s−1 than previously reported for the non-hydrated RuO2.89
Controlling the precise size and distribution of pores in novel electroactive materials may be an avenue for nanotechnology of supercapacitors to achieve more storage performance. However, care needs to be taken to attain a compromise between specific surface area of the electrodes for high capacitance and pore-size distribution for easy access of the electrolyte.
6. Effects of nanostructuring on thermoelectric materials
Solid-state cooling and power generation based on thermoelectric effects have potential applications in waste-heat recovery, air conditioning, and refrigeration. The conversion efficiency of such a thermoelectric device is highly dependent on the dimensionless figure of merit (ZT) of the constitute materials (Z = S2σ/κ, where S is the Seebeck coefficient, σ is the electrical conductivity and κ is the thermal conductivity). For thermoelectric devices to be competitive in the market, it is generally agreed that ZT of the thermoelectric materials should be >1, which means a perfect combination of high power factor (S2σ) and low thermal conductivity (κ). Materials researchers have investigated several systems of materials for decades, including typical narrow band semiconductors, oxides and cage-structured materials (skutterudites and clathrates).90–94 Yet, commonly used Bi2Te3-based materials have dominated in the performance at near room temperature with peak ZT values close to 1.Poudel et al.95 recently synthesized a p-type nanocrystalline BixSb2–xTe3 alloy by ball milling the bulk material and hot pressing the resulting nanoparticles into ingots. This results in a nanostructured material with highly crystalline, randomly oriented grains. Strong phonon scattering at the increased volume fraction of interfaces, gives a significant reduction in the thermal conductivity compared with the bulk alloy, and is largely responsible for the high peak ZT value of 1.4 at 373 K.
Another effective way to increase ZT value is to reduce the thermal conductivity by incorporating nano-sized particles into the matrix of thermoelectric materials on the basis of phonon-blocking effect of the nanostructures. Compared with the Bi2Te3 matrix, Bi2Te3 materials dispersed with nano-SiC show an increased Seebeck coefficient, decreased electrical conductivity, and reduced thermal conductivity in the measured temperature range of 323–523 K.96 The peak ZT was improved from 0.99 for the Bi2Te3 sample to 1.04 for 0.1 vol% SiC dispersed Bi2Te3 sample at 423 K. The lattice thermal conductivity was decreased due to enhanced defects scattering caused by the dispersion of nano-SiC as the impurity phases in Bi2Te3 matrix. In addition, much smaller grain sizes and amount of grain boundaries are introduced by the dispersion of nano-SiC, which results in the decrease of lattice and electronic thermal conductivity.
Further, CoSb3 nanoparticles are dispersed within the CoSb3 bulk material to improve the figure of merit.97,98 These incorporated nanoparticles are likely to add scattering centers that affect the phonons, without detrimental interference for the electronic conduction. Microstructure analysis of the bulk nanocomposites prepared by hot press shows that the bulk materials are composed of nano- and micro-grains. The nanocomposite structures are effective in reducing thermal conductivity more than electrical conductivity, hence improve thermoelectric performance. Peak ZT of 0.71 is obtained for the nanocomposite with 40 wt% nanopowder inclusions, which is about 54% increase compared to a sample without nanopowder inclusions.
Significant efforts have been made in recent years to improve the figure of merit of thermoelectric materials by making more exotic structures that exhibit reduced dimensionality and special-nanostructures including nanodot bulk materials, nanowire arrays91,92 and quantum dot superlattice thin films.93,94 Currently, the focus on thermoelectric materials research is to find bulk materials (both n-type and p-type) with a ZT value of the order of 2–3 with minimum losses due to contact resistance, radiation effects and the interdiffusion of the metals with low manufacturing costs.
7. Conclusions
In this review article, we have discussed various nano-size effects, highlighting few anomalous phenomena in thermodynamic and transport behaviour of materials relevant for energy conversion and storage. Nano-size results in enhanced energetics due to surface contributions, which increases the cell voltage in electrochemical systems. Metastable phases are seen to be stabilized at nano-size. Mesoscopic electrical conduction occurs due to the overlap of accumulated or depleted space charges at reduced interfacial spacings. Relevance of such interfacially controlled materials is briefly addressed in the context of fuel cells and solar cell applications. Increasing the interfacial conduction of oxide ion conductors at moderate temperature could lower the operation temperature of solid oxide fuel cells. Enhanced efficiency of hybrid solar cells is proposed by suppressing the charge recombination loss processes through optimization of the potential barrier at the contact of inorganic/organic conductors. Nanocrystallinity leads to unusual energy storage anomalies. Unlike the insertion reaction, enhanced reversible lithium storage has been achieved in transition metal oxides and fluorides through the conversion reaction as well as interfacial storage mechanisms due mainly to nanocrystallinity. Enhanced performance of supercapacitors reported in nano-sized metal oxides is a consequence of mesoporous morphology with increased surface area. More exotic nanostructures of thermoelectric materials are investigated currently to convert waste heat into electricity.Thus we have shown here, as we move from bulk materials towards nanostructured systems, that a number of exciting phenomena occur providing new opportunities for enhanced energy conversion and storage processes. Knowledge of and improved understanding of the fundamental phenomena associated with the anomalous behavior of materials at nano-size is very relevant to the scientific challenges we face for achieving sustainable and clean energy in the near future.
Acknowledgements
Part of the work reported here was carried out by the author during his stay at the Max Planck Institute for Solid State Research, Stuttgart, Germany. The author expresses sincere thanks to Joachim Maier, Seeram Ramakrishna, Eugene A. Kotomin, Janez Jamnik, Yuri F. Zhukovskii, Juergen Fleig and Hong Li for fruitful discussions.References
- V. S. Arunachalam and E. L. Fliescher, MRS Bull., 2008, 33, 264 CAS.
- S. M. Benson and F. M. Orr Jr., MRS Bull., 2008, 33, 297 CAS.
- F. Rosei, J. Phys.: Condens. Matter, 2004, 16, S1373 CrossRef CAS.
- E. L. Brus, J. Chem. Phys., 1984, 80, 4403 CrossRef CAS.
- J. Maier, Nat. Mater., 2005, 4, 805 CrossRef.
- J. Maier, Prog. Solid State Chem., 1995, 23, 171 CrossRef CAS.
- H. L. Tuller, Solid State Ionics, 2000, 131, 143 CrossRef CAS.
- J. Schoonman, Solid State Ionics, 2000, 135, 5 CrossRef CAS.
- Ph. Buffat and J.-P. Borel, Phys. Rev. A, 1976, 13, 2287 CrossRef CAS.
- M. R. Ranade, A. Navrotsky, H. Z. Zhang, J. F. Banfield, S. H. Elder, A. Zaban, P. H. Borse, S. K. Kulkarni, G. S. Doran and H. J. Whitfield, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 6476 CrossRef CAS.
- J. M. McHale, A. Auroux, A. J. Perrota and A. Novrotsky, Science, 1997, 277, 788 CrossRef CAS.
- A. Chatterjee, S. K. Pradhan, A. Datta, M. De and D. Chakravorty, J. Mater. Res., 1994, 9, 263 CrossRef CAS.
- A. A. Gribb and J. F. Banfield, Am. Mineral., 1997, 82, 717 CAS.
- G. Schimanke and M. Martin, Solid State Ionics, 2000, 136, 1235 CrossRef.
- P. Knauth, G. Schwitzgebel, A. Tschöpe and S. Villain, J. Solid State Chem., 1998, 140, 295 CrossRef CAS.
- K. T. Jacob, K. P. Jayadevan, R. M. Mallya and Y. Waseda, Adv. Mater., 2000, 12, 440 CrossRef CAS.
- A. Schroeder, J. Fleig, H. Drings, R. Wuerschum, J. Maier and W. Sitte, Solid State Ionics, 2004, 173, 95 CrossRef CAS.
- M. Holtzinger, J. Maier and W. Sitte, Solid State Ionics, 1996, 86, 1055 CrossRef.
- P. Balaya and J. Maier ( 2008, in preparation).
- J. Jamnik and J. Maier, Phys. Chem. Chem. Phys., 2003, 5, 5215 RSC.
- P. Balaya, A. J. Bhattacharyya, J. Jamnik, Yu.F. Zhukovskii, E. A. Kotomin and J. Maier, J. Power Sources, 2006, 159, 171 CrossRef CAS.
- O. Delmer, P. Balaya, L. Kienle and J. Maier, Adv. Mater., 2008, 20, 501 CrossRef CAS.
- J. Fleig, Solid State Ionics, 2002, 150, 181 CrossRef CAS.
- J. Lubomirsky, J. Fleig and J. Maier, J. Appl. Phys., 2002, 92, 6819 CrossRef CAS.
- N. Sata, K. Eberman, K. Eberl and J. Maier, Nature, 2000, 408, 946 CrossRef CAS.
- Y. G. Guo, J. S. Lee and J. Maier, Adv. Mater., 2005, 17, 2815 CrossRef CAS.
- I. Denk, J. Claus and J. Maier, J. Electrochem. Soc., 1997, 144, 3526 CAS.
- R. A. De Souza, J. Fleig, J. Maier, O. Kienle, Z. Zhang, W. Sigle and M. Rühle, J. Am. Ceram. Soc., 2003, 86, 922 Search PubMed.
- P. Balaya, J. Jamnik, J. Fleig and J. Maier, Appl. Phys. Lett., 2006, 88 Search PubMed 062109.
- P. Balaya, J. Jamnik, J. Fleig and J. Maier, J. Electrochem. Soc., 2007, 154, P69 CrossRef CAS.
- S. Kim and J. Maier, J. Electrochem. Soc., 2002, 149, J73 CrossRef CAS.
- C. N. Ginestra, R. Sreenivasan, A. Karthikeyan, S. Ramanathan and P. C. McIntyrea, Electrochem. Solid-State Lett., 2007, 10, B161 CrossRef CAS.
- D. K. -Merl, J. Lappalainen and H. L. Tuller, J. Electrochem. Soc., 2006, 153, J15 CrossRef.
- M. Grätzel, Nature, 2001, 414, 338 CrossRef CAS.
- B. A. Gregg, J. Phys. Chem. B, 2003, 107, 4688 CrossRef CAS.
- N. S. Lewis, Science, 2007, 315, 798 CrossRef CAS.
- H. J. Snaith and L. Schmidt-Mende, Adv. Mater., 2007, 19, 3187 CrossRef CAS.
- S. Gunes and N. S. Sariciftci, Inorg. Chim. Acta, 2008, 361, 581 CrossRef.
- E. Palomares E, J. N. Clifford, S. A. Haque, T. Lutz and J. R. Durrant, J. Am. Chem. Soc., 2003, 125, 425.
- A. Kay and M. Grätzel, Chem. Mater., 2002, 14, 2930 CrossRef CAS.
- A. N. M. Green, E. Palomares, S. A. Haque, J. M. Kroon and J. R. Durrant, J. Phys. Chem. B, 2005, 109, 12525 CrossRef CAS.
- S. Nakeda, Y. Saito, W. Kubo, T. Kitamura, Y. Wada and S. Yanagida, J. Phys. Chem. B, 2007, 107, 8607.
- K. Fujihara, A. Kumar, R. Jose, S. Ramakrishna and S. Uchida, Nanotechnology, 2007, 18, 365709 CrossRef.
- M. Law, L. E. Greene, J. C. Johnsons, R. Saykally and P. Yang, Nat. Mater., 2005, 4, 455 CrossRef CAS.
- R. R. Hawaldar, K. G. Kanade, K. R. Patel, S. D. Sathaye, U. P. Mulik and D. P. Amalnerkar, Mater. Chem. Phys., 2005, 91, 447 CrossRef CAS.
- M. C. Beard, K. P. Knutsen, P. Yu, J. M. Luther, W. K. Qing songMetzger, R. J. Ellinson and A. J. Nozik, Nano Lett., 2007, 7, 2507.
- W. Shockley and H. J. Queisser, J. Appl. Phys., 1961, 32, 510 CAS.
- A. S. Arico, P. Bruce, B. Scrosati, J. M. Tarascon and W. Van Schalkwijk, Nat. Mater., 2005, 4, 366 CrossRef CAS.
- A. K. Padhi, K. S. Nanjundaswamy and J. B. Goodenough, J. Electrochem. Soc., 1997, 144, 1188 CAS.
- A. S. Andersson and J. O. Thomas, J. Power Sources, 2001, 97–98, 498 CrossRef CAS.
- S.-Y. Chung, J.-T. Blocking and Y.-M. Ching, Nat. Mater., 2002, 1, 123 CrossRef CAS.
- N. Ravet, A. Abouimrane and M. Armand, Nat. Mater., 2003, 2, 702 CrossRef CAS.
- G. Rousse, J. R- Carvajal, S. Patoux and C. Masquelier, Chem. Mater., 2003, 15, 4082 CrossRef CAS.
- P. S. Herle, B. Ellis, N. Coombs and L. F. Nazar, Nat. Mater., 2004, 3, 147 CrossRef CAS.
- Yamada, H. Koizumi, S.-I. Nishimura, N. Sonoyama, R. Kanno, M. Yonemura, T. Nakamura and Y. Kobayashi, Nat. Mater., 2006, 5, 357 CrossRef CAS.
- Y.-S. Hu, Y.-G. Guo, R. Dominko, M. Gaberscek, J. Jamnik and J. Maier, Adv. Mater., 2007, 19, 1963 CrossRef CAS.
- R. Amin, P. Balaya and J. Maier, Electrochem. Solid-State Lett., 2007, 10, A13 CrossRef CAS.
- C. Delacourt, L. Laffont, R. Bouchet, C. Wurm, J.-B. Leriche, M. Morcrette, J.-M. Tarascon and C. Masquelier, J. Electrochem. Soc., 2005, 152, A913 CrossRef CAS.
- W. Ojczyk, J. Marzec, K. Swierczek, W. Zajac, M. Molenda, R. Dziembaj and J. Molenda, J. Power Sources, 2007, 173, 700 CrossRef CAS.
- D. Morgan, A. Van der Ven and G. Ceder, Electrochem. Solid-State Lett., 2004, 7, A30 CrossRef CAS.
- A. Manthiram, J. Choi and W. Choi, Solid State Ionics, 2006, 177, 2629 CrossRef CAS.
- M. S. Islam, D. J. Driscoll, C. A. J. Fisher and P. R. Slater, Chem. Mater., 2005, 17, 5085 CrossRef CAS.
- N. Ravet, A. Abouimrane and M. Armand, Nat. Mater., 2003, 2, 702 CrossRef CAS.
- R. Dominko, M. Bele, M. Gaberscek, M. Remskar, D. Hanzel, J. M. Goupil, S. Pejovnik and J. Jamnik, J. Power Sources, 2006, 153, 274 CrossRef CAS.
- K. Saravanan, M. V. Reddy, P. Balaya, H. Gong, B. V. R. Chowdari and J. J Vittal, J. Mater. Chem., 2008, submitted Search PubMed.
- J. Besenhard, Handbook of Battery Materials, Wiley-VCH, New York, 1999 Search PubMed.
- C. A. Vincent and B. Scrosati, Modern Batteries, Edward Arnold Publishers, London, 1997 Search PubMed.
- P. Poizot, S. Laruelle, S. Grugeon, L. Dupont and J. -M. Tarascon, Nature, 2000, 407, 496 CrossRef CAS.
- J. -M. Tarascon, S. Grugeon, M. Morcrette, S. Laruelle, P. Rozier and P. Poizot, C. R. Chim., 2005, 8, 9 CrossRef CAS.
- H. Li, G. Richter and J. Maier, Adv. Mater., 2003, 15, 736 CrossRef CAS.
- F. Badaway, N. Pereira, F. Cosandey and G. G. Amatucci, J. Electrochem. Soc., 2003, 150, A1209 CrossRef.
- H. Li, P. Balaya and J. Maier, J. Electrochem. Soc., 2004, 151, A1878 CrossRef CAS.
- M. V. Reddy, S. Madhavi, G. V. S. Rao and B. V. R. Chowdari, J. Power Sources, 2006, 162, 1312 CrossRef CAS.
- M. Unnithan and P. Balaya ( 2008, unpublished).
- P. Balaya, H. Li, L. Kienle and J. Maier, Adv. Funct. Mater., 2003, 13, 621 CrossRef CAS.
- N. Tsuda, K. Nasu, A. Fujimori and K. Siratori, Electronic Conduction in Oxides, Springer, New York, 2nd edn, 2000 Search PubMed.
- S. Laruelle, S. Grugeon, P. Poizot, M. Dolle, L. Dupont and J. -M. Tarascon, J. Electrochem. Soc., 2002, 149, A627 CrossRef CAS.
- Yu. Zhukovskii, P. Balaya, E. Kotomin and J. Maier, Phys. Rev. Lett., 2006, 96 Search PubMed 058302.
- Yu. F. Zhukovskii, P. Balaya, M. Dollé, E. A. Kotomin and J. Maier, Phys. Rev. B, 2007, 76, 235414 CrossRef.
- P. Liao, B. L. MacDonald, R. A. Dunlap and J. R. Dahn, Chem. Mater., 2008, 20, 454 CrossRef CAS.
- L. Y. Beaulieu, D. Larcher, R. A. Dunlap and J. R. Dahn, J. Electrochem. Soc., 2000, 147, 3206 CrossRef CAS.
- J. Schindall, IEEE Spectrum, 2007, 44, 42 CrossRef.
- J. M. Razal, J. N. Coleman, E. Muñoz, B. Lund, Y. Gogotsi, H. Ye, S. Collins, A. B. Dalton and R. H. Baughman, Adv. Funct. Mater., 2007, 17, 2918 CrossRef CAS.
- A. Rudge, J. Davey, I. Raistrick and S. Gottesfeld, J. Power Sources, 1994, 47, 89 CrossRef CAS.
- Y. G. Wang and Y. Y. Xia, Electrochim. Acta, 2006, 51, 3223 CrossRef CAS.
- J.-M. Luo, B. Gao and X.-G. Zhang, Mater. Res. Bull., 2008, 43, 1119 CrossRef CAS.
- A. A. F. Grupioni, E. Arashiro and T. A. F. Lassali, Electrochim. Acta, 2002, 48, 407 CrossRef CAS.
- J. P. Zheng and T. R. Jow, Electrochem. Solid-State Lett., 1999, 2, 359 CrossRef CAS.
- Y.-S. Hu, Y.-G. Guo, W. Sigle, S. Hore, P. Balaya and J. Maier, Nat. Mater., 2006, 5, 713 CrossRef CAS.
- M. Martin-Gonzalez, A. L. Prieto, R. Gronsky, T. Sands and A. M. Stacy, Adv. Mater., 2003, 15, 1003 CrossRef CAS.
- T. M. Tritt and M. A. Subramanian, MRS Bull., 2006, 31, 188.
- R. Venkatasubramanian, E. Siivola, T. Colpitts and B. O. Quinn, Nature, 2001, 413, 597 CrossRef CAS.
- T. C. Harman, P. J. Taylor, M. P. Walsh and B. E. LaForge, Science, 2002, 297, 2229 CrossRef CAS.
- T. M. Tritt, H. Bottner and L. Chen, MRS Bull., 2008, 33, 366 CAS.
- B. Poudel, Q. Hao, Y. Ma, Y. Lan, A. Minnich, B. Yu, X. Yan, D. Wang, A. Muto, D. Vashaee, X. Chen, J. Liu, M. S. Dresselhaus, G. Chen and Z. Ren, Science, 2008, 320, 634 CrossRef CAS.
- L.-D. Zhao, B.-P. Zhang, J.-F. Li, M.Z., W.-S. Liu and J. Liu, J. Alloys Compd., 2008, 455, 259 CrossRef CAS.
- P. N. Alboni, X. Ji, J. He, N. Gotherd, J. Hubbard and T. M. Tritt, J. Electron. Mater., 2007, 36, 711 CrossRef CAS.
- J. L. Mi, X. B. Zhao, T. J. Zhu and J. P. Tu, Appl. Phys. Lett., 2007, 91, 172116 CrossRef.
| This journal is © The Royal Society of Chemistry 2008 |