Dye-sensitized solar cells: A safe bet for the future.
Luís Moreira
Gonçalves
a,
Verónica
de Zea Bermudez
b,
Helena Aguilar
Ribeiro
a and
Adélio Magalhães
Mendes
*a
aLEPAE - Departamento de Engenharia Química, Faculdade de Engenharia, Universidade do Porto, R. Dr. Roberto Frias, 4200-465, Porto, Portugal. E-mail: mendes@fe.up.pt; Fax: +351 22 508 1449; Tel: +351 22 508 1695
bDepartamento de Química and CQ-VR, Universidade de Trás-os-Montes e Alto Douro, 5001-801, Vila Real, Portugal
First published on 24th October 2008
Abstract
This review describes the main features of dye-sensitized solar cells (DSCs) and highlights recent breakthroughs in this promising thin-film photovoltaic (PV) technology. After a brief presentation of the commercially available technologies, the general operation principles and the most relevant characteristics of DSCs are summarized. Recent major advances in high efficiency sensitizers, nanostructured semiconductors and robust electrolytes offer an opportunity for DSCs integration into the marketplace. With attractive features, like low-cost potential, simple processing, wide range of applicability – from low-power electronics to semi-transparent windowpanes for electricity generation – and good performance under typical operating conditions, these cells are one step from large-scale commercialization. We describe major strategies that are under way to make DSCs a key technology in the future PV paradigm.
 Luís Moreira Gonçalves Luís Moreira Gonçalves |
 Verónica de Zea Bermudez Verónica de Zea Bermudez |
 Helena Aguilar Ribeiro Helena Aguilar Ribeiro |
 Adélio Magalhães Mendes Adélio Magalhães Mendes |
Broader contextNow, more than ever before, energy is what makes our world continuously work. Currently, photovoltaics (PVs) account for no more than 0.04% of all energy production; however this is set to change. The PV market has shown an outstanding 33% growth in the last 10 years and is expected to grow by 25 to 30% yearly in the coming decades. This review describes the evolution and “state of the art” of promising recent PV technology called dye-sensitized solar cell (DSC). In this kind of cell, that mimics natural photosynthesis, photon harvesting and charge transport are performed by different elements. The components which comprise DSCs are reviewed along with the status of the technology in the context of the PV market, which is now making the transition from the laboratory to industry. |
PV and DSC position in the global energy market
Now, more than ever before, energy is what makes our world continuously work. World energy consumption is ca. 4.7 × 1020 J (450 quadrillion Btu) and is expected to grow about 2% each year for the next 25 years.1 Earth's resources upon which the world economy has sustained its grown are finite and governments are increasingly aware of this, particularly now that peak oil is on a near future.2 In order to maintain social and economic development, society is obliged to find a way of making a suitable transition to renewable fuels. This desired change in our energetic paradigm is also being hastened by environmental issues;3 mankind cannot afford to continue to progress by relying on sources of energy that release greenhouse gases.Nowadays, renewable sources comprise about 13% of all energy production and photovoltaics (PV) account for no more than 0.04% and most probably only in 2030 will that figure reach 1%.4 Solar PV energy costs are not yet competitive and continued PV grow is mainly based on government support,5 as is easily perceived by analyzing three major consumers: Germany, Japan and the USA (the three combined share about 90% of the world market).6 Nevertheless, PV solar cells are clearly very elegant and attractive devices for producing energy: cells are free from chemical and noise pollution; their power output is flexible, producing µW as well as MW; production can be done in situ, it is not dependent on the electrical grid, which makes them uniquely portable; they do not rely on reserves located abroad in geopolitically unstable countries and, of course, their source of energy – the Sun – is free and inexhaustible for the next few million years. Moreover, the sun's rays reaching the earth are enough to fulfill global energy demand more than 10 thousand times over, i.e. in theory one hour of sunlight is more than enough for a whole year of global consumption. Also, ad extremum, solar energy is the only way of respecting the second law of thermodynamics towards sustainable development, because it benefits from all the massive creation of entropy in the star's core. It is true that PVs do not work at night but that fact is minimized due to reasonable synchronization between peak production and peak consumption.
The PV market has had outstanding yearly growth, 33% growth per year since 1997, and it is expected to grow by 25 to 30% per year in the next decades.4 While traditional energy sources will become more expensive, PV will be much more competitive due not only to technology improvements but also to economies of scale, with a predicted learning factor of 18% till 2030.4 Some optimistic scenario predictions state that by 2100 solar power (PV and solar thermal generation combined) may be more than one half of our primary energy use (Fig. 1).7 This means than even among other renewable sources of energy, PV is likely to have the highest potential in the long-term.
 | ||
| Fig. 1 The exemplary path until 2050/2100 (source: German Advisory Council on Global Change7). | ||
Present PV market regarding all different technologies
The first modern PV solar cells, silicon (Si) p/n, were developed by Chapin et al. at Bell Laboratories in 1954.8 A few years later, they were already intensively used in space exploration. Si cells were the perfect solution for space energy production. Although several improvements in terms of efficiency and reliability occurred with time, considerable domestic use only begun in 1978 when NASA installed a 3.5 kW system in a 16 home Indian village in Papago Reservation, Arizona. Since, apart from regular use in power calculators, traffic signs, clocks and other small appliances, the use of solar cells has grown quite slowly although steadily.Mono and polycrystalline silicon (mc–Si and pc–Si, respectively) solar cells have totally dominated the terrestrial PV market so far (Fig. 2). Silicon is far from being the ideal material for PV conversion,9 so the ongoing situation may seem at first to be a bit surprising. The major reason for crystalline Si (c–Si) market dominance – besides Si natural abundance, low toxicity and a well-established processing technology – is that manufacturers have been supplied with rejected material from the high-tech semiconductor industry.
 | ||
| Fig. 2 Market distribution of PV technologies in 2005 (adapted from Pizzini et al.10). | ||
Apart from oxygen, Si is the most abundant element in the Earth's surface, it is generally found in quartz or sand as silicon dioxide (SiO2). This is turned into metallurgic grade Si (MGS) by a rather energy demanding process (it must be heated to over 2000 K). Such purity levels (ca. 98%) are widely used in many heavy industries but cannot be employed in semiconductor-based technologies. MGS is further purified into electronic grade Si (EGS), of a very high purity degree, by the Siemens process, again quite an energy demanding process. So far, the PV industry has been supplied with silicon scraps from the microelectronic industry on the assumption that the tolerated level of impurities is greater for PV cells (i.e. it is not a top-efficient device) like in microelectronic devices. This “intermediate” purity level can be roughly called “solar grade” Si (SGS). This means that purified Si is a fairly expensive feedstock due to the two costly purifying processes, and that it has been supplied cheaply to the PV industry as a “by-product” from electronic high-tech manufacturers. Logically, this situation has created an unwanted dependency that can only sustain itself if the electronic Si industry continues to grow at least at the same rate as the PV industry, which is not the case.10 The Si-dependent PV industry has known for quite some time that it has no other choice but to quickly find an autonomous source of SGS.11 This has been a dynamic research field in the past years within PV. Meanwhile the Si price has more than doubled since 200212 and, for instance, in Germany some production lines may be already working at half their capacity due to supply shortages.13
C–Si has a quite simple operating principle. The cell is constituted by three layers: the n-type, the p-type and the pn-junction (the connection formed between the previous two). The n-type layer is doped with elements from group V, normally phosphorous, because it has extra electrons and works as the donor (anode); the p-type layer, on the other hand, is doped with group III elements, commonly boron, and therefore has a lack of electrons to create “holes” and, consequently, becomes the receptor (cathode). Light reception occurs in the pn-junction, each photon generates an electron–hole pair by exciting electrons from the junction valence band to the conduction band (electrons are driven to the negative layer while “holes” are left as positive). This mechanism leads to different potentials in the anode and cathode, inducing an electron flow, and in doing so generates electricity. Champion cells achieve up to 24.4% efficiency under standard reporting conditions14 (1 sun irradiance, i.e. 1000 W m−2, AM 1.5, 298 K; all efficiencies presented here comply with these criteria).
The first amorphous Sicell (a–Si) appeared in 1976,15 right after the first oil shock, when a great amount of hope and expectation was set upon PV energy. It is odd that the main competitor to c–Si is also Si-based, especially as a–Si is very different from c–Si. Particularly, a–Si has a high absorption rate and therefore does not need a Si layer as thick as c–Si, which makes it a thin-film technology. Faster development of a–Si has been blocked by the Staebler–Wronski light induced degradation effect (named after its discoverers in 1977);16 such a photoelectronic effect may cause loss of cell efficiency of about 50% or more with light exposure, needless to say that this is a major stability issue to overcome. This effect has not been satisfactorily explained17 but so far researchers have managed to lower its disturbance to around 10% or less of losses using several techniques.
Besides c–Si and a–Si, Si is also used in other similar PV technologies not so commercially successful but still important, such as ribbon Si, thin-film c–Si, also known as nano or microcrystalline Si (a relevant example is crystalline silicone on glass (CSG)18) and heterostructure concepts, i.e. a combination of different Si technologies, e.g.c–Si/a–Si.
Most other PV technologies have the same working principle as c–Si. They are all based in one or more p–n junctions to absorb photons. It was mostly an empirical search within a large number of semiconductors that resulted in a few promising materials.19 A brief description of these commercial cells follows. Polycrystalline CdTe was one of the first PV thin-film materials proposed20, CdTe solar cells are typically hetero-junctions with CdS being the n-type component. CdTe has the adequate Eg (energy band gap) of 1.45 eV and a high optical absorption coefficient. A top 16.5% efficiency was achieved more than half a decade ago.21 Recent theoretical predictions point to a maximum efficiency of 17.6%,22 which means that future improvements have to be preferably aimed at reducing manufacturing costs. A troubling issue concerning these cells is CdTe toxicity.23,24 The first chalcopyrite solar cells were based on CuInSe2 (CIS),25 but it was realized later that incorporating Ga to produce CuInGaSe2 (CIGS) results in a widened Eg of 1.3 eV.26 CIGS solar cells together with a–Si and CdTe are the leading thin-film technologies. Of these three, CIGS cells appear to have the best future potential due to higher efficiencies,12 confirmed maximum efficiency of 19.2%21 and lower manufacturing energy consumption, which may even be the lowest for any PV technology. However, high quality cell fabrication is very complex and still challenging to undertake.27 Although not as much of concern as CdTe, CIGS have some inherent toxicity that may turn out to be a problem for mass production.23III–V semiconductor solar cells are very efficient but expensive devices, normally based on GaAs and InGaP, they can be used alone although better results are obtained in multijunction (also referred as tandem cells) with semiconductors showing different energy bandgaps, thus taking better advantage of the whole solar spectrum. Double and triple junction (J) devices are currently being commercialized; the most common 3J is GaInP/GaAs/Ge with a record efficiency of 32%.21 If higher efficiency is the ultimate goal, 4J, 5J or more junctions can be investigated and tested; for example a 6J cell (AlInGaP/InGaP/AlInGaAs/InGaAs/InGaNAs/Ge) has a predicted maximum efficiency of 57%.28 In space applications, where cost is not the major problem, multijunction cells have replaced Si cells. The problem lies in making an economically suitable transition for terrestrial purposes. Raw materials cost and high-purity demands make it almost impossible, and the only feasible solution appears to be a conjugation with concentrator systems. This takes advantage of the fact that these cells' efficiency may even increase with higher irradiance.28Molecular and polymer organic solar cells are simple PV devices that are made by organic semiconductors “sandwiched” between two electrodes. These cells are characterized by high optical absorption coefficients and low manufacturing costs. A great deal of attention has been given to these cells in recent times, as they are expected to play a key role in the future PV market, particularly now that the 5% efficiency barrier has been overcome.29,30
The supply of cheap raw materials is not exclusively a problem for the Si industry. CdTe, CIGS and III–V semiconductor cells face the same problem! Tellurium is not a widely available and cheap material, neither are gallium or indium. Indium is a by-product of zinc production and it is also used in flat-panel production, an exponentially growing industry, as indium–tin-oxide (ITO). Mass cell production may at first lead to a decrease in feedstock costs, but this is not expected to happen continuously. In the end non-abundant materials will be more expensive and so will significantly increase final product price.
Dye -sensitized solar cells ( DSC ) PV technology is a very promising alternative for low cost production of energy. State of the art DSCs achieve more than 11% energy conversion allied to good performance under any atmospheric condition and low irradiance.31
DSC is a successful attempt to create an anthropological analogous concept to photosynthesis. The photoreceptor and the charge carrier are different elements; it is an analogous situation to that of the photosynthetic process where chlorophyll absorbs photons but does not participate in charge transfer. This is contrary to conventional PV cells where a semiconductor assumes both functions. This separation of functions leads to lower purity demands on the raw materials side, and consequently makes DSCs a low cost alternative.
DSCs main advantages can be summarized as follows:
• Good performance under standard reporting conditions;
• Stable performance at non standard conditions of temperature, irradiation and solar incidence angle;
• Low cost;
• Available environmental-friendly raw materials;
• Semi-transparency and multi-color range possibilities.
This last factor should not be underrated as often happens. The manufacture of multi-colored cells is truly a competitive advantage of DSCs. This is achieved by changing the dye, either organic32 or inorganic.33 So the color possibilities are immense, as has been successfully shown.34 These cells can be used, for example, in power-producing windows by landscape architects to respect the natural scenery or for fashion reasons, and thus target a high-value market segment. It is also worth mentioning the crucial importance of a low cost PV power source to the population of developing countries, particularly in Africa, where sunshine is an abundant natural resource. Millions of people live without electricity for all their basic needs, and to provide them with this vital commodity, and so supply all that is normally taken for granted like refrigerators, radios or night illumination, is per se a stimulus more than enough to promote the development of this technology.
Evolution of PV market and DSC progressive importance
While wide band-gap semiconductors like TiO2 or ZnO are cheap and photochemically stable, making them suitable for PV applications, their spectral sensitivity is limited to UV. For this reason a chromophoric compound, the sensitizer, is adsorbed onto the semiconductor's surface expanding the absorption spectrum range, and thus increasing light harvesting efficiency.35Sensitization was first tried at the end of the XIX century with the advent of color photography using silver halides. Only one century later did sensitization started to be used for photoelectrochemical purposes. Significant pioneering works include Tributsch36 and Fujishima and Honda37 (photocatalytic splitting of water on a TiO2electrode) and Tsubomura et al.38 These works have more similarities to modern cells than two decades of difference would lead us to believe. A US patent submitted in 1977 already included almost all major characteristics of today's DSCs and is entitled Dye-sensitized solar cells.39Although previous works regarding TiO2sensitization by polypyridyl ruthenium complexes were published by Grätzel and co-workers40–42 in the 1980s, it was the 1991 article43 that originated the interest that still seems to be increasing every year (Fig. 3). The work reported a breakthrough efficiency of around 7% motivated by the innovative use of a nanoscopic TiO2 particle layer and a careful choice of a ruthenium complex as the light absorber.
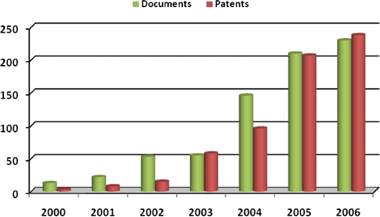 | ||
| Fig. 3 Number of published documents (articles, conference papers and reviews). Source Scopus search engine: search string “dye sensitized solar cells” in title, abstract and keywords. Filed patents history on DSCs. Source mailto:esp@cenet search engine with the same search string in title and abstract. | ||
Despite the current PV market status being dominated by Si (Fig. 2) research reports forecast an impressive growth of new emerging solar cell technologies. Within decades Si will loose its unquestionable leadership (Fig. 4). The average annual growth rate for non c–Si cells is estimated at 43% over the next 10 years, a tremendous rate of growth.44
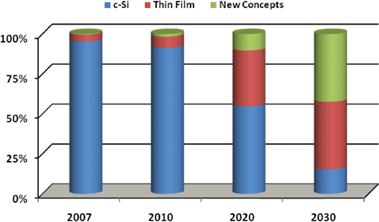 | ||
| Fig. 4 Relative market share of c–Si, thin film and new concepts over time (source: EPIA). | ||
It must be made clear that all these different technologies will coexist since they all have their pros and cons (Fig. 5). A number of factors like location, power demand, climatic conditions, panel orientation etc. must be taken into account before an accurate choice can be made. In the same way that no particular renewable source will solve our energy problems, no particular PV cells are expected to stand alone in the future.
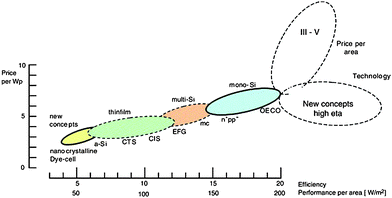 | ||
| Fig. 5 Area-related price and power output for various technologies (adapted from Hoffmann, 2006).4 | ||
Dye-sensitized solar cells
A typical DSC is composed of two sheets of glass coated with a transparent conductive oxide layer (TCO). One of the glass plates – the working electrode – is covered with a film of small dye-sensitized semiconductor particles; the other glass plate – the counter-electrode (CE) – is coated with a catalyst. Both plates are sandwiched together and the electrolyte, commonly a redox couple in an organic solvent, fills the gap between them.Light absorption is carried out by dye molecules, D; photons cause dye photoexcitation, D* (Fig. 6(1)) to rapidly release an electron to the semiconductor (Fig. 6(2)). The electron then percolates through the semiconductor, a process in which the injected electrons hop through the colloidal TiO2 particles and reach the collector (TCO). Afterwards, the electron goes through an outer circuit to reach the other TCO layer (at the counter-electrode), performing electrical work on the way (Fig. 6(3)). The electron is then transferred to the electrolyte where it reduces the oxidant species, Ox (Fig. 6(4)); subsequently, the reducing agent formed, Red, reduces the excited dye, D+, returning it to the ground state, D (Fig. 6(5)), and completing the circuit. Notice that all these dynamic processes are kinetic rather than thermodynamic-based; for example, after photoexcitation the electron can either be injected into the semiconductor, which is the desired pathway, or returned to ground state releasing energy. The electron is normally injected because the time scale of the process is in the femtosecond range while relaxation is in the picosecond range. A similar phenomenon prevents the diffusing electron from recombining either with the dye or the electrolyte, both microsecond-based processes.45 This is very similar to photosynthesis where, to prevent chlorophyll reduction, an electron is rapidly transferred from the excited reaction chlorophyll centre to a distant acceptor of lower energy.46
 | ||
| Fig. 6 A dye-sensitized solar cell sketch (A), an energy diagram and the corresponding reactions (B). | ||
To sum up, solar energy is converted into electricity primarily by a kinetic-based process, the generated voltage equals the difference between the Fermi level of the electron in the semiconductor and the redox potential of the electrolyte, while overall there are no chemical species consumed.
Dye
According to Grätzel, at the time of DSC initial development, the dye sensitizer was the device's weakest point.47 Not surprisingly, various research efforts were and are being made to develop the best performing dye. As stated before, the dye is the photoreceptor sensitizing the semiconductor, and so several requirements must be fulfilled. These include a broad absorption spectrum, adequate ground and excited energy states, long stability (it must endure at least 108redox turnovers corresponding to ∽20 years of operation48), no toxicity, good absorption to the semiconductor's surface, etc.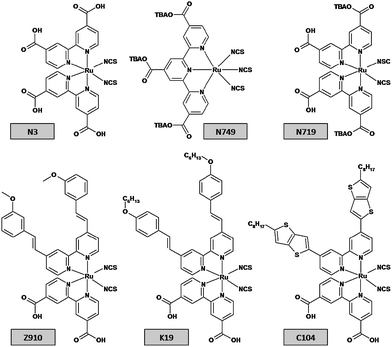 | ||
| Fig. 7 Six relevant ruthenium-based dyes used in DSCs. | ||
The first high-performance polypyridyl ruthenium complex was the so-called N3 [4,4′-dicarboxylic acid-2,2′-bipyridine)ruthenium(II)] reported in 1993 by Nazzeruddin et al.49 N3 results were only surpassed more than 5 years later by another ruthenium complex,50,51 the black dye [tri(isothiocyanato)-2,2′,2″-terpyridyl-4,4′,4″-tricarboxylate) ruthenium(II)], first introduced in 1997.52 Later, N3 returned to the top ranking position thanks to its combination with guanidinium thiocyanate, an additive that increased the cell open circuit voltage (VOC)53 (top row Fig. 7). To date, ruthenium complexes are the most successful dyes53,54 and the only ones so far to achieve over 10% efficiency under standard conditions. More recent research (bottom row Fig. 7) has focused on accomplishing a suitable balance of improved molar absorptivity and stability under thermal stress and light soaking55–57 by extending the π-conjugation of the hydrophobic ligands.58 C104 was reported very recently and presents noteworthy efficiency of 10.5%.59 Despite the high performances of these ruthenium dyes, other alternatives are currently being pursued.60 Due to its scarcity, ruthenium is a very expensive metal and hence requires an extra recycling fee. The most promising alternatives are organic dyes, natural or synthetic, which are considerably cheaper, though so far generally less stable and less efficient.
 | ||
| Fig. 8 Chl - Chlorophyll-a, where HT stands for hydrocarbon tail; a–c (compounds are zinc metalloporphyrins, these structures are analogous to that of chlorophyll): a η = 4.1%61; b η = 3.1%62; c η = 7.1%63. A–W (metal-free dyes): A η = 7.4%64; B η = 8.0%65; C η = 6.7%66; D η = 5.1%67; E η = 9.0%68,69; F η = 6.1%70; G η = 1.9%71; H η = 7.7%72; I η = 6.5%73; J η = 5.2%74; K η = 5.2%75; L η = 3.0%76; M η = 6.8%77; N η = 6.6%78; O η = 5.1%79; P η = 4.5%80; Q η = 2.7%81; R 0.6%82; S η = 6.2%83; T η = 2.6%84; U η = 1.9%85; V η = 4.5%86; W η = 6.3%87; X η = 6.6%88; Y η = 7.6%88. | ||
Chlorophyll (Chl) is the pigment responsible for light absorption in photosynthesis. It consists of porphyrin ring structures linked to a hydrocarbon tail.89 Not surprisingly, Chl-a has been extensively explored for PV applications, since the 1970s36,90 to the present,91 inclusively in DSCs.92,93 But Chl is far from being the only metalloporphyrin that has been tried in DSCs,61 and recent studies report structured guidelines for the identification of porphyrin analogues.94 However, these compounds cannot fully equal the best ruthenium dyes since they do not exhibit red light or near IR absorption.
Metal-free sensitizers, examples include cyanine,88,95merocyanine,81hemicyanine,85anthocyanine,82phthalocyanine,96indoline,70coumarin,72eosin Y,84perylene,97anthraquinone,98 polyene,77pentacene,99triphenylamine100,101 and other promising metal-free structures,64,66,75,102 have progressed enormously in the last few years and several structures (Fig. 8) have yielded efficiencies of around 8% and above.69
Co-sensitization, i.e., the use of several dyes with different spectral responses simultaneously, has the theoretical advantage of enhancing photoabsorption.95,103,104 However, inherent problems, such as energy or electron transfer from one dye to the other, have supressed its use.105
Semiconductor
Since research began, TiO2 has been the preferred semiconductor in DSCs, despite some promising properties offered by other metal oxides like ZnO, SnO2 and Nb2O5.106 Anatase, a crystalline form of TiO2, has been widely used because it has a high band gap energy (3.2 eV, and absorbs only below 388 nm) making it invisible to most of the solar spectrum, reducing the recombination rate of photoinjected electrons. Additionally, it has good thermal stability, is chemically inert, non-toxic and relatively cheap.107Rutile, another crystalline form of TiO2 can also be employed. However it has a higher dark current (Eg 3.0 eV) and so it is less effective (NREL tests point to a 30% decrease in the short-circuit current108) and also photon excitation within the band gap generates holes that act as oxidants making it less chemically stable.109 Some exceptionally good results have been obtained using mixtures, such as TiO2–ZrO2 (95 : 5)110 or ZnO–SnO2 (50 : 50).111The ideal semiconductor layer should have a nanostructured mesoscopic morphology, crucial for a high specific surface area. Logically, the objective is to obtain the maximum area available for dye adsorption using the minimum quantity of TiO2. The two deposition techniques generally used for this purpose are screen-printing and doctor-blading.112
TiO2 film morphology is a major variability factor in DSCs performance113 mainly because of its influence in the electron recombination rate. Zhu et al. concluded that recombination occurs close to the substrate (glass coated with a transparent conducting oxide (TCO)) and not throughout the whole titania matrix as one would perhaps think.114 For this reason, researchers use a compact115 or nanocrystalline116 TiO2 “blocking layer”; the latter seems to be particularly effective when using organic dyes.117 A simpler and yet effective method is by making the substrate undergo an initial TiCl4 treatment.118,119
The use of a light-scattering layer (LSL) is also quite common; it consists of larger titania particles that work as a photo-trapping system.113,119 It has been shown that, as expected, this layer performs photovoltaic work just as well.120 In fact, when using light scattering particles, Koo et al. observed efficiency increments of about 15%.121
A relatively new and exciting research field in semiconductor morphology is the use of nanostructures, namely nanotubes,122–125nanowires,126nanorods127,128 and inverse opals.129,130 The synthesis of oriented tubule nanostructures was first achieved with carbon in 1991.131 Knowledge of nanomaterials grew fast and now it is technically feasible to build nanostructures of metal oxides like ZnO132 and TiO2.133,134 These structures have been applied to DSC electrodes, replacing the semiconducting mesoporous layer. Despite nanowires having a constrained surface area electrode efficiency is increased because they provide a direct passageway for photoexcited electrons to get to the conducting substrate; this smart compromise between electronic conductivity and specific surface area available for dye adsorption has the potential to boost performance. Since the first pioneering applications of TiO2nanostructures to DSC by Uchida et al. and Adachi et al. in 2002135,136 these structures continue to show exciting results (9.3% efficiency was obtained with oriented anatase nanowires126) and they are particularly favorable in solvent-free ionic liquid electrolytes.137 Results have also demonstrated that highly oriented transparent TiO2nanotube arrays show enhanced photocurrent densities and high electron lifetimes.138
Electrolyte
The electrolyte is a crucial part of all DSCs. It is responsible for the inner charge carrier between electrodes – it is the hole-transport material. It endlessly regenerates the dye at the photo-electrode with the charge collected at the CE. The best results have always been obtained with the triiodide/iodide (I3−/I−) redox couple in an organic matrix, generally acetonitrile. The most noteworthy of the non-traditional electrolytes are room-temperature ionic liquids (RTILs), quasi-solid state and solid state. These electrolytes are progressively viscous enabling increased stability. They appear to solve problems such as dye desorption, solvent evaporation and sealing degradation, however, until now their performance has been consistently lower. A more viscous electrolyte diminishes regular charge diffusion and, therefore, requires higher concentration of the redox couple to maintain conductivity. Hence, a higher redox couple concentration creates new problems, or makes them meaningful (e.g., corrosion and direct reaction with the semiconductor). Balanced electrolyte development is the missing piece to remove complicated sealing and volatility issues while pursuing long-term high performance.Another issue to be considered when using I3−/I− is its concentration. Obviously, at low concentrations, conductivity will be insufficient and rapid reduction will not be ensured. On the other hand, when employed in high concentrations, apart from possible corrosion problems, iodide can substantially suppress cell efficiency by, increasing the recombination of I3− and injected electrons, and increasing the rate of light absorption by the redox couple. The spectroscopy and dynamics of I3− have been extensively studied in solution; its absorption spectrum consists of two broad bands centered at 360 and 290 nm.140 The suppression of the dark current may be achieved by additives such as tetrabutylammoniumhydroxide (TBAOH),1414-tert-butylpyridine (4TBP),142,1432-propylpyridine (2PP)144 or methylbenzimidazole (MBI).145 Additionally, these additives also enhance the cell's long-term stability.145
As stated before, other redox couples have been tested, and theoretically a well designed change in the electrolyte formulation could increase the Voc by up to 300 mV.146 In 1997, Argazzi et al. tested some phenothiazine compounds but IPCEs were persistently low.147 Oskam et al. provided evidence that neither (SeCN)2/SeCN− nor (SCN)2/SCN− were able to produce results as good as I3−/I− with TiO2,148 though encouraging results were obtained when using SnO2 as the charge carrier.149 In 2005, Wang et al. concluded that with certain organic dyes the redox couple Br3−/Br− may be more effective,84 a claim that may interestingly influence near-future works. Nonetheless, probably the most tested and most viable alternative to date is the use of cobalt complexes. Several complexes of Co(II)/Co(III) have been tried.150–152 Compared to iodide, their advantage is that they are non-volatile, non-corrosive and have the benefit of being easy for molecular modifications.151 However, with present technology the current exchange rate at the counter-electrode is much smaller and leads to voltage losses.152
|
|
||||
|---|---|---|---|---|
| Year | R | X− | η (%) | Reference |
| 1996 | (CH2)5CH3 | I− | – | 160 |
| 2002 | (CH2)4CH3 | I− | 5.3 | 165 |
| 2003 | (CH2)4CH3 : CH2CH3 (13 : 7) | I− : N3− | 6.6 | 159 |
| 2003 | (CH2)2CH3 | I− | 7.0 | 166 |
| 2004 | (CH2)4CH3 : CH2CH3 (13 : 7) | I− : NCS− | 7.0 | 167 |
| 2004 | (CH2)2CH3 | SeCN − | 7.5 | 168 |
| 2005 | CH2)2CH3 : CH2CH3 (1 : 1) | I− : C(CN)3− | 7.4 | 169 |
| 2006 | (CH2)2CH3 | B(CN)4− | 7.0 | 170 |
| 2007 | (CH2)2CH3 : CH2CH3 (13 : 7) | I− : B(CN)4− | 7.6 | 171 |
| 2008 | CH2CH3 : CH2CH3 : CH3 (16 : 12 : 12) | B(CN)4− : I− : I− | 8.2 | 163 |
In the first works with p-type semiconductors solid state DSCs (Table 2), which appeared in 1995 from Tennakone et al.172 and simultaneously O'Regan et al.173, the overall efficiency was lower than 1%. In these pioneering works, electrolytes were totally inorganic but soon afterwards organic materials were tried.174,175 In 1998, Tennakone's group replaced the initially used cyanidine dyes, derived from flower pigments, by ruthenium complex dyes obtaining exciting results176 and breaking the 2% efficiency barrier.177
CuSCN and CuI have been almost exclusively the p-type inorganic semiconductors used in solid state DSCs, CuBr178 being a notable exception. So far, performances by CuI have been higher than CuSCN, because deposition methods for copper thiocyanate are not yet satisfactory. However, instability has been frequently reported in CuI-based cells.179 This turns out to be rather ironic since solving instability issues with DSCs was the original reason for experimenting solid state cells. Of the several organic materials tried (in contrast to inorganic compounds they have high chemical tenability) spiro-MeOTAD has been one of the most important. It was initially presented by Grätzel and co-workers in 1998175 and presently attains conversion yields above 4%.180
| Year | Dye | Electrolyte | η (%) | Reference |
|---|---|---|---|---|
| 1995 | cyanidine | CuI | 0.8 | 172 |
| 1998 | N3 | spiro-MeOTAD | 0.8 | 175 |
| 1999 | N3 | CuI | 2.4 | 177 |
| 2001 | N719 | spiro-MeOTAD | 2.6 | 181 |
| 2002 | N719 | spiro-MeOTAD | 3.2 | 182 |
| 2003 | N3 | CuI | 3.8 | 183 |
| 2005 | Z907 | spiro-MeOTAD | 4.0 | 184 |
| 2005 | indoline | spiro-MeOTAD | 4.1 | 180 |
| 2005 | N3 | LiI/Hydroxypropionitrile | 5.5 | 185 |
| 2008 | N719 | poly(N-alkyl-4-vinyl-pyridine) iodide | 5.6 | 186 |
Conducting substrates
The most commonly used substrates for DSCs are made of coated glass with a transparent conducting oxide (TCO). Suitable TCO must have high electrical conductivity (an order of magnitude of 104 S cm−1) to efficiently collect all the generated photocurrent, and high transparency (better than 80% visible wavelengths). Among a whole variety of TCOs, the most widely used is fluorine doped tin dioxide (SnO2:F or FTO) due to its thermal steadiness and low cost (the two TCO layers may be up to 60% of the cell's total price153). Indium tin oxide (In2O3:Sn or ITO) has also been extensively used because it has higher specific conductivity. However, it does not remain stable at high temperatures and hence it is specifically used in flexible cell technologies in which sintering temperatures are generally lower (typically 200 °C or less).187 Recently, Goto et al.188 reported a DSC with a multi-layer FTO and ITO substrate with increased heat resistance. The above mentioned TCOs are the most widely used materials in the remaining PV industry for the same reasons.Flexible substrates, either plastics or metals (Table 3), are advantageous when considering mass production, because flexible materials allow a low-cost roll-to-roll manufacture. Plastic substrates, examples include polyethylene naphthalate (PEN)189 or polyethylene terephthalate (PET)190, are advantageous in terms of malleability, weight and handling. However, since most polymers cannot withstand the regular sintering high temperature, the sintering step had to be changed to low-temperature alternatives.191 Metal sheets have been progressively tried as substrates: they have excellent electrical and thermal conductivities and low price. Metals that form a non-conducting layer during the high temperature sintering process (steel, Al or Co) cannot be used without a TCO layer or else they are liable to corrosion by the redox couple species. In contrast, metals that form a conducting layer (stainless steel, W or Ti) can be used by themselves and are very promising alternatives.192 Recently Onoda et al.193 showed Ti superiority even when compared to FTO.
Conclusions
Since the 1991 breakthrough, all aspects related to DSCs have been subject to improvement. Currently, there are more than 100 research groups working in DSC research around the world (countries include, among others, Switzerland, the Netherlands, the UK, Spain, Germany, Sweden, Finland, Portugal, China, Japan, South Korea, Australia, New Zealand, South Africa and the USA).As outlined previously,204 energy conversion efficiency (Fig. 9) is controlled by four elements: (i)light harvesting efficiency (augmented by dyes with an enhanced near infrared response, thus increasing Isc); (ii) charge injection efficiency; (iii) electron transport and collection efficiency in the electrodes (diminished internal resistance should augment the FF – fill factor); and (iv) hole transport and collection efficiency in the electrolyte. There is no scientific deterrent to a potential 15% efficiency result.58 Possible efficiency boosts may come about by: the complex use of a cocktail dye; better control of undesirable electron recombination between the percolated electron and the redox couple; or from the application of nanotechnologies in the development of nanostructured semiconductors. The possibilities are immense and promising.
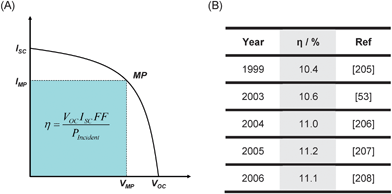 | ||
| Fig. 9 (A) Typical I–V curve of PV solar cells. The overall efficiency (η) of the PV cell is the product of integral photocurrent density (ISC – current obtained at short-circuit conditions, divided by the cell area), the open-circuit photovoltage (VOC) and the cell fill factor (FF) (defined as VMPJMP / VocJsc, where MP stands for maximum power point) divided by the intensity of the incident light (Pincident). (B) Some DSC top efficiency results (standard reported conditions of 1000 W m−2, 25 °C and air mass 1.5). | ||
Along with efficiency, long-term stability, particularly at high temperatures, has been a major challenge in the last decade.209 However, most if not all problems have feasible solutions on the horizon.210 DSCs have been tested to identify and solve the last remaining critical issues; latest endurance results under light soaking for thousands of hours report no significant loss of performance211 (recent experiments were performed in a 500 W DSC panel power station212).
 | ||
| Fig. 10 A DSC lightweight and portable mobile phone charger (courtesy of G24i). | ||
In the end it will be the positive combination of efficiency and stability which will determine the commercial success of DSCs. The start of significant market exploitation is on the way. Companies such as Aisin (Japan), Greatcell (Switzerland), Dyesol (Australia) and G24 Innovations (UK), a Konarka (USA) subsidiary, are manufacturing and commercializing – or intend to commercialize – dye-sensitized cellsFig. 10; and new companies will most certainly appear in the very near future. Notably, the Swiss company Swatch is planning to launch a DSC powered watch.213 Fortunately for the environment a great deal of optimism is moving the DSC concept forward.
Acknowledgements
Luís Gonçalves and Helena Aguilar Ribeiro wish to acknowledge the Portuguese Foundation for Science and Technology (FCT) for their PhD and Postdoctoral grants (SFRH/BD/36791/2007 and SFRH/BPD/36992/2007, respectively).References
- International Energy Outlook 2008, Energy Information Administration, 2008, http://www.eia.doe.gov/oiaf/ieo/index.html.
- Newsletter no. 83, The Association for the Study of Peak Oil and Gas, Nov 2007, http://www.aspo-ireland.org/index.cfm/page/newsletter.
- Climate Change 2007, Fourth Assessment Report, Intergovernmental Panel on Climate Change, 2007, http://www.ipcc.ch/ipccreports/assessments-reports.htm.
- W. Hoffmann, Sol. Energy Mater. Sol. Cells, 2006, 90, 3285–3311 CrossRef CAS.
- Renewables in Global Energy Supply, International Energy Agency, 2007, http://www.iea.org/Textbase/publications/free_new_DESC.asp?PUBS_ID=1596.
- Renewable Energy - Market and Policy Trends in IEA Countries, International Energy Agency, 2004, http://www.iea.org/Textbase/publications/free_new_DESC.asp?PUBS_ID=1263.
- World in Transition - Towards Sustainable Energy Systems, German Advisory Council on Global Change, 2003, http://www.wbgu.de/wbgu_jg2003_kurz_engl.html.
- D. Chapin, C. Fuller and G. Pearson, J. Appl. Phys., 1954, 25, 676–677 CAS.
- A. Goetzberger, J. Luther and G. Willeke, Sol. Energy Mater. Sol. Cells, 2002, 74, 1–11 CrossRef CAS.
- S. Pizzini, M. Acciarri and S. Binetti, Phys. Status Solidi A, 2005, 202, 2928–2942 CrossRef CAS.
- P. Woditsch and W. Koch, Sol. Energy Mater. Sol. Cells, 2002, 72, 11–26 CrossRef CAS.
- N. Dhere, Sol. Energy Mater. Sol. Cells, 2007, 91, 1376–1382 CrossRef CAS.
- D. Hopwood, Refocus, 2007, vol. 8, 54–56, 58–59 Search PubMed.
- J. Zhao, A. Wang, M. Green and F. Ferrazza, Appl. Phys. Lett., 1998, 73, 1991–1993 CrossRef CAS.
- D. Carlson and C. Wronski, Appl. Phys. Lett., 1976, 28, 671–673 CrossRef CAS.
- D. Staebler and C. Wronski, Appl. Phys. Lett., 1977, 31, 292–294 CrossRef CAS.
- T. Hinuma, H. Kasai, H. Tanimoto, M. Yamanaka, I. Sakata and H. Mizubayashi, Mater. Sci. Eng., A, 2006, 442, 302–306 CrossRef.
- M. Green, P. Basore, N. Chang, D. Clugston, R. Egan, R. Evans, D. Hogg, S. Jarnason, M. Keevers, P. Lasswell, J. O'Sullivan, U. Schubert, A. Turner, S. Wenham and T. Young, Sol. Energy, 2004, 77, 857–863 CrossRef CAS.
- A. Goetzberger and V. Hoffmann, Photovoltaic Solar Energy Generation, Springer-Verlag, Berlin/Heidelberg, 2005, xii, p. 3http://www.springer.com/physics/optics/book/978-3-540-23676-4 Search PubMed.
- D. Cusano, Solid-State Electron., 1963, 6, 217–218 CrossRef CAS.
- M. Green, K. Emery, Y. Hishikawa and W. Warta, Prog. Photovoltaics Res. Appl., 2008, 16, 435–440 Search PubMed.
- A. Morales-Acevedo, Sol. Energy Mater. Sol. Cells, 2006, 90, 2213–2220 CrossRef CAS.
- D. Morgan, C. Shines, S. Jeter, M. Blazka, M. Elwell, R. Wilson, S. Ward, H. Price and P. Moskowitz, Toxicol. Appl. Pharmacol., 1997, 147, 399–410 CrossRef CAS.
- S. Cho, D. Maysinger, M. Jain, B. Roder, S. Hackbarth and F. Winnik, Langmuir, 2007, 23, 1974–1980 CrossRef CAS.
- L. Kazmerski, F. White and G. Morgan, Appl. Phys. Lett., 1976, 29, 268–270 CrossRef CAS.
- B. Dimmler and H. Schock, Prog. Photovoltaics Res. Appl., 1996, 4, 425–433 Search PubMed.
- A. Delahoy, L. Chen, M. Akhtar, B. Sang and S. Guo, Sol. Energy, 2004, 77, 785–793 CrossRef CAS.
- M. Bosi and C. Pelosi, Prog. Photovoltaics Res. Appl., 2007, 15, 51–68 Search PubMed.
- W. Ma, C. Yang, X. Gong, K. Lee and A. Heeger, Adv. Funct. Mater., 2005, 15, 1617–1622 CrossRef CAS.
- J. Kim, K. Lee, N. Coates, D. Moses, T. Nguyen, M. Dante and A. Heeger, Science, 2007, 317, 222–225 CrossRef CAS.
- G. Tulloch, J. Photochem. Photobiol., A, 2004, 164, 209–219 CrossRef CAS.
- H. Otaka, M. Kira, K. Yano, S. Ito, H. Mitekura, T. Kawata and F. Matsui, J. Photochem. Photobiol., A, 2004, 164, 67–73 CrossRef CAS.
- M. Grätzel, Inorg. Chem., 2005, 44, 6841–6851 CrossRef.
- K. Millington, K. Fincher and L. King, Sol. Energy Mater. Sol. Cells, 2007, 91, 1618–1630 CrossRef CAS.
- C. Kelly and G. Meyer, Coord. Chem. Rev., 2001, 211, 295–315 CrossRef CAS.
- H. Tributsch, Photochem. Photobiol., 1972, 16, 261–269 CAS.
- A. Fujishima and K. Honda, Nature, 1972, 238, 37–38 CAS.
- H. Tsubomura, M. Matsumura, Y. Nomura and T. Amamiya, Nature, 1976, 261, 402–403 CAS.
- T. Skotheim, US Pat., 4 190 950, 1980 Search PubMed.
- J. Desilvestro, M. Grätzel, L. Kavan, J. Moser and J. Augustynski, J. Am. Chem. Soc., 1985, 107, 2988–2990 CrossRef CAS.
- P. Liska, N. Vlachopoulos, M. Nazeeruddin, P. Comte and M. Grätzel, J. Am. Chem. Soc., 1988, 110, 3686–3687 CrossRef CAS.
- N. Vlachopoulos, P. Liska, J. Augustynski and M. Grätzel, J. Am. Chem. Soc., 1988, 110, 1216–1220 CrossRef CAS.
- B. O'Regan and M. Grätzel, Nature, 1991, 353, 737–740 CrossRef CAS.
- Global Market: Current & Next Generation Solar Cell & Related Material – Market Outlooks, Fuji-Keizai USA, 2007 Search PubMed.
- N. Rossier-Iten, PhD Thesis, École Polytechnique Fédérale de Lausanne #3457, p. 13, 2006.
- A. Kay, PhD Thesis, École Polytechnique Fédérale de Lausanne #1214, p. 15, 1994.
- R. Grunwald and H. Tributsch, J. Phys. Chem. B, 1997, 101, 2564–2575 CrossRef.
- A. Hagfeldt and M. Grätzel, Acc. Chem. Res., 2000, 33, 269–277 CrossRef CAS.
- M. Nazeeruddin, A. Kay, I. Rodicio, R. Humphry-Baker, E. Mueller, P. Liska, N. Vlachopoulos and M. Grätzel, J. Am. Chem. Soc., 1993, 115, 6382–6390 CrossRef CAS.
- M. Grätzel, Prog. Photovoltaics Res. Appl., 2000, 8, 171–185 Search PubMed.
- M. Nazeeruddin, P. Pechy, T. Renouard, S. Zakeeruddin, R. Humphry-Baker, P. Comte, P. Liska, L. Cevey, E. Costa, V. Shklover, L. Spiccia, G. Deacon, C. Bignozzi and M. Grätzel, J. Am. Chem. Soc., 2001, 123, 1613–1624 CrossRef CAS.
- M. Nazeeruddin, P. Péchy and M. Grätzel, Chem. Commun., 1997, 1705–1706 RSC.
- M. Grätzel, J. Photochem. Photobiol., C, 2003, 4, 145–153 CrossRef CAS.
- T. Renouard, R. Fallahpour, M. Nazeeruddin, R. Humphry-Baker, S. Gorelsky, A. Lever and M. Grätzel, Inorg. Chem., 2002, 41, 367–378 CrossRef CAS.
- P. Wang, S. Zakeeruddin, J. Moser, M. Nazeeruddin, T. Sekiguchi and M. Grätzel, Nat. Mater., 2003, 2, 402–407 CrossRef CAS.
- P. Wang, S. Zakeeruddin, J. Moser, R. Humphry-Baker, P. Comte, V. Aranyos, A. Hagfeldt, M. Nazeeruddin and M. Grätzel, Adv. Mater., 2004, 16, 1806–1811 CrossRef CAS.
- P. Wang, C. Klein, R. Humphry-Baker, S. Zakeeruddin and M. Grätzel, J. Am. Chem. Soc., 2005, 127, 808–809 CrossRef CAS.
- J. Kroon, N. Bakker, H. Smit, P. Liska, K. Thampi, P. Wang, S. Zakeeruddin, M. Grätzel, A. Hinsch, S. Hore, U. Würfel, R. Sastrawan, J. Durrant, E. Palomares, H. Pettersson, T. Gruszecki, J. Walter, K. Skupien and G. Tulloch, Prog. Photovoltaics Res. Appl., 2007, 15, 1–18 Search PubMed.
- F. Gao, Y. Wang, J. Zhang, D. Shi, M. Wang, R. Humphry-Baker, P. Wang, S. Zakeeruddin and M. Grätzel, Chem. Commun., 2008, 2635–2637 RSC.
- F. Kong, S. Dai and K. Wang, Adv. Optoelectron., 2007, 2007, 75384 Search PubMed.
- M. Nazeeruddin, R. Humphry-Baker, D. Officer, W. Campbell, A. Burrell and M. Grätzel, Langmuir, 2004, 20, 6514–6517 CrossRef CAS.
- P. Reddy, L. Giribabu, C. Lyness, H. Snaith, C. Vijaykumar, M. Chandrasekharam, M. Lakshmikantam, J. Yum, K. Kalyanasundaram, M. Grätzel and M. Nazeeruddin, Angew. Chem., Int. Ed., 2007, 46, 373–376 CrossRef CAS.
- W. Campbell, K. Jolley, P. Wagner, K. Wagner, P. Walsh, K. Gordon, L. Schmidt-Mende, M. Nazeeruddin, Q. Wang, M. Gratzel and D. Officer, J. Phys. Chem. C, 2007, 111, 11760–11762 CrossRef CAS.
- H. Choi, J. Lee, K. Song, S. Kang and J. Ko, Tetrahedron, 2007, 63, 3115–3121 CrossRef CAS.
- S. Kim, J. Lee, S. Kang, J. Ko, J. Yum, S. Fantacci, F. DeAngelis, D. DiCenso, M. Nazeeruddin and M. Grätzel, J. Am. Chem. Soc., 2006, 128, 16701–16707 CrossRef CAS.
- I. Jung, J. Lee, K. Song, K. Song, S. Kang and J. Ko, J. Org. Chem., 2007, 72, 3652–3658 CrossRef CAS.
- T. Horiuchi, H. Miura and S. Uchida, J. Photochem. Photobiol., A, 2004, 164, 29–32 CrossRef CAS.
- T. Horiuchi, H. Miura, K. Sumioka and S. Uchida, J. Am. Chem. Soc., 2004, 126, 12218–12219 CrossRef CAS.
- S. Ito, S. Zakeeruddin, R. Humphry-Baker, P. Liska, R. Charvet, P. Comte, M. Nazeeruddin, P. Péchy, M. Takata, H. Miura, S. Uchida and M. Grätzel, Adv. Mater., 2006, 18, 1202–1205 CrossRef CAS.
- T. Horiuchi, H. Miura and S. Uchida, Chem. Commun., 2003, 3036–3037 RSC.
- S. Ferrere and B. Gregg, New J. Chem., 2002, 26, 1155–1160 RSC.
- K. Hara, M. Kurashige, Y. Dan-oh, C. Kasada, A. Shinpo, S. Suga, K. Sayama and H. Arakawa, New J. Chem., 2003, 27, 783–785 RSC.
- Z. Wang, Y. Cui, K. Hara, Y. Dan-oh, C. Kasada and A. Shinpo, Adv. Mater., 2007, 19, 1138–1141 CrossRef CAS.
- K. Hara, Y. Tachibana, Y. Ohga, A. Shinpo, S. Suga, K. Sayama, H. Sugihara and H. Arakawa, Sol. Energy Mater. Sol. Cells, 2003, 77, 89–103 CrossRef CAS.
- J. Thomas, J. Lin, Y. Hsu and K. Ho, Chem. Commun., 2005, 4098–4100 RSC.
- H. Choi, J. Lee, K. Song, K. Song, S. Kang and J. Ko, Tetrahedron, 2007, 63, 1553–1559 CrossRef CAS.
- K. Hara, M. Kurashige, S. Ito, A. Shinpo, S. Suga, K. Sayama and H. Arakawa, Chem. Commun., 2003, 252–253 RSC.
- T. Kitamura, M. Ikeda, K. Shigaki, T. Inoue, N. Anderson, X. Ai, T. Lian and S. Yanagida, Chem. Mater., 2004, 16, 1806–1812 CrossRef CAS.
- Z. Wang, F. Li and C. Huang, J. Phys. Chem. B, 2001, 105, 9210–9217 CrossRef CAS.
- K. Sayama, S. Tsukagoshi, K. Hara, Y. Ohga, A. Shinpou, Y. Abe, S. Suga and H. Arakawa, J. Phys. Chem. B, 2002, 106, 1363–1371 CrossRef CAS.
- K. Sayama, K. Hara, H. Sugihara, H. Arakawa, N. Mori, M. Satsuki, S. Suga, S. Tsukagoshi and Y. Abe, Chem. Commun., 2000, 1173–1174 RSC.
- N. Cherepy, G. Smestad, M. Grätzel and J. Zhang, J. Phys. Chem. B, 1997, 101, 9342–9351 CrossRef CAS.
- S. Li, K. Jiang, K. Shao and L. Yang, Chem. Commun., 2006, 2792–2794 RSC.
- Z. Wang, K. Sayama and H. Sugihara, J. Phys. Chem. B, 2005, 109, 22449–22455 CrossRef CAS.
- Z. Wang, F. Li, C. Huang, L. Wang, M. Wei, L. Jin and N. Li, J. Phys. Chem. B, 2000, 104, 9676–9682 CrossRef CAS.
- J. Yum, P. Walter, S. Huber, D. Rentsch, T. Geiger, F. Nuesch, F. DeAngelis, M. Gratzel and M. Nazeeruddin, J. Am. Chem. Soc., 2007, 129, 10320–10321 CrossRef CAS.
- Q. Yao, L. Shan, F. Li, D. Yin and C. Huang, New J. Chem., 2003, 27, 1277–1283 RSC.
- X. Ma, J. Hua, W. Wu, Y. Jin, F. Meng, W. Zhan and H. Tian, Tetrahedron, 2008, 64, 345–350 CrossRef CAS.
- I. Fleming, Nature, 1967, 216, 151–152 CrossRef CAS.
- C. Tang and A. Albrecht, J. Chem. Phys., 1975, 62, 2139–2149 CrossRef CAS.
- M. Mabrouki, A. Oueriagli, A. Outzourhit, E. Ameziane, S. Hotchandani and R. LeBlanc, Phys. Status Solidi A, 2002, 191, 345–354 CrossRef CAS.
- Y. Amao, Y. Yamada and K. Aoki, J. Photochem. Photobiol., A, 2004, 164, 47–51 CrossRef CAS.
- Y. Amao and K. Kato, Electrochim. Acta, 2007, 53, 42–45 CrossRef CAS.
- M. Balanay, C. Dipaling, S. Lee, D. Kim and K. Lee, Sol. Energy Mater. Sol. Cells, 2007, 91, 1775–1781 CrossRef CAS.
- K. Sayama, S. Tsukagoshi, T. Mori, K. Hara, Y. Ohga, A. Shinpou, Y. Abe, S. Suga and H. Arakawa, Sol. Energy Mater. Sol. Cells, 2003, 80, 47–71 CrossRef CAS.
- L. Giribabu, V. Kumar, V. Reddy, P. Reddy, S. Rao, S. Jang, J. Yum, M. Nazeeruddin and M. Grätzel, Sol. Energy Mater. Sol. Cells, 2007, 91, 1611–1617 CrossRef CAS.
- S. Ferrere, A. Zaban and B. Gregg, J. Phys. Chem. B, 1997, 101, 4490–4493 CrossRef CAS.
- C. Li, X. Yang, R. Chen, J. Pan, H. Tian, H. Zhu, X. Wang, A. Hagfeldt and L. Sun, Sol. Energy Mater. Sol. Cells, 2007, 91, 1863–1871 CrossRef CAS.
- G. Senadeera, P. Jayaweera, V. Perera and K. Tennakone, Sol. Energy Mater. Sol. Cells, 2002, 73, 103–108 CrossRef.
- M. Liang, W. Xu, F. Cai, P. Chen, B. Peng, J. Chen and Z. Li, J. Phys. Chem. C, 2007, 111, 4465–4472 CrossRef CAS.
- H. Tian, X. Yang, R. Chen, R. Zhang, A. Hagfeldt and L. Sun, J. Phys. Chem. C, 2008, 112, 11023–11033 CrossRef CAS.
- K. Hara, T. Sato, R. Katoh, A. Furube, T. Yoshihara, M. Murai, M. Kurashige, S. Ito, A. Shinpo, S. Suga and H. Arakawa, Adv. Funct. Mater., 2005, 15, 246–252 CrossRef CAS.
- W. Zhao, Y. Hou, X. Wang, B. Zhang, Y. Cao, R. Yang, W. Wang and X. Xiao, Sol. Energy Mater. Sol. Cells, 1999, 58, 173–183 CrossRef CAS.
- J. Clifford, E. Palomares, M. Nazeeruddin, R. Thampi, M. Grätzel and J. Durrant, J. Am. Chem. Soc., 2004, 126, 5670–5671 CrossRef.
- A. Kay, PhD Thesis, École Polytechnique Fédérale de Lausanne #1214, p. 99, 1994.
- K. Sayama, H. Sugihara and H. Arakawa, Chem. Mater., 1998, 10, 3825–3832 CrossRef CAS.
- K. Kalyanasundaram and M. Grätzel, Coord. Chem. Rev., 1998, 177, 347–414 CrossRef CAS.
- S. Deb, Sol. Energy Mater. Sol. Cells, 2005, 88, 1–10 CrossRef CAS.
- N. Park, J. Lagemaat and A. Frank, J. Phys. Chem. B, 2000, 104, 8989–8994 CrossRef CAS.
- A. Kitiyanan and S. Yoshikawa, Mater. Lett., 2005, 59, 4038–4040 CrossRef CAS.
- K. Tennakone, G. Kumara, I. Kottegoda and V. Perera, Chem. Commun., 1999, 15–16 RSC.
- R. Plass, PhD Thesis, École Polytechnique Fédérale de Lausanne #3017, p. 38, 2004.
- Z. Wang, H. Kawauchi, T. Kashima and H. Arakawa, Coord. Chem. Rev., 2004, 248, 1381–1389 CrossRef CAS.
- K. Zhu, E. Schiff, N. Park, J. Lagemaat and A. Frank, Appl. Phys. Lett., 2002, 80, 685–687 CrossRef CAS.
- J. Hart, D. Menzies, Y. Cheng, G. Simon and L. Spiccia, C. R. Chim., 2006, 9, 622–626 CrossRef CAS.
- S. Ito, K. Ishikawa, C. Wen, S. Yoshida and T. Watanabe, Bull. Chem. Soc. Jpn., 2000, 73, 2609–2614 CrossRef CAS.
- A. Burke, S. Ito, H. Snaith, U. Bach, J. Kwiatkowski and M. Grätzel, Nano Lett., 2008, 8, 977–981 CrossRef CAS.
- S. Ito, P. Liska, P. Comte, R. Charvet, P. Pechy, U. Bach, L. Schmidt-Mende, S. Zakeeruddin, A. Kay, M. Nazeeruddin and M. Grätzel, Chem. Commun., 2005, 4351–4353 RSC.
- S. Ito, T. Murakami, P. Comte, P. Liska, C. Grätzel, M. Nazeeruddin and M. Grätzel, Thin Solid Films, 2008, 516, 4613–4619 CrossRef CAS.
- Z. Zhang, S. Ito, B. O'Regan, D. Kuang, S. Zakeeruddin, P. Liska, R. Charvet, P. Comte, M. Nazeeruddin, P. Péchy, R. Humphry-Baker, T. Koyanagi, T. Mizuno and M. Grätzel, Z. Phys. Chem., 2007, 221, 319–327 CrossRef CAS.
- H. Koo, J. Park, B. Yoo, K. Yoo, K. Kim and N. Park, Inorg. Chem., 2008, 361, 677–683 CAS.
- G. Mor, K. Shankar, M. Paulose, O. Varghese and C. Grimes, Nano Lett., 2006, 6, 215–218 CrossRef CAS.
- K. Zhu, N. Neale, A. Miedaner and A. Frank, Nano Lett., 2007, 7, 69–74 CrossRef CAS.
- M. Wei, Y. Konishi, H. Zhou, H. Sugihara and H. Arakawa, J. Electrochem. Soc., 2006, 153, A1232–A1236 CrossRef CAS.
- J. Park, T. Lee and M. Kang, Chem. Commun., 2008, 2867–2869 RSC.
- M. Adachi, Y. Murata, J. Takao, J. Jiu, M. Sakamoto and F. Wang, J. Am. Chem. Soc., 2004, 126, 14943–14949 CrossRef CAS.
- J. Jiu, S. Isoda, F. Wang and M. Adachi, J. Phys. Chem. B, 2006, 110, 2087–2092 CrossRef CAS.
- L. Greene, B. Yuhas, M. Law, D. Zitoun and P. Yang, Inorg. Chem., 2006, 45, 7535–7543 CrossRef.
- C. Huisman, J. Schoonman and A. Goossens, Sol. Energy Mater. Sol. Cells, 2005, 85, 115–124 CAS.
- C. Kuo and S. Lu, Nanotechnology, 2008, 19, 095705 CrossRef.
- S. Iijima, Nature, 1991, 354, 56–58 CrossRef CAS.
- M. Huang, S. Mao, H. Feick, H. Yan, Y. Wu, H. Kind, E. Weber, R. Russo and P. Yang, Science, 2001, 292, 1897–1899 CrossRef CAS.
- T. Kasuga, M. Hiramatsu, A. Hoson, T. Sekino and K. Niihara, Langmuir, 1998, 14, 3160–3163 CrossRef CAS.
- J. Fan, S. Boettcher and G. Stucky, Chem. Mater., 2006, 18, 6391–6396 CrossRef CAS.
- S. Uchida, R. Chiba, M. Tomiha, N. Masaki and M. Shirai, Electrochemistry, 2002, 70, 418–420 CAS.
- M. Adachi, I. Okada, S. Ngamsinlapasathian, Y. Murata and S. Yoshikawa, Electrochemistry, 2002, 70, 449–452 CAS.
- M. Law, L. Greene, J. Johnson, R. Saykally and P. Yang, Nat. Mater., 2005, 4, 455–459 CrossRef CAS.
- D. Kuang, J. Brillet, P. Chen, M. Takata, S. Uchida, H. Miura, K. Sumioka, S. Zakeeruddin and M. Grätzel, ACS Nano, 2008, 2, 1113–1116 CrossRef CAS.
- H. Nusbaumer, PhD Thesis, École Polytechnique Fédérale de Lausanne #2955, p. 24, 2004.
- M. Zanni, J. Greenblatt, A. Davis and D. Neumark, J. Chem. Phys., 1999, 111, 2991–3003 CrossRef CAS.
- H. Tributsch, Coord. Chem. Rev., 2004, 248, 1511–1530 CrossRef CAS.
- M. Yanagida, T. Yamaguchi, M. Kurashige, K. Hara, R. Katoh, H. Sugihara and H. Arakawa, Inorg. Chem., 2003, 42, 7921–7931 CrossRef CAS.
- G. Boschloo, L. Haggman and A. Hagfeldt, J. Phys. Chem. B, 2006, 110, 13144–13150 CrossRef CAS.
- A. Kay and M. Grätzel, Sol. Energy Mater. Sol. Cells, 1996, 44, 99–117 CrossRef CAS.
- E. Figgemeier and A. Hagfeldt, Int. J. Photoenergy, 2004, 6, 127–140 CrossRef CAS.
- T. Hamann, R. Jensen, A. Martinson, H. Ryswyk and J. Hupp, Energy Environ. Sci., 2008, 1, 66–78 RSC.
- R. Argazzi, C. Bignozzi, T. Heimer, F. Castellano and G. Meyer, J. Phys. Chem. B, 1997, 101, 2591–2597 CrossRef CAS.
- G. Oskam, B. Bergeron, G. Meyer and P. Searson, J. Phys. Chem. B, 2001, 105, 6867–6873 CrossRef CAS.
- B. Bergeron, A. Marton, G. Oskam and G. Meyer, J. Phys. Chem. B, 2005, 109, 937–943 CrossRef CAS.
- H. Nusbaumer, S. Zakeeruddin, J. Moser and M. Grätzel, Chem.–Eur. J., 2003, 9, 3756–3763 CrossRef CAS.
- S. Sapp, C. Elliott, C. Contado, S. Caramori and C. Bignozzi, J. Am. Chem. Soc., 2002, 124, 11215–11222 CrossRef CAS.
- P. Cameron, L. Peter, S. Zakeeruddin and M. Grätzel, Coord. Chem. Rev., 2004, 248, 1447–1453 CrossRef CAS.
- T. Ma, X. Fang, M. Akiyama, K. Inoue, H. Noma and E. Abe, J. Electroanal. Chem., 2004, 574, 77–83 CrossRef CAS.
- Y. Saito, W. Kubo, T. Kitamura, Y. Wada and S. Yanagida, J. Photochem. Photobiol., A, 2004, 164, 153–157 CrossRef CAS.
- W. Campbell, A. Burrell, D. Officer and K. Jolley, Coord. Chem. Rev., 2004, 248, 1363–1379 CrossRef CAS.
- S. Ito, M. Nazeeruddin, P. Liska, P. Comte, R. Charvet, P. Péchy, M. Jirousek, A. Kay, M. Zakeeruddin and M. Grätzel, Prog. Photovoltaics Res. Appl., 2006, 14, 589–601 Search PubMed.
- A. Stanley, B. Verity and D. Matthews, J. Photochem. Photobiol., C, 1998, 52, 141–154 CAS.
- M. Gorlov and L. Kloo, Dalton Trans., 2008, 2655–2666 RSC.
- P. Wang, S. Zakeeruddin, J. Moser and M. Grätzel, J. Phys. Chem. B, 2003, 107, 13280–13285 CrossRef CAS.
- N. Papageorgiou, Y. Athanassov, M. Armand, P. Bonhote, H. Pettersson, A. Azam and M. Grätzel, J. Electrochem. Soc., 1996, 143, 3099–3108 CAS.
- W. Kubo, T. Kitamura, K. Hanabusa, Y. Wada and S. Yanagida, Chem. Commun., 2002, 374–375 RSC.
- M. Gorlov, H. Pettersson, A. Hagfeldt and L. Kloo, Inorg. Chem., 2007, 46, 3566–3575 CrossRef CAS.
- Y. Bai, Y. Cao, J. Zhang, M. Wang, R. Li, P. Wang, S. Zakeeruddin and M. Grätzel, Nat. Mater., 2008, 7, 626–630 CrossRef CAS.
- D. Kuang, S. Uchida, R. Humphry-Baker, Shaik Zakeeruddin and M. Grätzel, Angew. Chem., 2008, 120, 1949–1953 CrossRef.
- P. Wang, S. Zakeeruddin, I. Exnar and M. Grätzel, Chem. Commun., 2002, 2972–2973 RSC.
- P. Wang, S. Zakeeruddin, P. Comte, I. Exnar and M. Grätzel, J. Am. Chem. Soc., 2003, 125, 1166–1167 CrossRef CAS.
- P. Wang, S. Zakeeruddin, R. Humphry-Baker and M. Grätzel, Chem. Mater., 2004, 16, 2694–2696 CrossRef CAS.
- P. Wang, S. Zakeeruddin, J. Moser, R. Humphry-Baker and M. Grätzel, J. Am. Chem. Soc., 2004, 126, 7164–7165 CrossRef CAS.
- P. Wang, B. Wenger, R. Humphry-Baker, J. Moser, J. Teuscher, W. Kantlehner, J. Mezger, E. V. Stoyanov, S. Zakeeruddin and M. Grätzel, J. Am. Chem. Soc., 2005, 127, 6850–6856 CrossRef.
- D. Kuang, P. Wang, S. Ito, S. Zakeeruddin and M. Grätzel, J. Am. Chem. Soc., 2006, 128, 7732–7733 CrossRef CAS.
- D. Kuang, C. Klein, Z. Zhang, S. Ito, J. Moser, S. Zakeeruddin and M. Grätzel, Small, 2007, 3, 2094–2102 CrossRef CAS.
- K. Tennakone, G. Kumara, A. Kumarasinghe, K. Wijayantha and P. Sirimanne, Semicond. Sci. Tech., 1995, 10, 1689–1693 Search PubMed.
- B. O'Regan and D. Schwartz, Chem. Mater., 1995, 7, 1349–1354 CrossRef CAS.
- J. Hagen, W. Schaffrath, P. Otschik, R. Fink, A. Bacher, H. Schmidt and D. Haarer, Synth. Met., 1997, 89, 215–220 CrossRef CAS.
- U. Bach, D. Lupo, P. Comte, J. Moser, F. Weissortel, J. Salbeck, H. Spreitzer and M. Grätzel, Nature, 1998, 395, 583–585 CrossRef.
- K. Tennakone, G. Kumara, I. Kottegoda, K. Wijayantha and V. Perera, J. Phys. D: Appl. Phys., 1998, 31, 1492–1496 CrossRef CAS.
- K. Tennakone, V. Perera, I. Kottegoda and G. Kumara, J. Phys. D: Appl. Phys., 1999, 32, 374–379 CrossRef CAS.
- K. Tennakone, G. Senadeera, D. Silva and I. Kottegoda, Appl. Phys. Lett., 2000, 77, 2367–2369 CrossRef CAS.
- B. Li, L. Wang, B. Kang, P. Wang and Y. Qiu, Sol. Energy Mater. Sol. Cells, 2006, 90, 549–573 CrossRef CAS.
- L. Schmidt-Mende, U. Bach, R. Humphry-Baker, T. Horiuchi, H. Miura, S. Ito, S. Uchida and M. Grätzel, Adv. Mater., 2005, 17, 813–815 CrossRef.
- J. Krüger, R. Plass, L. Cevey, M. Piccirelli, M. Grätzel and U. Bach, Appl. Phys. Lett., 2001, 79, 2085–2087 CrossRef CAS.
- J. Krüger, R. Plass, M. Grätzel and H. Matthieu, Appl. Phys. Lett., 2002, 81, 367–369 CrossRef CAS.
- Q. Meng, K. Takahashi, X. Zhang, I. Sutanto, T. Rao, O. Sato, A. Fujishima, H. Watanabe, T. Nakamori and M. Uragami, Langmuir, 2003, 19, 3572–3574 CrossRef CAS.
- M. Grätzel, MRS Bull., 2005, 30, 23–27 CAS.
- H. Wang, H. Li, B. Xue, Z. Wang, Q. Meng and L. Chen, J. Am. Chem. Soc., 2005, 127, 6394–6401 CrossRef CAS.
- J. Wu, S. Hao, Z. Lan, J. Lin, M. Huang, Y. Huang, P. Li, S. Yin and T. Sato, J. Am. Chem. Soc., 2008, 130, 11568–11569 CrossRef CAS.
- R. Gordon, MRS Bull., 2000, 25, 52–57 CAS.
- K. Goto, T. Kawashima and N. Tanabe, Sol. Energy Mater. Sol. Cells, 2006, 90, 3251–3260 CrossRef CAS.
- S. Ito, N. Ha, G. Rothenberger, P. Liska, P. Comte, S. Zakeeruddin, P. Pechy, M. Nazeeruddin and M. Grätzel, Chem. Commun., 2006, 4004–4006 RSC.
- T. Murakami, Y. Kijitori, N. Kawashima and T. Miyasaka, J. Photochem. Photobiol., A, 2004, 164, 187–191 CrossRef CAS.
- D. Gutierrez-Tauste, I. Zumeta, E. Vigil, M. Hernandez-Fenollosa, X. Domenech and J. Ayllon, J. Photochem. Photobiol., A, 2005, 175, 165–171 CrossRef.
- Y. Jun, J. Kim and M. Kang, Sol. Energy Mater. Sol. Cells, 2007, 91, 779–784 CrossRef CAS.
- K. Onoda, S. Ngamsinlapasathian, T. Fujieda and S. Yoshikawa, Sol. Energy Mater. Sol. Cells, 2007, 91, 1176–1181 CrossRef CAS.
- T. Wei, C. Wan and Y. Wang, Appl. Phys. Lett., 2006, 88, 103122–103123 CrossRef.
- J. Halme, MSc Thesis, Helsinki University of Technology, p. 53, 2002 and references within.
- N. Papageorgiou, W. Maier and M. Grätzel, J. Electrochem. Soc., 1997, 144, 876–884 CAS.
- T. Kitamura, M. Maitani, M. Matsuda, Y. Wada and S. Yanagida, Chem. Lett., 2001, 30, 1054–1055 CrossRef.
- T. Murakami and M. Grätzel, Inorg. Chim. Acta, 2008, 361, 572–580 CrossRef CAS.
- K. Imoto, K. Takahashi, T. Yamaguchi, T. Komura, J. Nakamura and K. Murata, Sol. Energy Mater. Sol. Cells, 2003, 79, 459–469 CrossRef CAS.
- K. Suzuki, M. Yamaguchi, M. Kumagai and S. Yanagida, Chem. Lett., 2003, 32, 28–29 CrossRef CAS.
- N. Ikeda, K. Teshima and T. Miyasaka, Chem. Commun., 2006, 1733–1735 RSC.
- J. Krüger, PhD Thesis, École Polytechnique Fédérale de Lausanne #2793, p. 17, 2003.
- E. Olsen, G. Hagen and S. Lindquist, Sol. Energy Mater. Sol. Cells, 2000, 63, 267–273 CrossRef CAS.
- S. Yanagida, C. R. Chim., 2006, 9, 597–604 CrossRef CAS.
- M. Grätzel, Curr. Opin. Colloid Interface Sci., 1999, 4, 314–321 CrossRef CAS.
- M. Grätzel, J. Photochem. Photobiol., A, 2004, 164, 3–14 CrossRef CAS.
- M. Nazeeruddin, F. DeAngelis, S. Fantacci, A. Selloni, G. Viscardi, P. Liska, S. Ito, B. Takeru and M. Grätzel, J. Am. Chem. Soc., 2005, 127, 16835–16847 CrossRef CAS.
- Y. Chiba, A. Islam, Y. Watanabe, R. Komiya, N. Koide and L. Han, Jpn J. Appl. Phys., 2006, 45, L638–L640 CrossRef CAS.
- A. Hinsch, J. Kroon, R. Kern, I. Uhlendorf, J. Holzbock, A. Meyer and J. Ferber, Prog. Photovoltaics Res. Appl., 2001, 9, 425–438 Search PubMed.
- M. Grätzel, C. R. Chim., 2006, 9, 578–583 CrossRef.
- P. Wang, C. Klein, R. Humphry-Baker, M. Zakeeruddin and M. Grätzel, Appl. Phys. Lett., 2005, 86, 123508 CrossRef.
- S. Dai, J. Weng, Y. Sui, S. Chen, S. Xiao, Y. Huang, F. Kong, X. Pan, L. Hu, C. Zhang and K. Wang, Inorg. Chim. Acta, 2008, 361, 786–791 CrossRef CAS.
- G. Marsh, Refocus, 2008, 9, 58–62 CrossRef.
| This journal is © The Royal Society of Chemistry 2008 |