Formation of stoichiometrically 18O-labelled oxygen from the oxidation of 18O-enriched water mediated by a dinuclear manganese complex—a mass spectrometry and EPR study†
Katrin
Beckmann‡
b,
Hannes
Uchtenhagen‡
a,
Gustav
Berggren
a,
Magnus F.
Anderlund
a,
Anders
Thapper
a,
Johannes
Messinger§
b,
Stenbjörn
Styring
*a and
Philipp
Kurz
*ac
aDepartment of Photochemistry and Molecular Science, Ångström Laboratory, Uppsala University, Box 523, 751 20, Uppsala, Sweden. E-mail: stenbjorn.styring@fotomol.uu.se; Fax: (+46) 18 471 6844
bMax-Planck-Institut für Bioanorganische Chemie, Stiftstraße 34–36, 45470, Mülheim an der Ruhr, Germany
cInstitut für Anorganische Chemie, Christian-Albrechts-Universität zu Kiel, Max-Eyth-Straße 2, 24118, Kiel, Germany. E-mail: phkurz@ac.uni-kiel.de; Fax: (+49) 431 880 1520
First published on 18th September 2008
Abstract
Oxygen formation was detected for the oxidations of various multinuclear manganese complexes by oxone (HSO5−) in aqueous solution. To determine to what extent water was the source of the evolved O2, H218O isotope-labelling experiments coupled with membrane inlet mass spectrometry (MIMS) were carried out. We discovered that during the reaction of oxone with [Mn2(OAc)2(bpmp)]+ (1), stoichiometrically labelled oxygen (18O2) was formed. This is the first example of a homogeneous reaction mediated by a synthetic manganese complex where the addition of a strong chemical oxidant yields 18O2 with labelling percentages matching the theoretically expected values for the case of both O-atoms originating from water. Experiments using lead acetate as an alternative oxidant supported this finding. A detailed investigation of the reaction by EPR spectroscopy, MIMS and Clark-type oxygen detection enabled us to propose potential reaction pathways.
Introduction
Mankind becomes more and more aware of the dangerous consequences of a potential climate change on earth. The observed rise in globally-averaged temperatures since the mid-20th century is very likely due to the increase in anthropogenic greenhouse gas concentrations in the atmosphere, mainly caused by the extensive combustion of fossil fuels.1 In consequence, researchers have turned their attention to the development of alternative methods to generate the large amounts of energy needed to meet global demand without the use of oil, natural gas or coal.Artificial photosynthesis, the transformation of solar energy into chemical energy, enjoys increasing attention in this context as a concept for alternative energy production.2–9 If successful, the product of such a process would be a “solar fuel” that could be oxidised by atmospheric oxygen to generate energy. Therefore the fuel, whatever it may be, will necessarily be formed in a reduction reaction for which reducing equivalents are needed.
| 2H2O→O2 + 4H+ + 4e− | (1) |
We, and others, have focused our attention on the possibility to use the oxidation of water (eqn (1)) as a possible electron source for artificial photosynthesis. This would provide both an unlimited energy source (solar energy) and raw material (water) for fuel production. We have adopted a biomimetic approach as we aim to copy the reactivity of the natural water oxidising complex (WOC) of photosystem II (PS II), the enzyme able to oxidise water in oxygenic photosynthesis using the energy of absorbed sunlight.8–10 The WOC is the only known example of an efficient homogeneous water oxidation catalyst. Its catalytic site for water oxidation is composed of a tetranuclear manganese complex and a calcium ion closely interacting with its protein surroundings.11–15 Numerous manganese compounds have been synthesized to mimic the manganese cluster of PS II. However, there is not one example of a synthetic manganese compound known today able to achieve true catalyticwater oxidation in homogeneous solution.16–19
Recently, we have carried out a systematic screening of di- and tetranuclear manganese complexes for their ability to act as catalysts for oxygen evolution from aqueous solutions. We studied the reactions of these compounds with various oxidation agents to identify conditions under which oxygen was formed.20
Several new reactions were found for which oxygen evolution was detected. We concluded that the few reactions reported so far in the literature21–23 were far from unique but rather examples of general reactivity patterns. It appears that many polynuclear manganese complexes are efficient catalysts for: (i) disproportionation reactions of H2O2 or organic peroxides, and (ii) oxygen evolution resulting from the reactions with oxygen transfer agents.
Here, we have carried out an investigation for some of the new reactions to learn more about possible oxygen formation mechanisms. The focus of our attention was on the question to what extent water was the source of the evolved oxygen. We identified one manganese mediated O2 forming reaction where most likely both oxygen atoms originate from the bulk water, indicating that water oxidation according to eqn (1) occurred. This process was studied in detail for various reaction conditions by the use of H218O labelling combined with membrane inlet mass spectrometry (MIMS), EPR spectroscopy and polarographic oxygen detection.
Results and discussion
Four of the complexes studied in our first investigation (1–4)20 are presented in Scheme 1. They differ in oxidation states (MnII, MnIII or MnIV), coordination number (6 or 7), nuclearity (di- or tetranuclear) and nature of the ligands (O-/N-ligands), and thus feature a wide range of chemical properties. In the original screening we found that all four complexes produced oxygen when oxidised by hydrogen peroxomonosulfate (oxone, HSO5−). However, no oxygen was detected when any of the complexes were reacted with CeIV or photochemically generated [RuIII(bipy)3]3+ (both non-oxygen-transferring oxidation agents), in contrast to recent reports.22,25 In consequence we suggested20 that oxygen transfer, resulting in manganyl intermediates, appears as key step on the way to oxygen formation, as also proposed in the literature for other manganese mediated reactions.21,26![Structures of the studied complexes: [Mn2(OAc)2(bpmp)]+ (1), [(Mn2(OTf)(OH2)(tphpn))2O2]3+ (2), [Mn2(OH2)2(mgbpen)2]2+ (3), [(Mn2O2(OH2)2(terpy)2]3+ (4) and [Mn2Cl2(OH2)2(bpmp)]+ (5). Abbreviation of ligands according to ref. 17 and 24.](/image/article/2008/EE/b811806j/b811806j-s1.gif) | ||
| Scheme 1 Structures of the studied complexes: [Mn2(OAc)2(bpmp)]+ (1), [(Mn2(OTf)(OH2)(tphpn))2O2]3+ (2), [Mn2(OH2)2(mgbpen)2]2+ (3), [(Mn2O2(OH2)2(terpy)2]3+ (4) and [Mn2Cl2(OH2)2(bpmp)]+ (5). Abbreviation of ligands according to ref. 17 and 24. | ||
Oxygen evolution detected by MIMS
To test this proposal, we carried out the oxidations of complexes 1–4 with oxone in presence of 18O-enriched water inside a cell that was connected via a Teflon-membrane to the vacuum of an isotope ratio mass spectrometer. The O2 formed in the solution passes through the Teflon membrane and oxygen molecules of different isotopic composition are separated and then simultaneously recorded in individual Faraday cups.22,27–29 A typical trace showing the parallel detection of16O2, 16O18O and 18O2 is shown in Fig. 1.![Oxygen evolution traces for the reaction of oxone with 3 detected by membrane inlet mass spectrometry (MIMS). [3] = 2 mM in argon-purged water (10% enriched in H218O); injection of 10 eq. HSO5− per Mn centre at t = 0 min. Oxygen concentrations used to calculate the data of Table 1 were taken for the time of maximal [O2] as indicated.](/image/article/2008/EE/b811806j/b811806j-f1.gif) | ||
| Fig. 1 Oxygen evolution traces for the reaction of oxone with 3 detected by membrane inlet mass spectrometry (MIMS). [3] = 2 mM in argon-purged water (10% enriched in H218O); injection of 10 eq. HSO5− per Mn centre at t = 0 min. Oxygen concentrations used to calculate the data of Table 1 were taken for the time of maximal [O2] as indicated. | ||
The fractions of the different oxygen isotopes formed were calculated from the MIMS traces for peak oxygen concentrations as indicated by arrows in Fig. 1. Results for all studied reactions are shown in Table 1. Especially important here is the ratio calculated in column 4 between the fraction of 18O-labelled water added (column 2) and Φ18O, the fraction of 18O atoms found in the evolved oxygen (column 3).
| Reaction | H218O added (%) | Φ 18O detecteda | Φ 18O / H218O added | O2 species detected (%) | ||
|---|---|---|---|---|---|---|
| 16O2 | 16O18O | 18O2 | ||||
| a Fraction of 18O atoms in the evolved O2 (%): Φ18O = ([18O2] + ½[16O18O])/[O2]total). b As expected for both oxygen atoms of the formed O2 originating from water. | ||||||
| 1 + HSO5− | 10 | 10.05 | 1.005 | 80.8 | 18.1 | 1.0 |
| 2 + HSO5− | 10 | 5.6 | 0.56 | 89.2 | 10.4 | 0.4 |
| 3 + HSO5− | 10 | 4.4 | 0.44 | 91.1 | 8.8 | 0.03 |
| 4 + HSO5− | 10 | 4.3 | 0.43 | 93.6 | 8.2 | 0.2 |
| 5 + HSO5− | 10 | 9.8 | 0.98 | 81.5 | 17.4 | 1.1 |
| 1 + PbIV | 10 | 8.3 | 0.83 | 84.2 | 14.9 | 0.8 |
| Theor. ratiob | 10 | 81.0 | 18.0 | 1.0 | ||
For reactions of complexes 2–4 with oxone, Φ18O is about half the added H218O percentage. This indicates that one oxygen atom of the produced O2 does not originate from the bulk water but from another source, most probably from the oxidant. This is not unexpected, as oxone is known to act as oxygen transfer agent to manganese.21,30 It has been suggested that the formation of singly 18O-labelled oxygen is the consequence of oxygen formation via a two step process: in a first step manganyl or manganese oxyl-radical intermediates are formed by the reaction of the manganese complexes with HSO5−. Water or hydroxide anions from the bulk then attack these intermediates in a second step to form O2.21,26 In this way maximally one of the oxygen atoms of the evolved O2 carries the label from H218O.
Reaction of complex 1 with oxone
Very interestingly, the initial phase of the reaction of 1 with oxone produced stoichiometrically 18O- enriched dioxygen. Fig. 2 shows the evolution of oxygen for 10% 18O- enriched water as detected by MIMS (top) and a parallel experiment where oxygen production was monitored by a Clark electrode (bottom). The measurements demonstrate that after injection of the oxone there was a burst of O2 formation with a much higher percentage of 18O2 formed than observed for the other complexes studied (e.g. as shown in Fig. 1). Analysis of the MIMS measurement revealed that the labelling percentage closely matched the theoretically expected value for a reaction where bothoxygen atoms in the formed O2 originate from the bulk H2O (Table 1). The reaction phase producing stoichiometrically labelled oxygen generates a peak concentration of about 50 µM O2 in solution for the standard reaction conditions. Consequently, the formation of oxygen only involves a fraction of the amount of 1 in solution and is not catalytic. However, as discussed later, the oxygen formation might be underestimated as side reactions could consume a considerable amount of the formed O2.![Oxygen evolution traces for reactions of oxone with 1 detected by MIMS (top) and Clark electrode (bottom). [1] = 2 mM in H2O/MeCN at 3 : 1; injection of 10 eq. HSO5− per Mn centre at t = 0 min, water 10% enriched in H218O was used in the MIMS experiment (top).](/image/article/2008/EE/b811806j/b811806j-f2.gif) | ||
| Fig. 2 Oxygen evolution traces for reactions of oxone with 1 detected by MIMS (top) and Clark electrode (bottom). [1] = 2 mM in H2O/MeCN at 3 : 1; injection of 10 eq. HSO5− per Mn centre at t = 0 min, water 10% enriched in H218O was used in the MIMS experiment (top). | ||
Thus, in the reaction of oxone with 1, water is most likely the source of both oxygen atoms of the evolved O2, which shows that oxidation of bulk water to molecular oxygen occurs. This has never been observed for a homogeneous manganese mediated reaction before. Other manganese compounds, including 2–4 studied here under identical reaction conditions, produce mostly unlabelled or singly labelled oxygen in reactions with oxone.21,30 The overall incorporation of the 18O label for these compounds is 60% or less (Table 1, column 4). Consequently, the traces of 18O2 are clearly substoichiometric for the respective enrichments – a result which is in accordance with previous studies on related compounds.21,22,30
Oxidation of 1 by oxone monitored by EPR spectroscopy
EPR spectroscopy is a powerful method to characterise manganese oxidation states.17,31,32 We added oxone to solutions of 1 under conditions identical to those of the O2 evolution experiments and stopped the reactions by freezing in liquid N2 at different times after the addition of HSO5−. Subsequently, the EPR spectra of the reaction mixtures were recorded (Fig. 3).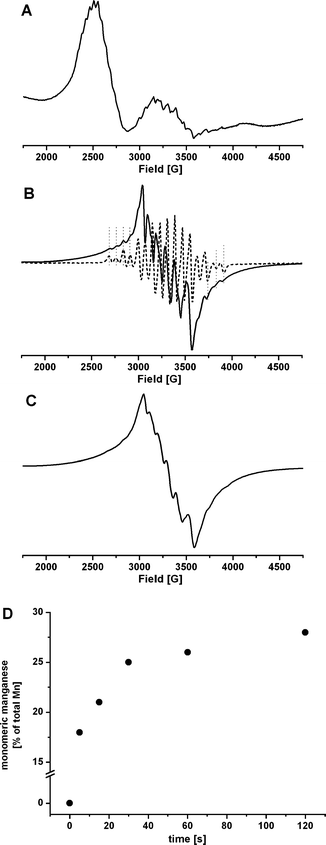 | ||
| Fig. 3 EPR spectra for 1 (2 mM in H2O/MeCN at 3 : 1): (A) before the addition of oxidant; (B) solid line: 5 s after the addition of oxone (10 eq. per Mn centre), dashed line: 1 oxidised to the Mn2III,IV state (see Fig. 4 for details); (C) 1 min after the addition of oxone (10 eq.); (D) quantification of the six-line EPR signal from monomeric manganese species over time after the addition of oxone. EPR measurement conditions: temperature: 12 K (A), 5 K (B and C); microwave frequency: 9.58 GHz; modulation amplitude: 10 G; microwave power: 2 mW (A), 200 µW (B and C). | ||
Before the oxidant was added, the complex showed its well-known Mn2II,II EPR spectrum of 11 lines, centred at g≈ 2.5 and best observed at temperatures of around 12 K (Fig. 3A).31 During the initial phase of the reaction with oxone, this signal disappeared very fast and could not be detected any more even for samples with a reaction time of only 5 s. Instead the EPR spectrum recorded after a reaction time of 5 s is a mixture of two other components (Fig. 3B). It contains a six-line signal that dominates the central part of the spectrum. This originates from monomeric manganese species (see below). Additionally, the wings of the EPR spectrum reveal the presence of an underlying 16-line EPR signal. This spectrum is about 1220 G wide and characteristic for a di-µ-oxo- dimanganese core in the Mn2III,IV oxidation state.33 The oxidation of 1 to a Mn2III,IV species has been observed before34 and a Mn2III,IV EPR spectrum of 1 is shown as dotted line in Fig. 3B for comparison. Furthermore, µ-oxo- dimanganese species have also been detected for oxidised samples of 1 by EXAFS.35
After a reaction time of 1 min, the EPR spectrum is dominated by the large six-line EPR spectrum characteristic for monomeric MnII or MnIV (Fig. 3C). However, a quantification of the six-line signal showed that 2 min after the addition of the oxidant, only 30% of 1 was transformed into monomeric manganese species (Fig. 3D). As no other EPR signals could be observed, a large part of the complex must have been transformed into EPR invisible forms like Mn2III,III / Mn2IV,IV complexes or solid manganese oxides.
Influence of the solution pH
Oxone is synthesised as a mixed salt, 2KHSO5·KHSO4·K2SO4, and solutions of this salt are very acidic (pH ≈ 2) due to the presence of the hydrogen sulfate and hydrogen peroxomonosulfate anions. Additions of oxone are therefore always additions of an oxidation agent and an acid at the same time.To estimate the stability of 1 under acidic conditions, 10 eq. of KHSO4 were added to a solution of 1, resulting in an acidic solution of pH 2.8. Significant and fast decomposition of the complex was observed by EPR as a broadened six-line spectrum was recorded (Fig. S1†). We determined that a fraction of 60% of 1 is destroyed to monomeric manganese species within 1 min under these conditions.
When the reaction between oxone and 1 was carried out at less acidic conditions (125 mM acetate buffer, pH = 4.5) the six-line signal was not detected. Instead a characteristic Mn2III,IV 16-line spectrum with 25 ± 5 % of the compound in the Mn2III,IV oxidation state was found (Fig. 4).
 | ||
| Fig. 4 EPR spectrum of 1 (2 mM) in aqueous acetate buffer (125 mM, pH = 4.5, 25% MeCN) 1 min after an addition of oxone (10 eq. per Mn centre). EPR measurement conditions: temperature: 5 K; microwave frequency: 9.58 GHz; modulation amplitude: 10 G; microwave power: 200 µW. | ||
We conclude that a higher pH stabilizes the complex against oxidation damage, thereby allowing the observation of its Mn2III,IV form. However, employing acetate or phosphate buffers also prevents the formation of oxygen: no evolution of O2 was detected for reactions of 1 with even 50 eq. of oxone if the pH of the solution was kept at values above pH ≈ 3 by either of these buffers.
Multiple additions of oxone
As described above, a single addition of 10 eq. of oxone to 1 dissolved in 18O-labelled water/acetonitrile mixtures resulted in a short burst of stoichiometrically 18O-labelled O2 (Fig. 2). Consecutive additions of more HSO5− initiated the beginning of longer, very productive phases of oxygen production, lasting 4 to 6 min (Fig. 5).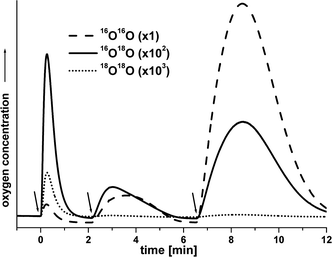 | ||
| Fig. 5 Sequential oxone additions (each 10 eq. per Mn centre) to a solution of 1 (2 mM in H2O/MeCN at 3 : 1, water 7% enriched in H218O). The arrows indicate the time of addition. | ||
MIMS analysis of these phases revealed that the oxygen formed for the second oxidant addition yields clearly less 18O-labeled oxygen species. The isotope label in the O2 formed after the third oxidant addition is similar to the isotopic distribution of oxygen species at natural 18O abundance, not reflecting the 18O enrichment of the water. (Fig. 5, Φ18O for 7% 18O enriched water: 1st addition: 7.3%, 2nd addition: 1.1%, 3rd addition: 0.4%). Therefore, the O2 formed after the second or third addition of oxone was produced by different processes than the O2 of the initial burst, which showed stoichiometric 18O labelling with both oxygen atoms originating from the bulk water.
Similar reactivity was observed when we increased the amount of oxone added in the first addition from 10 to 50 eq. per 1 (not shown). In this case, the reaction also proceeded in two phases: first an initial burst of oxygen, followed by a time span of about 30 s with no O2 evolution and then again a productive second phase, which lasted more than 10 min.
These results indicate that the initial reaction of 1 with oxone converts most of the complex to manganese species with entirely different properties. At least one of the reaction products appears to be able to catalyse the known disproportionation of oxone into sulfate and O2.36 As the oxone bears no 18O label, the product of this process is 16O2, which explains the lack of 18O label for the third peak of Fig. 5.
Reaction of oxone with manganese oxides
From the results of our EPR investigation we concluded that a large fraction of 1 is rapidly destroyed in the presence of oxone at the strongly oxidising and very acidic reaction conditions (Fig. 3D). Monomeric Mn2+ or Mn4+ ions or manganese oxides appear as likely decomposition products. We therefore investigated reactions of MnCl2 solutions or MnO2 suspensions with HSO5− to see if the disproportionation of oxone was catalysed by these compounds as well. We found that neither produced detectable amounts of O2 (not shown). The reaction of HSO5− with a suspension of Mn2O3, however, caused a slow but steady evolution of oxygen. In the presence of H218O, no labelled O2 was formed (Fig. S2†), so here the O2 is most likely the product of a disproportionation reaction of oxone catalysed by Mn2O3 particles. These observations also show that the 18O2 formed from labelled water during the reaction of 1 (Fig. 2) did not originate from reactions of Mn2+, MnO2 or Mn2O3 with oxone.Reaction of oxone with the analogous chloride complex 5
We investigated the reaction of 537 (Scheme 1), a chloride derivative of 1, with oxone, to judge whether the observed oxygen evolution is related to the presence of acetate ligands. This does not seem to be the case as complex 5 showed virtually identical reaction behaviour compared to 1. Oxygen formation was observed for reactions of 5 with oxone at similar rates as found for 1. In addition, and more importantly, it was found that stoichiometrically labelled O2 is the product of the reaction of HSO5− with 5 as well (Table 1). Complex 5 also showed the two O2 forming reaction phases observed for 1 if 50 eq. of HSO5− per Mn centre were added. These observations indicate that 1 and 5 most likely form O2via similar intermediates and manganese acetate complexes are not crucial for the key steps of the oxygen forming reaction pathway.CO2 formation, oxygen consumption
Our MIMS instrument allows monitoring of the masses 32, 34, 36, 40, 44, 46 and 48 (O2, Ar, CO2) simultaneously. We were therefore also able to follow the formation of carbon dioxide during reactions. It was found that large amounts of CO2 evolved for reactions of oxone with all complexes 1–5. The evolution of carbon dioxide was also detected for the reaction of oxone with Hbpmp, the heptadentate ligand of complexes 1 and 5, on its own (Hbpmp: 2,6-bis[[N,N-di(2-pyridylmethyl)amino]methyl]-4-methylphenol).To identify the source of the evolved CO2, the reaction was also carried out using complex 1*, identical to 1, but bearing singly 13C-enriched acetate ligands, H312C13COO−. The fraction of 13CO2 produced with oxone was significantly increased for reactions of 1* when compared to 1 (not shown), but far less than expected if the decarboxylation reaction of acetate was the only source of carbon dioxide. CO2 could also be formed from reactions of the Hbpmp ligand, especially if very reactive SO4·− radicals are formed from oxone in manganese mediated reactions as reported in the literature.38,39 Therefore, it seems that both acetate and the Hbpmp ligand in the reaction mixture undergo oxidative decompositions yielding CO2 in the presence of oxone.
Such decomposition reactions take place viaorganic radical intermediates, which are known to be very reactive towards molecular oxygen. Consequently, radical species as products of the organic decomposition processes are likely to consume parts of the initially formed O2. This was indeed found as a reaction of Hbpmp with oxone in an oxygen saturated solution resulted in a slow consumption of parts of the dissolved O2 (Fig. S3†). As a consequence, the amount of oxygen formed by the reaction of 1 with HSO5− (Fig. 2) is most likely larger than detected, but a part of the formed O2 is scavenged by reactions with the organic radicals.
Oxygen formation using PbIV as oxidation agent
We observed that O2 was formed from water as product of the reaction of 1 and 5 with oxone at very acidic conditions (Fig. 2, Table 1). However, we reported earlier that no oxygen was evolved if 1 was reacted with CeIV at pH ≈ 2.20 This is a surprising result: the labelling pattern shows that oxone does not act as oxygen atom source for the first reaction. Therefore oxone's well-known ability to act as oxygen donating agent cannot explain the differences in reactivity between the two oxidants. However, the key difference between the oxidation agents CeIV and oxone might be the ability of the latter to act as a two-electron oxidant. To test this hypothesis, we studied the reaction of 1, 3 and 4 with PbIV, another two-electron oxidation agent used at mildly acidic conditions (pH ≈ 4 for Pb(AcO)4).36 Additionally, PbIV has not been reported to act as an oxygen atom donor in oxidation reactions of this type.We detected the formation of oxygen if acetonitrile solutions of Pb(AcO)4 were added to 1 dissolved in H2O/MeCN. In contrast to this, no oxygen formation was detected for the reaction of Pb(AcO)4 with either complex 3 or 4. In all experiments where Pb(AcO)4 was used, the reaction mixture became a dark red-brown suspension within seconds after the addition of lead tetraacetate, as hydrolysis of PbIV caused the formation of insoluble PbO2.36 Most of the lead was therefore only available for the reaction for a very short time.
In the presence of H218O, oxygen enriched in 18O2 evolved. Our MIMS analysis of the isotope labelling pattern revealed the formation of a large percentage of 18O2 from H218O (Fig. 6, top). However, the 18O labelling of the O2 product was not entirely complete: for water enriched 10% in 18O, 8.3% labelling was found for the evolving oxygen (Table 2). The number is much too high to be explained by single labelling alone. From this observation we conclude that the 18O label from water is incorporated, but not stoichiometrically, as found before for the reaction of 1 with oxone. The fact that oxygen was produced at all in the reaction of 1 with PbIV is important as it provides very strong evidence that the ability of the oxidant to act as two-electron acceptor is critical for the initial phase of the oxygen forming reaction mediated by 1.
![Oxygen evolution traces for reactions of Pb(OAc)4 with 1 and 3 detected by MIMS (top, curves for 18O2 shown) and Clark electrode (bottom). [1] = [3] = 2 mM in H2O/MeCN at 3 : 1; injection of 10 eq. Pb(OAc)4 per Mn centre at t = 0 min, water 10% enriched in H218O was used in the MIMS experiments (top).](/image/article/2008/EE/b811806j/b811806j-f6.gif) | ||
| Fig. 6 Oxygen evolution traces for reactions of Pb(OAc)4 with 1 and 3 detected by MIMS (top, curves for 18O2 shown) and Clark electrode (bottom). [1] = [3] = 2 mM in H2O/MeCN at 3 : 1; injection of 10 eq. Pb(OAc)4 per Mn centre at t = 0 min, water 10% enriched in H218O was used in the MIMS experiments (top). | ||
Exchange reactions of the 18O label
It has been mentioned in previous studies40–42 that potential exchange reactions between the 18O label of the water and other molecules containing oxygen atoms could occur, thereby making the analysis of 18O labelling experiments less conclusive. There are relatively few studies of these exchange reactions. A spectroscopic study concerning the 18O exchange of oxone with bulk water established that oxygen exchange of HSO5− is too slow to influence the labelling percentages observed here.42We also considered the possibility that an oxygen transfer from oxone to 1 initially generates a manganese 16O-oxo species which is able to exchange with the 18O of the bulk water. This has been extensively discussed and studied before for similar cases and cannot be ruled out completely here.40–42 However, it appears very unlikely to us that this reaction pathway can explain our data for the following reasons:
• All other manganese complexes for which 18O labelling was studied (here and previously21,22,30) show incomplete labelling, indicating slow (or no) exchange on the experimental timescale. There is no obvious reason why only an oxo species formed after oxidation of compound 1 should undergo rapid exchange while all other compounds exchange their oxo-16O atoms slowly or not at all.
• There are critical differences in the reactivity of 1 when compared to that of compound 3 or 4. For reactions of the compounds with oxone we found stoichiometrically 18O-enriched dioxygen for 1, but only single labelling with 3 or 4. This could have been explained by different exchange rates for the oxo-ligands coordinated to the manganese. But then we detect the formation of almost stoichiometrically labelled dioxygen for the oxidations of 1 by PbIV and no O2 for the reaction of lead with 3 or 4. Taken together, these results strongly suggest that 1 is able to form oxygen from water after two 2e−oxidations without the need of an O-atom donor. In contrast, for 3 or 4 the use of an oxygen atom donor is a pre-requisite for oxygen formation.
• If 18O exchange occurred on a timescale of seconds, the ratio 16O2 : 16O18O : 18O2 should change for different time points of the oxygen evolution shown in Fig. 2. This is not the case.
• Even faster 18O exchange rates (k≈ 1000 s−1) have so far only been reported for MnV-oxo species of manganese porphyrin compounds.43 Such fast exchange rates could explain the stoichiometric 18O labelling detected here. But as the oxygen exchange mechanism established for MnV![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O porphyrin compounds is fundamentally different from those suggested for dinuclear manganese complexes,42 we do not think that the very fast exchange rates established for these compounds necessarily apply to possible oxygen exchange reactions of 1. For dinuclear manganese systems more similar to our case, exact rates for oxygen exchange reactions of manganese oxo compounds have so far not been determined.21,41
O porphyrin compounds is fundamentally different from those suggested for dinuclear manganese complexes,42 we do not think that the very fast exchange rates established for these compounds necessarily apply to possible oxygen exchange reactions of 1. For dinuclear manganese systems more similar to our case, exact rates for oxygen exchange reactions of manganese oxo compounds have so far not been determined.21,41
A synthetic pathway to prepare a Mn2IV,IV oxo-species of 1 is currently not known. Therefore a direct measurement of the oxygen exchange rate for this system is not feasible at the moment—and might be generally impossible if the Mn2IV,IV oxo-species is the reactive species to produce O2. Nevertheless, considering the arguments just presented, we do not consider fast 16O/18O exchange as a very likely explanation for the observed stoichiometric 18O labelling.
Conclusions
We suggest a network of reaction pathways, presented in Scheme 2, as a possible explanation of the observed reactivity of 1 with strong oxidation agents in aqueous solution. | ||
| Scheme 2 Reaction pathways suggested for the reaction of compound 1 with HSO5− or PbIV. Species directly observed by EPR or MIMS are marked by frames. | ||
The central pathway shows the reactions leading to O2 formation. We propose they are the same for both oxone and PbIV. It is known that 1 can be reversibly oxidised by two electrons to the Mn2III,III complex. In the presence of water, the Mn2III,III species exchanges bridging acetates for aqua- or oxo-ligands.44 A second two-electron oxidation by oxone or PbIV would result in a di-µ-oxo Mn2IV,IV species, not observed so far for 1, but well documented in the literature for similar complexes.17,32 From our own work we would expect such a species to be very electron-poor (E > + 1 V vs. Fc0/+)44 and therefore thermodynamically able to oxidise coordinated water or hydroxide to molecular oxygen. From the presented data we can not determine whether oxygen is formed from a single Mn2IV,IV compound or in a reaction involving two Mn2IV,IV species. In any case, both oxygen atoms originate from the bulk water as they show the 18O isotope labelling of the water in solution. Oxygen transfer from the oxidising agent does not appear to be involved in the reaction.
Parallel to this O2 producing route various other reactions occur. They are also depicted in Scheme 2, but were only studied in detail for reactions with oxone. Redox disproportionation of the Mn2III,III compound or comproportion of the Mn2III,III and Mn2IV,IV species could yield the [Mn2III,IV(µ-O)2] complex detected by EPR (upper half of Scheme 2). Decomposition reactions of the complexes, the bpmp− ligand and/or acetate molecules could be responsible for the high detected concentrations of monomeric manganese species and CO2 (lower half of Scheme 2). Small Mn2O3 particles, known to form if MnII is oxidised at acidic conditions,45 are another likely reaction product. These could act as efficient catalysts for the disproportionation of HSO5−, leading to completely unlabelled O2, which was observed for additions of larger amounts of oxone. Our results do not allow the identification of the 18O2 producing manganese species and we therefore cannot rule out that manganese oxide particles might act as water oxidation site. But the fact that we do not observe stoichiometrically labelled 18O2 for any of the other studied reactions (compounds 2–4, MnO2, Mn2O3) makes this an unlikely route of 18O2 formation.
In conclusion, we have identified a first example of a reaction mediated by a synthetic manganese complex where two water molecules might be oxidised to form O2 in homogeneous solution. However, very fast 16O/18O exchange reactions of manganese oxo intermediates could also explain the presented data, but seem very unlikely for the reasons given above. Unfortunately, the compound undergoes decomposition under the reaction conditions, so that a fast degradation is observed and as a result the reaction is not catalytic.
Even so, this reaction presents a very important proof of principle: our data strongly suggest that synthetic manganese compounds like 1 are able to mediate the oxidation of two water molecules to molecular oxygen.
Experimental
Syntheses
Complexes 1–5 (Scheme 1) were synthesized according to published procedures.24,46–49 The synthesis of 1*, bearing 13C labelled acetate ligands was carried out in analogy to that of 1 with the use of H312C13COOH (99% 13C enriched, HOAc*) as the only acetate source:Oxygen detection
The polarographic signal from a Clark electrode (Hansatech) was recorded using the CalMeter software by Calmetric. The volume at the beginning of each experiment was 1 mL, thermostated at 20 °C and continuously stirred. The volume above the sample solution was reduced to ∼100 µL with a plunger to minimize gas exchange processes. Complexes and manganese(III) oxide (325 Mesh, Sigma) were freshly dissolved before each experiment in mixtures of 25% acetonitrile in water or 125 mM acetate or phosphate buffers. Samples (final manganese concentration 4 mM) were made anaerobic with argon before freshly prepared, nitrogen-purged solutions of the oxidants were injected (5–100 µL). Stock solutions of the oxidants were: oxone: 1 M KHSO5/0.5 M KHSO4/0.5 M K2SO4 in H2O; lead tetraacetate: 300 mM Pb(AcO)4 in MeCN. For further details compare ref. 20.MIMS
A magnetic sector field mass spectrometer (Thermo Finnigan, Delta plusXP) was used to separate 16O2 (m/z = 32), 16O18O (m/z = 34), 18O2 (m/z = 36) as well as 12C16O2 (m/z = 44), 12C16O18O (m/z = 46), 12C18O2 (m/z = 48) and argon (m/z = 40).27–29 A Teflon membrane (PTFE 12.5 µm) seamlessly resting on a porous Teflon support and permeable only for gases, separated the high vacuum (8 × 10−8 mbar) from the solutions in the sample chamber (volume 150 µL) which was equipped with stirring and thermostated to 20 °C. Solutions of 1–5 (150 µL, 4 mM Mn) were filled into the reaction chamber. Degassing was achieved through the gas consumption by the adjacent vacuum. Nitrogen-purged solutions of the oxidants were injected and the resulting gas evolution was detected online with the simultaneous measurement of argon (m/z = 40) serving as a control.EPR spectroscopy
EPR spectra at liquid helium temperatures were recorded on Bruker eleXsys E500 or E580 spectrometers equipped with Oxford-900 cryostats and ITC-4 temperature controllers. Bruker ER 4116DM or SHQE4122 EPR cavities were used. Analysis of the spectra was performed using the Bruker Xepr 2.1 software. Samples for six-line signal quantifications where prepared by mixing an 8 mM stock solution of compound 1 in MeCN with the adequate aqueous solution of oxone, KHSO4 or sulfuric acid, respectively, in EPR tubes in a ratio of 1 : 3. Samples were frozen and stored in liquid nitrogen. Signal intensities where quantified by comparing their integral to that of compound 1 subjected to 50 eq. of sulfuric acid, resulting in a maximal release of monomeric Mn. The signals were recorded at 5 K and a microwave power of 200 µW. For the quantification of the Mn2III,IV 16-line signal, 180 µL of a 2 mM solution of compound 1 in 125 mM sodium acetate buffer (pH 4.7) in 25% MeCN, 75% H2O were added to 20 µL of a 100 mM solution of oxone in an EPR tube. The signal intensities were quantified by comparison to the signal of a 1 mM solution of the Mn2III,IV-reference complex [(cyclamMnO)2](ClO4)3.50 The signals were recorded at 20 K and a microwave power of 20 µW.Acknowledgements
This work was supported by the Swedish Energy Agency, the Knut and Alice Wallenberg Foundation, NEST-STRP SOLAR-H (EU contract no. 516510), Deutsche Forschungsgemeinschaft (grant Me 1629/2-4) and the Max-Planck-Gesellschaft. The authors thank Dr Ping Huang and Dat Tran from Uppsala University for EPR and Clark electrode measurements, respectively. Yoonhee Cho synthesised compound 5 and a sample of [(cyclamMnO)2](ClO4)3 was prepared by Dr Nizamuddin Shaikh. We also thank Prof. William H. Armstrong from Boston College, MA, USA, for a sample of compound 2.References
- IPCC, Fourth Assessment Report, Geneva, Switzerland, 2007 Search PubMed.
- E. Amouyal, Sol. Energy Mater. Sol. Cells, 1995, 38, 249–276 CrossRef CAS.
- N. Armaroli and V. Balzani, Angew. Chem., Int. Ed., 2007, 46, 52–66 CrossRef CAS.
- A. J. Bard and M. A. Fox, Acc. Chem. Res., 1995, 28, 141–145 CrossRef CAS.
- M. Kirch, J. M. Lehn and J. P. Sauvage, Helv. Chim. Acta, 1979, 62, 1345–1384 CrossRef CAS.
- N. S. Lewis and D. G. Nocera, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 15729–15735 CrossRef CAS.
- T. J. Meyer, Acc. Chem. Res., 1989, 22, 163–170 CrossRef CAS.
- L. Sun, L. Hammarström, B. Åkermark and S. Styring, Chem. Soc. Rev., 2001, 30, 36–49 RSC.
- W. Lubitz, E. J. Reijerse and J. Messinger, Energy Environ. Sci., 2008, 1, 15–31 RSC.
- R. Lomoth, A. Magnuson, M. Sjödin, P. Huang, S. Styring and L. Hammarström, Photosynth. Res., 2006, 87, 25–40 CrossRef CAS.
- K. N. Ferreira, T. M. Iverson, K. Maghlaoui, J. Barber and S. Iwata, Science, 2004, 303, 1831–1838 CrossRef CAS.
- M. Haumann, C. Müller, P. Liebisch, L. Iuzzolino, J. Dittmer, M. Grabolle, T. Neisius, W. Meyer-Klaucke and H. Dau, Biochemistry, 2005, 44, 1894–1908 CrossRef CAS.
- B. Loll, J. Kern, W. Saenger, A. Zouni and J. Biesiadka, Nature, 2005, 438, 1040–1044 CrossRef CAS.
- J. Yano, J. Kern, K. Sauer, M. J. Latimer, Y. Pushkar, J. Biesiadka, B. Loll, W. Saenger, J. Messinger, A. Zouni and V. K. Yachandra, Science, 2006, 314, 821–825 CrossRef CAS.
- F. A. Armstrong, Philos. Trans. R. Soc. London, Ser. B, 2008, 363, 1263–1270 CrossRef CAS.
- N. A. Law, M. T. Caudle and V. L. Pecoraro, Adv. Inorg. Chem., 1999, 46, 305–440 Search PubMed.
- S. Mukhopadhyay, S. K. Mandal, S. Bhaduri and W. H. Armstrong, Chem. Rev., 2004, 104, 3981–4026 CrossRef CAS.
- W. Ruttinger and G. C. Dismukes, Chem. Rev., 1997, 97, 1–24 CrossRef CAS.
- M. Yagi and M. Kaneko, Chem. Rev., 2001, 101, 21–35 CrossRef CAS.
- P. Kurz, G. Berggren, M. F. Anderlund and S. Styring, Dalton Trans., 2007, 4258–4261 RSC.
- H. Y. Chen, R. Tagore, G. Olack, J. S. Vrettos, T. C. Weng, J. Penner-Hahn, R. H. Crabtree and G. W. Brudvig, Inorg. Chem., 2007, 46, 34–43 CrossRef CAS.
- A. K. Poulsen, A. Rompel and C. J. McKenzie, Angew. Chem., Int. Ed., 2005, 44, 6916–6920 CrossRef CAS.
- Y. Shimazaki, T. Nagano, H. Takesue, B. H. Ye, F. Tani and Y. Naruta, Angew. Chem., Int. Ed., 2004, 43, 98–100 CrossRef.
- C. Baffert, M. N. Collomb, A. Deronzier, S. Kjaergaard-Knudsen, J. M. Latour, K. H. Lund, C. J. McKenzie, M. Mortensen, L. Nielsen and N. Thorup, Dalton Trans., 2003, 1765–1772 RSC.
- R. Tagore, H. Y. Chen, H. Zhang, R. H. Crabtree and G. W. Brudvig, Inorg. Chim. Acta, 2007, 360, 2983–2989 CrossRef CAS.
- M. Lundberg, M. R. A. Blomberg and P. E. M. Siegbahn, Inorg. Chem., 2004, 43, 264–274 CrossRef CAS.
- J. Clausen, K. Beckmann, W. Junge and J. Messinger, Plant Physiol., 2005, 139, 1444–1450 CrossRef CAS.
- J. Messinger, M. Badger and T. Wydrzynski, Proc. Natl. Acad. Sci. U. S. A., 1995, 92, 3209–3213 CAS.
- L. Konermann, J. Messinger and W. Hillier, in Biophysical Techniques in Photosynthesis Research, vol. II, Series Advances in Photosynthesis and Respiration, vol. 26, ed. T. J. Aartsma and J. Matysik, Springer, Dordrecht, 2008 Search PubMed.
- J. Limburg, J. S. Vrettos, L. M. Liable-Sands, A. L. Rheingold, R. H. Crabtree and G. W. Brudvig, Science, 1999, 283, 1524–1527 CrossRef CAS.
- P. Huang, P. Kurz and S. Styring, Appl. Magn. Reson., 2007, 31, 301–320 CrossRef CAS.
- A. J. Wu, J. E. Penner-Hahn and V. L. Pecoraro, Chem. Rev., 2004, 104, 903–938 CrossRef CAS.
- C. Hureau, L. Sabater, E. Anxolabehere-Mallart, M. Nierlich, M. F. Charlot, F. Gonnet, E. Riviere and G. Blondin, Chem.–Eur. J., 2004, 10, 1998–2010 CrossRef CAS.
- P. Huang, J. Högblom, M. F. Anderlund, L. Sun, A. Magnuson and S. Styring, J. Inorg. Biochem., 2004, 98, 733–745 CrossRef CAS.
- A. Magnuson, P. Liebisch, J. Högblom, M. F. Anderlund, R. Lomoth, W. Meyer-Klaucke, M. Haumann and H. Dau, J. Inorg. Biochem., 2006, 100, 1234–1243 CrossRef CAS.
- N. Wiberg, A. Holleman and E. Wiberg, Lehrbuch der Anorganischen Chemie, Walter de Gruyter, Berlin, New York, 1995 Search PubMed.
- Y. Sasaki, T. Akamatsu, K. Tsuchiya, S. Ohba, M. Sakamoto and Y. Nishida, Polyhedron, 1998, 17, 235–242 CrossRef.
- G. P. Anipsitakis and D. D. Dionysiou, Environ. Sci. Technol., 2004, 38, 3705–3712 CrossRef CAS.
- G. P. Anipsitakis, D. D. Dionysiou and M. A. Gonzalez, Environ. Sci. Technol., 2006, 40, 1000–1007 CrossRef CAS.
- M. J. Baldwin, N. A. Law, T. L. Stemmler, J. W. Kampf, J. E. Penner-Hahn and V. L. Pecoraro, Inorg. Chem., 1999, 38, 4801–4809 CrossRef CAS.
- A. S. Larsen, K. Wang, M. A. Lockwood, G. L. Rice, T. J. Won, S. Lovell, M. Sadilek, F. Turecek and J. M. Mayer, J. Am. Chem. Soc., 2002, 124, 10112–10123 CrossRef CAS.
- J. Limburg, J. S. Vrettos, H. Y. Chen, J. C. de Paula, R. H. Crabtree and G. W. Brudvig, J. Am. Chem. Soc., 2001, 123, 423–430 CrossRef CAS.
- J. T. Groves, J. B. Lee and S. S. Marla, J. Am. Chem. Soc., 1997, 119, 6269–6273 CrossRef CAS.
- G. Eilers, C. Zettersten, L. Nyholm, L. Hammarström and R. Lomoth, Dalton Trans., 2005, 1033–1041 RSC.
- S. Y. Zhang, Z. W. Chen, S. Tan, J. Wang and S. H. Jin, Nanostruct. Mater., 1997, 8, 719–723 CrossRef CAS.
- M. K. Chan and W. H. Armstrong, J. Am. Chem. Soc., 1990, 112, 4985–4986 CrossRef CAS.
- H. Y. Chen, R. Tagore, S. Das, C. Incarvito, J. W. Faller, R. H. Crabtree and G. W. Brudvig, Inorg. Chem., 2005, 44, 7661–7670 CrossRef CAS.
- Y. Nishida and M. Nasu, Z. Naturforsch., B, 1991, 46b, 231–234.
- L. Sun, M. K. Raymond, A. Magnuson, D. LeGourriérec, M. Tamm, M. Abrahamsson, P. Huang-Kenéz, J. Mårtensson, G. Stenhagen, L. Hammarström, S. Styring and B. Åkermark, J. Inorg. Biochem., 2000, 78, 15–22 CrossRef CAS.
- K. J. Brewer, A. Liegeois, J. W. Otvos, M. Calvin and L. O. Spreer, J. Chem. Soc., Chem. Commun., 1988, 1219–1220 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: additional oxygen traces and EPR spectra. See DOI: 10.1039/b811806j |
| ‡ The first two authors contributed equally to the work as main experimental investigators. |
| § Current address: Department of Chemistry, Umeå University, 90187 Umeå, Sweden. |
| This journal is © The Royal Society of Chemistry 2008 |