Hydrogen storage in liquid organic heterocycles
Robert H.
Crabtree
Yale University, Chemistry Department, 225 Prospect Street, P.O. Box 208107, New Haven, CT 06520-8107, USA. E-mail: robert.crabtree@yale.edu
First published on 11th June 2008
Abstract
Hydrogen storage in liquid organic heterocycles is feasible thermodynamically and is attractive in terms of simplicity, safety, scalability, heat management and economy, but extensive catalyst development is needed to bring it to fruition.
 Robert H. Crabtree | Educated at Oxford and Sussex Universities and CNRS Natural Products Institute in Paris. Now, a Professor of Chemistry at Yale, he works extensively on catalysis, both organometallic and bioinorganic. Appointed Dow lecturer at Berkeley, Sabatier Lecturer at Toulouse, and will be Osborn Lecturer at Strasbourg and Mond Lecturer in the UK. He has been awarded ACS and RSC prizes for organometallic chemistry. |
In his 1908 book, Worlds in the Making, Svante Arrhenius1 expanded on his 1896 prediction that industrial CO2 production would eventually raise average global temperature. Doubling atmospheric CO2 would, he believed, cause a 4 °C rise, a value within the presently accepted range. Perhaps because he was writing from a cool northern city, however, he felt this rise would be a good thing.
We often hear lamentations that the coal stored up in the earth is wasted by the present generation without any thought of the future… We may find a kind of consolation that here, as in every other case, there is good mixed with the evil. By the influence of the increasing percentage of carbonic acid in the atmosphere, we may hope to enjoy ages with more equable and better climates, especially as regards the colder regions of the earth, ages when the earth will bring forth more abundant crops than at present, for the benefit of a rapidly propagating mankind. 1a
Few informed individuals still retain this optimistic view of climate change, now much more often considered deeply problematic.2 Average energy consumption for the planet as a whole has been estimated at 2 kW h per person,3 more than 80% based on fossil fuel, leading to an injection of ca. 1013 kg of CO2 per year into the atmosphere.
Efforts to alleviate CO2 production confront several problems. Costs are obvious, prompt and local while benefits are putative, delayed and global. It can be little wonder that the research budgets in this field are dwarfed by those for health and defense. As the reality of the situation sinks in and climate disruption becomes more obvious, climate change and alternative energy may well become the dominant scientific problems of the century.
While a number of technical solutions appear plausible in principle, the challenge of applying them on the required global scale is daunting. For instance, to take just one likely scenario, in a world based on nuclear power, transport would require either light, highly efficient storage batteries or else some transportable fuel that can be safely stored on board. In either case the principle is the same—electrical energy is stored in chemical form.
Taking a global view, transport is an increasingly energy-intensive area, particularly with China and India rapidly industrializing. Hydrogen has been suggested as a possible energy carrier using an internal combustion engine (ICE) or fuel cell for the motive power. Efficiency in an ICE is limited by the physics of the Carnot cycle to approximately 25% while fuel cells escape from this limitation and can have efficiencies above 50%.4 H2 is currently generated mainly from fossil fuel with release of CO2, so the relatively widespread demonstration transport vehicles, usually advertised as ‘green’, do not yet achieve the stated goal. Both nuclear-to-H2 and solar-to-H2 schemes have been proposed to remedy this defect. Assuming H2 can be generated efficiently by a CO2-free route or with CO2 sequestration, very hard problems in themselves,5–7 we would next need a method for hydrogen storage in vehicles.
Numerous reports have treated this problem3,4,7,8 so it will only be necessary to summarize the major prior H2storage approaches. These are physical: (i) high pressure (HP) tanks; (ii) cryogenic methods; or chemical: (iii) reversible absorption in metal or main group hydrides; (iv) reversible absorption on solids; and (v) storage in the form of metals that can liberate H2 from H2O. Current plans for future vehicles employ one of three approaches, either (i), (ii) or (iii).
For a fuel-efficient automobile, 4–8 kg of H2 need to be stored to match current consumer needs and expectations.3,4 High pressure tanks are heavy and voluminous and may pose practical problems. Just this month (Feb 2008) a major interstate highway in the writer's state was closed down for the whole day as a result of an otherwise minor mishap without gas release involving a commercial vehicle carrying high pressure H2. Nevertheless, this is a cheap and readily reversible storage method and it is available today.
Given the low critical temperature, 33 K, cryogenic storage requires very low temperatures, implying a large energy loss from the liquefaction step. Apart from the weight penalty of the cooling module, this method is not ideal for vehicles used only intermittently because energy input is constantly required to maintain cooling.
Chemical methods are advantageous in binding H2 at ambient temperature and pressure but can suffer from lack of complete and easy reversibility. The H2 release enthalpies that appear to be most appropriate for automobile H2storage lie in the range 15–25 kJ mol−1 and correspond to release temperatures in the range of 0–100 °C. Carbon-based solids9 such as single walled nanotubes10 (SWNTs) or microporous metal–organic frameworks11 have shown attractive performance in room temperature hydrogen absorption but the performance still needs to be much improved before practical application can be envisaged. Metal hydrides12,13 such as LaNi5, Mg2Ni or MgH2 have also been very extensively studied. Although they have many attractive properties, the fact that they tend to employ relatively high atomic weight elements means that few such metal hydrides are close to meeting the very ambitious criteria set by the US Department of Energy (DOE): 9% hydrogen by weight by 2015. For comparison, one H stored per C atom leads to a gravimetric capacity of 7.1% but LaNi5H6 stores only 1.37%.
This gravimetric criterion has focused attention on the light atoms of the Periodic Table, notably Li and B and on compounds that hold more than one H per non-hydrogen atom. One compound that fulfils both points is BH3NH3 (ammonia–borane, or AB)14 and related materials.15 From its molecular weight and polarity, AB would be a gas except for the strong proton–hydride interactions present in the solid (m pt, 109 °C).16 If it were to lose all its H2 (19.6% theoretical capacity) it would form boron nitride, an unpromising material for the regeneration step needed for any storage/release cycle.
Thermal H2 release from BH3NH3 is straightforward, and catalytic methods can also be successfully applied. Acid-catalyzed, as well as transition metal-catalyzed release have recently been reported.14CatalyticAB dehydrogenation can give BH3NH2BH2NH3, (H2NBH2)n (n = 3,5), (HBNH)3 or (HBNH)n polymer. The regeneration step is not at all straightforward, however.17
A related strategy based on light element salts involves borohydrides,18,19 amidoborates20 or aluminohydrides. For example, Zuettel and coworkers have investigated LiBH4, a hydride salt containing 18 mass% of hydrogen. Hydrogen desorption was catalyzed with SiO2 and 13.5 mass% of hydrogen was liberated over the range 200–350 °C; LiBH4regeneration proved challenging, however.21 Bogdanovic and Schwickardi19 demonstrated reversible hydrogen storage with Ti-doped NaAlH4, where the Ti acts as the catalyst. The LiBH4–LiNH2 system in which the principal phase present is Li4BH4(NH2)3F, has also been proposed.22
Perhaps not sufficiently considered in current approaches is scalability. Any new technology would have to be applied on a vast scale to make any impact on global climate. Assuming a material having a 10% gravimetric capacity, 102 kg of material would plausibly be needed for each of an assumed 109 vehicles worldwide for a total of 1011 kg of storage material. On this basis, elements that are not available on this scale may be unrealistic candidates. According to the US Geological Survey Mineral Resources Program,23 the 2007 world production values for selected elements are: La, < 1.6 × 106 kg; B, 9 × 108 kg; Ni, 1.6 × 109 kg; Mg, 4 × 109 kg; Li, 2.5 × 1010 kg. In other cases, the element or elements may be available in quantity but the challenges for production of such vast quantities of absorbent may have been underestimated. This does not detract from the scientific value of work on such materials, of course, particularly where new principles emerge.
Liquid storage materials and heat management strategy
Whatever the storage material, liquids have significant engineering advantages over solids. They can be readily pumped not just for distribution and delivery but also within the vehicle during operation. This means that instead of heating the whole storage tank, only a small aliquot would be pumped into the catalytic dehydrogenation chamber for heating to reaction temperature at any given time. This is also a safety consideration in a collision, where it would be undesirable to have a large mass of hot, reactive—perhaps even pyrophoric—material present in the vehicles involved. Once dehydrogenated, the spent storage material would pass back into the fuel tank and a partition would move across the tank to allow the spent material that is being pumped in to displace, but not mix with, the fresh material that is being pumped out. At the fuel station, the spent material would be off-loaded and the fresh material substituted in a similar manner. Trucks that deliver fresh material to the fuel stations would return the spent material for recharging with hydrogen. The liquid strategy also employs a simple, light fuel tank, as today, not a heavy duty tank capable of taking high pressure and temperature as would be needed if the whole storage bed had to be heated.The properties of hydrogen make it most suitable for handling within commercial facilities by trained personnel. A liquid storage material thus has the further advantage that there would be no free hydrogen in the public sphere. H2 has an exceptionally high diffusivity, leading to an enhanced risk from leaks. It can cause embrittlement of metals, generating greater potential for leaks and complicating the engineering. A hydrogen flame is essentially invisible thus presenting greater dangers than a flame from other fuels. As an illustration, a standard method of detecting H2 flames is to advance cautiously with a piece of paper in an outstretched hand, the flame being located when the piece of paper begins to burn.
An even more compelling argument for the liquid strategy is its heat management benefits. When any storage material is hydrogenated, large amounts of heat are necessarily generated. If this happens in a solid bed of storage material, the exotherm will require dissipation by cooling while the vehicle stands at the fuelling station. Not only is this an energy loss to the global energy balance of the strategy, but it means filling the vehicle will be prolonged, and incompatible with the level of patience usually encountered among the driving public, as well as detracting from the profitability of the fuel station. If cooling is applied via refrigeration to speed the filling process, the energy input required would further degrade the energy balance of the system as a whole. This contrasts with the liquid strategy in which filling the vehicle takes no more time than today and involves no exotherm. If the hydrogenation step occurs in a commercial facility on a very large scale, as in the liquid strategy, then the exotherm is produced in one place, where it can be at least partially recovered, rather than in fuel stations throughout the city where it will typically be lost. Fig. 1 illustrates the liquid strategy in more detail.
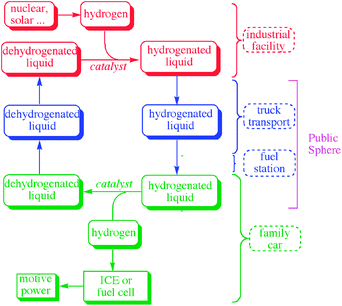 | ||
| Fig. 1 The liquid strategy showing how the presence of free hydrogen is avoided in the public sphere. The exotherm of rehydrogenation of the spent storage material can also be more efficiently recovered if produced in a central facility on a large scale. | ||
Organic heterocycle strategy
The possibility of storage of hydrogen in organic compounds has been widely excluded from consideration because reversible, low temperature H2 release has not been thought feasible. For example, according to a standard review of the field.3The second important criterion is reversibility of hydrogen uptake and release. This criterion excludes all covalent carbon hydrogen compounds as hydrogen is only released from carbon hydrogen compounds if they are heated to temperatures above 800 °C or if the carbon is oxidized.
While this was a plausible argument based on much of the data available at the time (2004), this analysis does not consider the twin possibilities of catalytic H2 release and of chemical modification of the “carbon hydrogen compounds” to favor low temperature H2 release.
Catalysis was first considered for alkane–arene pairs, such as decalin–naphthalene, but the endothermicity of the release step is such that elevated temperatures are required for the thermodynamics to become favorable. Saito and coworkers24 proposed ‘liquid film state’ conditions, a nonequilibrium technique which allows much higher hydrogen production rates than in a batch reaction. Similarly, a pulse-spray mode reactor has been adopted by Ichikawa and coworkers.25 The highest rate, an impressive 3800 mmol g−1Pt min−1, was obtained in the dehydrogenation of cyclohexane over Pt/alumite heated at 375 °C with a cyclohexane feed of 190 mmol min−1 with 3.5 mmol pulses at 1.0 s intervals. A bimetallic Pt–Rh catalyst showed higher activity than a simple Pt catalyst on the same support.
The release temperatures are still rather high and it would be useful to lower the endothermicity of release and thus bring down the equilibrium release temperature.
Taking the cyclohexane–benzene pair as a model, the endothermicity of H2 release is such that a temperature of ca. 600 K is required to bring the reaction to a ΔG value of zero. At this point the unfavorable enthalpy is exactly compensated by the favorable entropy of H2 release. We refer to this temperature as Td, the point at which ΔG = 0. Intermediate dehydrogenation products (e.g.cyclohexene) that do not benefit from the aromaticity of benzene are even more strongly disfavored than the final arene. The overall reaction could still be accessible if these intermediates were sufficiently stabilized by binding to the catalyst. Even in very endothermic cases, product formation is experimentally possible, however. For example, alkane dehydrogenation to alkenes and free H2 has been observed with numerous homogeneous catalysts by us and others even at temperatures of 90–150 °C. These reactions are driven by reflux of the alkane, because the H2 is swept out of the solvent and the equilibrium continually displaced.26
The organic heterocycle H2storage strategy, first proposed by Alan Cooper and Guido Pez at Air Products, appeared in a series of key patents.27,28 Our later, but independent computational work on this problem, largely in collaboration with Eric Clot and Odile Eisenstein,29 identified some general trends for design of the heterocycles to favor low temperature H2 release. In summary, the storage step involves catalytic hydrogenation of an aromatic heterocycle to give the corresponding hydrogenated product. Release is effected by heating the hydrogenated form in the presence of a catalyst. Both directions must therefore be viable, implying that the endothermicity of the dehydrogenation has to be moderate. This endothermicity translates into a temperature Td at which ΔG = 0 and the results are therefore discussed in terms of Td.
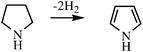
The all-carbon systems (e.g., cyclohexane–benzene) have a Td that is far too high but introduction of nitrogen atoms into the organic ring dramatically favors the thermodynamics of H2 release: in extreme cases, Td can now go below 50 K. The most important design consideration for a low Td is a move to a 5-membered ring, when aromatic stabilization can be achieved after cleavage of only four C–H bonds (eqn (1)), not six as for cyclohexane–benzene, always provided a NH or NR is present in the 1-position to permit aromaticity. Cyclopentane–cyclopentadiene shows no such advantage.
 | (1) |
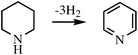
Incorporation of N into a six-membered ring also favors dehydrogenation because the N–H bond that is now broken is weaker than the C–H bond it replaced. In addition, C–H bonds adjacent to a N atom are also weakened relative to a C–H bond in a pure carbocycle. Nitrogen substituents are also effective.
 | (2) |
The thermodynamic data calculated {DFT(B3PW91)} by Eric Clot and Odile Eisenstein allow useful structure-activity trends to be identified. Scheme 1 shows the Td values for a number of key cases. The most effective way of lowering Td is moving to a 5-membered ring, with incorporation of N-substituents and N ring atoms in a 1,3-arrangement being somewhat less effective strategies. As an aside, the data also suggest that the long known resistance of certain azoles (e.g., imidazole) to hydrogenation is the result of thermodynamic rather than purely kinetic factors.
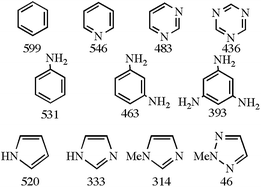 | ||
| Scheme 1 Thermodynamic hydrogen release temperatures (temperature {K} at which ΔG = 0) for selected model compounds by DFT(B3PW91) calculations of Clot and Eisenstein.22 | ||
In their patent, Pez and coworkers28 demonstrate reversible hydrogenation–dehydrogenation of heterocyclic liquids. For example, N-ethyl carbazole is hydrogenated with 72 atm H2 and a Pd catalyst at 160 °C to form a mixture of isomers of the fully hydrogenated species (eqn (3)). Dehydrogenation gave pure H2 with Ru at 50–197 °C and at least 5 cycles can be run without HSM degradation.
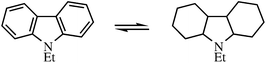 | (3) |
The carbazole fulfils some of the thermodynamic requirements of Scheme 1 in that the nitrogen is a substituent to two rings and a member of the central 5-membered ring.
In this vision, a nitrogen-containing organic liquid is preferred as a hydrogen storage material (HSM) on several counts. Both C and N are available in very large amounts, with 2007 world production figures for nitrogen23 of 1.2 × 1011 kg and just counting carbon in the form of coal, 5 × 1014 kg (World Coal Institute).30 The material must not only be readily available but also be distributed economically to users. The Sallan Foundation31 estimates the infrastructure costs of the distribution system for petroleum based transport fuels at several hundred billion dollars. A liquid organic HSM can plausibly be distributed via the existing gasoline infrastructure with minimal modification, saving vast capital costs. Ideally the HSM would be minimally volatile, minimally toxic, and biodegradable.
One advantage of the heterocyclic liquid strategy is that the liquid can in principle be repurified when necessary, so that the inevitable non-regenerable fraction of the HSM does not build up on the vehicle—a weight and capacity penalty. For this step, vacuum distillation may be desirable and if so, the components will need to have appreciable volatility.
Efficiency of HSM regeneration will be a key point because any shortfall would raise purification costs and require HSM to be made up with newly manufactured material with a consequent economic penalty. Catalysts that can meet the severe selectivity and activity requirements must be further developed. Since it is not yet clear which HSM to choose, the HSM and the catalyst will have to be optimized together.
Limitations
Gravimetric capacity for typical HSMs of the type proposed are in the range from 6 to 8%, not as good as BH3NH3. To meet the more aggressive DOE goals, more than one H would need to be stored per heavy atom, for example C(CH2NH2)4–C(CN)4 has an 11.7% capacity. The volumetric capacity for typical heterocycles is also satisfactory, although in energy terms not as good as gasoline, because only some of the hydrogen atoms of the HSM end up as H2O and all the carbon remains uncombusted. The situation for gasoline, denoted (CH2)n, undergoing complete combustionversus the same (CH2)n, but now acting as an HSM with the hydrogen released ultimately undergoing oxidation with air, can be represented as follows.| (CH2)n + 1.5nO2 = nCO2 + nH2O | (4) |
| (CH2)n + 0.25nO2 = (CH)n + 0.5nH2O | (5) |
In general terms, the exothermicity of a combustion reaction of H2 or of a hydrocarbon is simply proportional to the amount of O2 consumed. Comparison of the O2 used in the two equations above suggests that the energy content of an organic HSM is ca. 17% of the value that the same material would have if completely combusted as a normal fuel. Of course, complete combustion would form CO2, contrary to the requirement for carbon dioxide abatement.
Design of the HSM requires attention to numerous points including (i) toxicity; (ii) thermal stability against undesired decomposition pathways; (iii) safety in accidental release such as in automobile collisions; (iv) biodegradability; (v) thermodynamic tendency to liberate H2; (vi) kinetic facility for reversible H2 release; (vii) cheap manufacture on a 1011 kg scale. Points (i)–(vii) severely restrict the types of materials that can be used, not only for the liquid organic strategy but for all others as well.
Azaheterocycles can be of low toxicity, 1-decyl pyridinium chloride is used as a mouthwash in almost all common formulations and is thus in intimate contact with humans on a daily basis as well as being released into the environment on a large scale. Thermal decomposition is a serious issue and work will be needed to understand the thermolysis pathways of candidate HSMs and find ways to guard against them by suitable design. Liquid organic HSMs can be of low volatility and have a high flash point, minimizing accidental release problems, and they may be biodegradable, particularly in their hydrogenated form, thanks to their heteroatom content. Quantitative structure-activity relationships (QSAR), now being developed both for toxicity and for biodegradation of N-heterocycles.32
A HSM that readily releases H2 is of necessity hard to hydrogenate. ‘Virtual H2storage’ could avoid this step by using the electrical power source not to produce free H2 but to directly reduce the HSM electrocatalytically, but precedent is lacking. Likewise, a direct fuel cell could allow the conversion of the fresh HSM to motive power without H2 production. With air as the oxidant, the driving force of eqn (5) would be greatly enhanced and the high Td problem circumvented.
Conclusion
The liquid heterocycle strategy is worth greater emphasis because it has a number of advantages of simplicity, safety, scalability, heat management, and economy. Catalyst development is needed for further progress, however, since catalytic heterocycle dehydrogenation is a neglected topic.Acknowledgements
I thank Eric Clot and Odile Eisenstein for their many insightful contributions to this and other problems of mutual interest, Peter Hall (Strathclyde) for the direct fuel cell idea, the referees for useful suggestions and DOE and NSF for funding our catalysis work.References
- S. Arrhenius, Worlds in the Making, (transl. by H. Borns), Harper Bros., London & New York, 1908; p. 62 Search PubMed.
- “Climate Change 2007”, Fourth IPCC Assessment Report, http://www.ipcc.ch/ipccreports/index.htm Search PubMed.
- A. Zuettel, Naturwissenschaften, 2004, 91, 157–172 CrossRef.
- L. Sclapbach and A. Zuettel, Nature, 2001, 414, 353 CrossRef.
- J. A. Turner, Science, 2004, 305, 972 CrossRef CAS.
- R. F. Service, Science, 2004, 305, 962 CrossRef CAS.
- W. Grochala and P. P. Edwards, Chem. Rev., 2004, 104, 1283 CrossRef CAS.
- A. W. C. van den Berg and C. O. Areán, Chem. Commun., 2008, 6, 668–681 RSC.
- (a) Z. Yang, Y. Xia and R. Mokaya, J. Am. Chem. Soc., 2007, 129, 1673 CrossRef CAS; (b) H. M. Cheng, Q. H. Yang and C. Liu, Carbon, 2001, 39, 1447 CrossRef CAS.
- H. G. Schimmel, G. J. Kearley and M. G. Nijkamp, Chem.–Eur. J., 2003, 9, 4764 CrossRef CAS.
- N. L. Rosi, J. Eckert, M. Eddaoudi, D. T. Vodak, J. Kim, M. O'Keeffe and O. M. Yaghi, Science, 2003, 300, 1127 CrossRef CAS.
- C. M. Jensen and K. J. Gross, Appl. Phys. A: Mater. Sci. Process., 2001, 72, 213 CrossRef CAS.
- S. I. Orimo, Y. Nakamori, J. R. Eliseo, A. Zuettel and C. M. Jensen, Chem. Rev., 2007, 107, 4111 CrossRef CAS.
- (a) R. J. Keaton, J. M. Blacquiere and R. T. Baker, J. Am. Chem. Soc., 2007, 129, 1844 CrossRef CAS; (b) F. H. Stephens, R. T. Baker, M. H. Matus, D. J. Grant and D. A. Dixon, Angew. Chem., Int. Ed., 2007, 46, 746 CrossRef CAS.
- D. A. Dixon and M. Gutowski, J. Phys. Chem. A, 2005, 109, 5129 CrossRef CAS.
- W. T. Klooster, T. F. Koetzle, P. E. M. Siegbahn, T. B. Richardson and R. H. Crabtree, J. Am. Chem. Soc., 1999, 121, 6337 CrossRef CAS.
- S. Hausdorf, F. Baitalow, G. Wolf and F. O. R. L. Mertens, Int. J. Hydrogen Energy, 2008, 33, 608 CrossRef CAS.
- Y. Nakamori and S. Orimo, J. Alloys Compd., 2004, 370, 271 CrossRef CAS.
- B. Bogdanovic and M. Schwickardi, J. Alloys Compd., 1997, 253, 1 CrossRef.
- Z. Xiong, C. K. Yong, G. Wu, P. Chen, W. Shaw, A. Karkamkar, T. Autrey, M. O. Jones, S. R. Johnson, P. P. Edwards and W. I. F. David, Nat. Mater., 2008, 7, 138 CrossRef CAS.
- A. Zuettel, P. Wenger, S. Rentsch, P. Sudan, Ph. Mauron and Ch. Emmenegger, J. Power Sources, 2003, 118, 1 CrossRef CAS.
- (a) F. E. Pinkerton, G. P. Meisner, M. S. Meyer, M. P. Baloghand and M. D. Kundrat, J. Phys. Chem. B, 2005, 109, 6 CrossRef CAS; (b) M. Aoki, K. Miwa, T. Noritake, G. Kitahara, Y. Nakamori, S. Orimo and S. Towata, Appl. Phys. A: Mater. Sci. Process., 2005, 80, 1409 CrossRef CAS.
- US Geological Survey, Washington, DC, USA, http://minerals.usgs.gov/.
- N. Meng, S. Shinoda and Y. Saito, Int. J. Hydrogen Energy, 1997, 22, 361 CrossRef CAS.
- N. Kariya, A. Fukuoka, T. Utagawa, M. Sakuramoto, Y. Goto and M. Ichikawa, Appl. Catal., A, 2003, 247, 247 CrossRef CAS.
- (a) K. Krogh-Jespersen, M. Czerw, N. Summa, K. B. Renkema, P. D. Achord and A. S. Goldman, J. Am. Chem. Soc., 2002, 124, 11404 CrossRef CAS; (b) T. Aoki and R. H. Crabtree, Organometallics, 1993, 12, 294 CrossRef CAS.
- G. P. Pez, A. R. Scott, A. C. Cooper and H. Cheng, US Pat., 7101530, September 5, 2006 and prior patents cited.
- G. P. Pez, A. R. Scott, A. C. Cooper, H. Cheng, F. C. Wilhelm and A. H. Abdourazak, US Pat., 7351395, 2008.
- E. Clot, O. Eisenstein and R. H. Crabtree, Chem. Commun., 2007, 22, 2231–2233 RSC.
- World Coal Institute, London, UK, http://www.worldcoal.org/.
- Sallan Foundation, New York City, USA, http://www.sallan.org/newviews/archives/2007/05/000617.php.
- (a) J. Chen, Y. Liao, Y. Zhao, L. Wang, G. Lu and T. Zhao, Bull. Environ. Contam. Toxicol., 1996, 57, 77 CrossRef CAS; (b) B. Philipp, M. Hoff, F. Germa, B. Schink, D. Beimborn and V. Mersch-Sundermann, Environ. Sci. Technol., 2007, 41, 1390 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2008 |