Exceptionally high CO2storage in covalent-organic frameworks: Atomistic simulation study†
Ravichandar
Babarao
and
Jianwen
Jiang
*
Department of Chemical & Biomolecular Engineering, National University of Singapore, Singapore 117576. E-mail: chejj@nus.edu.sg; Fax: +65 6516-5083; Tel: +65 6779-1936
First published on 11th June 2008
Abstract
Atomistic simulations have been performed on CO2storage in covalent-organic frameworks (COFs) including 3D (COF-102, COF-103, COF-105, and COF-108), 2D (COF-6, COF-8, COF-10) and 1D (COF_NT) structures. Compared to 2D and 1D COFs, 3D COFs have substantially larger free volume, porosity and surface area. As a counterbalance of low framework density and large porosity, COF-105 and COF-108 show exceptionally high storage capacity, even surpassing the experimentally measured highest capacity in MOF-177. COF-6 exhibits the largest isosteric heat and Henry constant due to the presence of constricted pores, but the lowest saturation capacity. COF_NT has adsorption behavior similar to a carbon nanotube. Different adsorption capacities in COFs are attributed to the interplay of various complex factors such as framework density, free volume, porosity and surface area. Gravimetric and volumetric capacities at 300 K and 30 bar correlate well with these factors. The molecular-based structure–function correlations are useful to predict capacity and screen COFs for CO2storage.
With the rapid increase of population and energy consumption, a huge amount of greenhouse gas CO2 has been released into the atmosphere and caused a severe impact on global warming.1 Development of cost-effective processes for CO2 sequestration is one of the pressing issues in environmental protection. Among a handful of techniques, adsorption by porous adsorbents is energetically efficient and technically feasible. Towards this end, it is crucial to choose a suitable adsorbent with a high capacity for CO2storage.
There has been extensive interest over the past few years in a new family of porous materials, named metal-organic frameworks (MOFs).2 With well-defined pore dimension and high thermal stability, MOFs have profound applications for gas storage, separation, catalysis, etc. MOFs are easily tunable by varying either the organic linkers or metal oxides. Experimental study has shown that CO2storage in some MOFs is remarkably higher than in zeolites and carbonaceous materials.3 In coordination with experiments, a number of atomistic simulations for gas adsorption in MOFs have been reported. Simulation is a vital tool to gain insight into the microscopic mechanism and subsequently to provide guidelines for rational design of new materials. For instance, adsorption of light gases in MOFs was simulated and compared with available experimental data.4 Among three different types of nanoporous structures (IRMOF1, silicalite and carbon schwarzite), IRMOF1 was demonstrated to have the largest capacity for CO2 and CH4 adsorption.5 A series of MOFs were investigated for CO2storage and the structure–function correlations were proposed for the molecular screening of MOFs toward high-efficacy CO2storage.6 The effect of framework charge was found to be insignificant in determining CO2 adsorption in MOFs.6,7
Very recently, Côtéet al.8,9 and El-Kaderi et al.10 synthesized crystalline, porous, covalent-organic frameworks (COFs) solely from light elements like B, C, O and H. Consisting of the organic-linkers covalently bonded with boron-oxide clusters, COFs have salient features such as high thermal stability, large surface area and porosity. These boron-oxide clusters can be regarded as analogous to the metal-oxide clusters in MOFs. Composed of light elements, however, COFs have even lower density than MOFs.
Intriguingly, COFs can exist as 3D, 2D and 1D structures as shown in Fig. 1. Co-condensation of boronic acid with hexahydroxytriphenylene results in 2D COF-6, -8 and -10.9 COF-6 and -8 have a pore size of 8.6 and 16.4 Å, respectively; whereas COF-10 has a pore size of 31.7 Å. These 2D COF structures resemble the layered graphite composed of graphene sheets. The inter-layer distances in COF-6, -8 and -10 are 3.399, 3.630 and 3.526 Å, respectively. Alternatively, joining triangular and tetrahedral nodes leads to 3D COF-102, -103, -105 and -108.10 COF-102 has a cubic structure with a lattice constant of 27.18 Å and a crystal density 0.41 g cm−3. The largest cavity in the center of COF-102 is 5.66 Å from the nearest H atoms. COF-103 is identical to COF-102, except that the tetrahedral C atoms are replaced by Si atoms. COF-105 and COF-108 were reported to have the lowest density (as low as 0.17 g cm−3), even lower than the highly porous materials MOF-177 (0.42 g cm−3). COF-108 has two types of cavities namely 9.34 and 15.46 Å from the center C atom excluding the van der Waals radii. In general, the pores in 3D COFs are not spherical but fully accessible to all the edges and faces in the framework. Similar to carbon nanotube, armchair or zig-zig 1D COF nanotube (COF_NT) could be constructed by rolling a COF layer in a particular direction. Mazzoni and coworkers tested the stability of COF_NTs by examining the structural and electronic properties using first-principles calculations.11 We have constructed an hexagonal COF_NT bundle to explore its capacity for CO2storage. Each COF_NT has a diameter of 16.8 Å and the van der Waals gap (distance between two neighboring nanotubes) is 3.2 Å. There exist three types of energetically favorable adsorption sites within the bundle; the annular layer and center inside the nanotube and the interstitial channels between the nanotubes.
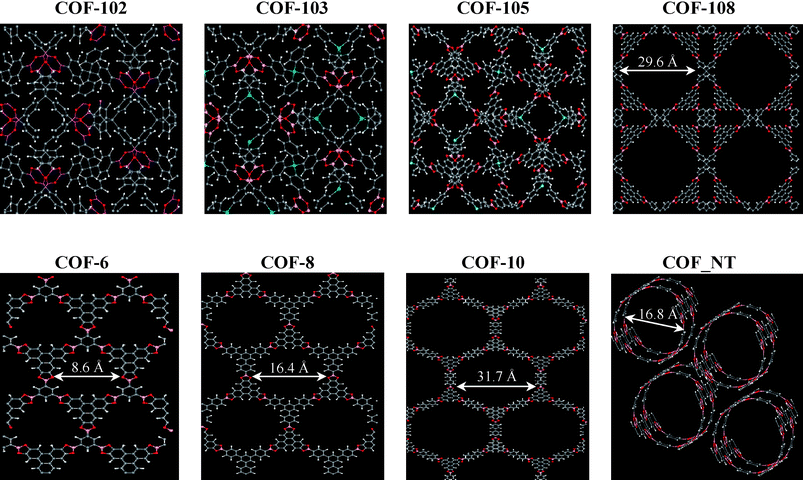 | ||
| Fig. 1 Atomic structures of COF-102, COF-103, COF-105, COF-108, COF-6, COF-8, COF-10 and COF_NT. The structures are not drawn to scale. B: pink, C: grey, O: red, Si: cyan, H: white. | ||
Currently there are scarce experimental or simulation studies of gas adsorption in COFs. Ar and N2 isotherms were measured experimentally in different COFs.9,10 H2storage at 77 and 300 K was determined in COF-1 using the spillover technique.12 Adsorption isotherms of Ar, CH4 and H2 in COF-102, -103, -105 and -108 were simulated, in which COF-102 and COF-103 were found to show a greater affinity for CH4 due to the compact atomic structure.13 In this work, we have systematically investigated CO2storage in a variety of 3D, 2D and 1D COFs using atomistic simulations. The adsorption capacities were determined over a wide range of pressure at room temperature. In order to establish the structure–function relations, both gravimetric and volumetric capacities at 30 bar were correlated with the framework density, free volume, porosity and accessible surface area.
CO2 was represented by the elementary physical model fitted to the experimental vapor–liquid equilibrium data of bulk CO2.14 The partial charges were +0.6645e on the C atom and −0.33225e on the O atom (e = 1.6022 × 10−19C is the elementary charge). The C–O bond was assumed to be rigid at 1.161 Å, while the ∠OCO bond was flexible and governed by a harmonic potential ½kθ(θ − θ0)2. The force constant kθ/kB is 153355.79 (K rad−2) and the equilibrium angle θ0 is 180°. CO2–CO2 intermolecular interaction was modeled as the additive pair-wise site–site Lennard–Jones (LJ) and coulombic potentials. The Lorentz–Berthelot combining rules were used to calculate the LJ cross parameters. The dispersive interactions of the atoms in COFs were modeled by the universal force field.15 The atomic charges in COFs were fitted to the electrostatic potentials calculated from the B3LYP density functional theory (DFT) with 6-31+G(d) basis set. All the DFT computations were carried out using the Gaussian 03 suite of programs.16 Fig. S1 in the ESI† gives the cleaved clusters used in the DFT calculations and the atomic charges.
Most simulation studies of gas adsorption use the grand canonical Monte Carlo (GCMC) method at fixed chemical potential, volume and temperature. The chemical potential has to be converted into pressure for comparison with experimental data, and this is usually implemented using empirical equation of state or additional simulations. Here we have used the Gibbs ensemble Monte Carlo (GEMC) simulation at a given pressure.17,18 Two simulation boxes are used; one for adsorbent and other for bulk adsorbate. The total number of adsorbate molecules is fixed, but molecules can be transferred from one box to the other. The volume of the adsorbent is fixed, while the bulk phase is allowed to change at fixed bulk pressure. The advantage of this method is one can directly obtain the uptake at a desired bulk pressure, as well as the bulk density and enthalpy. The latter are needed to calculate the excess adsorption and the heat of adsorption. In our case, the simulation box representing COF adsorbent contained (1 × 1 × 1) to (2 × 2 × 8) unit cells with the periodic boundary conditions exerted in all three directions. A spherical cutoff length of 15.0 Å was used to calculate the Lennard–Jones interactions with the tail correction added. The coulombic interactions were evaluated by the Ewald sum with an infinity dielectric constant in the surrounding. Four types of trial moves were randomly attempted including displacement and rotation in each phase, swapping between the two phases, and volume change of the bulk phase. The number of trial moves in a typical simulation was 2 × 107, though additional trial moves were used at high coverages. The first 107 moves were used for equilibration, and the second 107 moves to obtain ensemble averages.
Experimentally determined adsorption is usually reported in the excess amount Nex, while simulation gives the absolute amount Nab. The excess amount Nex is the total amount of adsorbate in the framework pores minus the amount that would be present if the pores were filled with the bulk adsorbate. To convert from Nab to Nex, Nex = Nab − ρbVfree was used, where ρb is the density of bulk adsorbate and Vfree is the free volume estimated using a non-adsorbing species (He) as a probe.5 The ratio of Vfree to the occupied volume Vtotal gives the porosity of adsorbent. The accessible surface area Asurf was estimated using CO2 (kinetic diameter of 3.30 Å) to roll over the framework surface. In addition, canonical ensemble simulations were performed to estimate the isosteric heat of adsorption and Henry constant at infinite dilution. All simulations were performed at 300 K using in-house developed code.
To characterize the adsorbent structure, the framework density ρf, free volume Vfree, porosity ϕ and accessible surface area Asurf for each COF were calculated. Table 1 lists these properties as well as the isosteric heat q0st and Henry constant KH, which reflect the affinity with the framework at infinite dilution. The 3D COFs, particularly COF-105 and COF-108, have the lowest ρf and largest Vfree, ϕ and Asurf; followed by 1D COF_NT and then 2D COFs. COF-102 and COF-103 have similar structures except that Si atoms replace the tetrahedral C atoms in COF-102. The presence of Si atoms in COF-103 slightly increases the affinity of CO2 with the framework and in turn leads to a higher q0st and KH. Though ρf and ϕ of COF-105 and COF-108 are nearly the same, q0st is higher in COF-105. This is due to the relatively more compact atomic packing in COF-105 than in COF-108 that enhances the interaction strength with CO2. In 2D COFs of layered structures, q0st and KH are sensitive to the inter-layer distance. With a short distance (3.399 Å), ρf, q0st and KH in COF-6 are significantly larger than all other COFs attributed to the overlap of attractive space between CO2 and the framework. With increasing distance in COF-8 (3.630 Å) and COF-10 (3.526 Å), q0st and KH decrease. In 1D COF_NT, q0st and KH are comparable to or slightly larger than in 3D COFs, but lower than in 2D COFs.
| ρ f/g cm−3 | V free/cm3 g−1 | ϕ | A surf/m2 g−1 | q 0st/kJ mol−1 | K H /mmol cm−3kPa−1 | |
|---|---|---|---|---|---|---|
| COF-102 | 0.41 | 2.02 | 0.85 | 5172 | 16.54 | 0.013 |
| COF-103 | 0.39 | 2.20 | 0.86 | 5366 | 18.13 | 0.014 |
| COF-105 | 0.18 | 5.22 | 0.94 | 6636 | 21.98 | 0.012 |
| COF-108 | 0.17 | 5.59 | 0.95 | 6298 | 14.22 | 0.003 |
| COF-6 | 1.07 | 0.47 | 0.50 | 1288 | 32.79 | 0.517 |
| COF-8 | 0.71 | 0.86 | 0.61 | 1911 | 24.81 | 0.043 |
| COF-10 | 0.48 | 1.62 | 0.78 | 2214 | 24.14 | 0.034 |
| COF_NT | 0.49 | 1.59 | 0.78 | 3509 | 20.91 | 0.022 |
Fig. 2 shows the excess adsorption isotherms of CO2 in COFs. At low pressures, there is a steep rise in 2D COFs as a consequence of the constricted pores within the layered structures, particularly, in COF-6. At high pressures, however, the saturation capacities in 2D COFs are lower compared to 3D COFs because of smaller available free volumes. COF_NT has a close saturation capacity with COF-10 as they possess approximately the same free volume. In 3D COFs, COF-102 and COF-103 show higher adsorption at low pressures than COF-105 and COF-108. This is due to the compact packing of atoms, which in turn increases the strength of interaction of CO2 with the adsorbent. This has been recently observed from the simulation study of CH4 adsorption in COF-102 and COF-103.13 The framework density decreases from 0.39 g cm−3 in COF-103 to 0.18 g cm−3 in COF-105 and 0.17 g cm−3 in COF-108. In contrast, the porosity increases from 0.86 to 0.94 and 0.95, the free volume increases significantly from 2.20 to 5.22 and 5.59 cm3 g−1, and the surface area increases from 5366 to 6636 and 6298 m2 g−1. As a consequence, COF-105 and COF-108 have the largest capacity for CO2 adsorption at high pressures compared to other COFs, although the adsorption is lower at low pressures. Since the pores in an adsorbent are almost fully filled at high pressures and adsorption approaches saturation, adsorbent with a larger free volume has more space to accommodate sorbate molecules and hence exhibits a higher capacity. At 30 bar, the capacities in COF-105 and COF-108 are 82 and 96 mmol g−1, respectively. These values are two to three fold greater than in MOF-177, which was reported experimentally to be the highest as of 33 mmol g−1.3 Using the framework density shown in Table 1, the gravimetric capacity is converted into the volumetric capacity. While COF-105 and COF-108 have significantly higher gravimetric capacities than other COFs, in a volumetric basis the capacities are close due to the cancelling effect of the framework density.
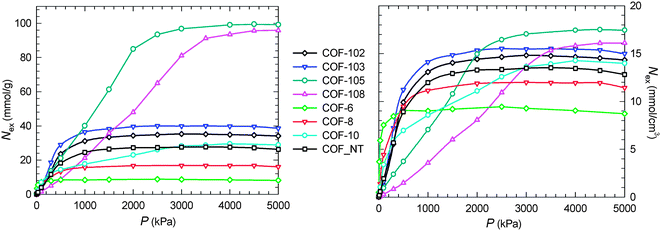 | ||
| Fig. 2 Gravimetric (left) and volumetric (right) isotherms of CO2 adsorption in COF-102, COF-103, COF-105, COF-108, COF-6, COF-8, COF-10 and COF_NT. Symbols are from simulation and the lines are to guide the eye. | ||
In order to identify the favorable locations of CO2 adsorption in COFs, the density distribution contours in the xy plane of COF-108, COF-6 and COF_NT at 1000 kPa are shown in Fig. 3. The contours are generated by accumulating the centers-of-mass of CO2 molecules in 100 equilibrium configurations. In COF-108, CO2 adsorption occurs preferentially around the carbon–oxygen–boron rings where CO2 interacts strongly with the framework. At higher pressures (not shown), adsorption also occurs in the central cavity. As the inter-layer distance in COF-6 is short, CO2 molecules predominantly intercalate the constricted central pores. In COF_NT, CO2 is adsorbed at the annular layer and the center inside the nanotube as well as into the interstitial channels among the nanotubes. This observation found here is similar to gas adsorption in carbon nanotubes at high pressures.18
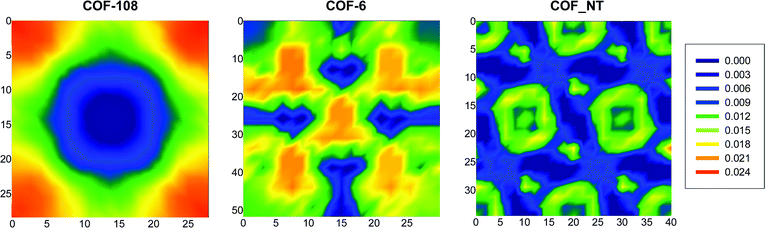 | ||
| Fig. 3 Density distribution contours for the center-of-mass of CO2 molecules in COF-108, COF-6 and COF_NT at 1000 kPa. | ||
The ability of COFs for CO2storage is quantitatively accessed by examining the capacity at 30 bar as a function of the framework density ρf, free volume Vfree, porosity ϕ and accessible surface area Asurf. In Fig. 4a, the gravimetric capacity Ngraex drops inversely proportional to the framework density ρf, whereas the volumetric capacity Nvolex drops linearly. Both can be correlated well with ρf
| Ngraex = −5.58 + 16.55/ρf | (1.1) |
| Nvolex = 17.10 − 7.05ρf | (1.2) |
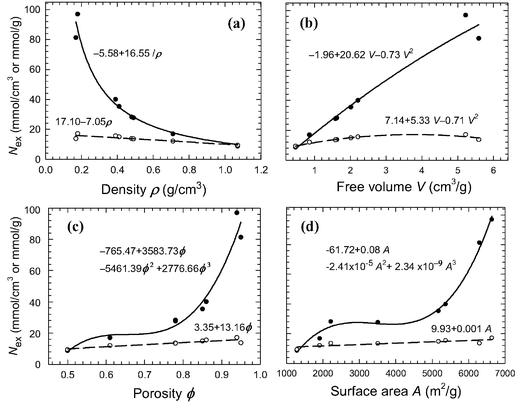 | ||
| Fig. 4 CO2 capacities at 30 bar as a function of (a) framework density (b) free volume (c) porosity (d) accessible surface area. Solid circles and curves: gravimetric capacity, open circles and dashed curves: volumetric capacity. | ||
In Fig. 4b, while Ngraex rises with the free volume Vfree, Nvolex first rises and then tends to drop slightly. The relations of Ngraex and Nvolex with Vfree are
| Ngraex = −1.96 + 20.62Vfree − 0.73V2free | (2.1) |
| Nvolex = 7.14 + 5.33Vfree − 0.71V2free | (2.2) |
In Fig. 4c, both Ngraex and Nvolex rise with the porosity ϕ following
| Ngraex = −765.47 + 3583.73ϕ − 5461.39ϕ2 + 2776.66ϕ3 | (3.1) |
| Nvolex = 3.35 + 13.16ϕ | (3.2) |
Similar to Fig. 4c, both Ngraex and Nvolex in Fig. 4d rises with the accessible surface area Asurf. As a function of Asurf, Ngraex and Nvolex can be correlated as
| Ngraex = −61.72 + 0.08Asurf − 2.41 × 10−5A2surf + 2.34 × 10−9A3surf | (4.1) |
| Nvolex = 9.93 + 0.001Asurf | (4.2) |
Therefore, good correlations exist between Ngraex and Nvolex with the framework density, free volume, porosity and accessible surface area. Ngraex and Nvolex can be enhanced by decreasing the framework density, increasing free volume, porosity or accessible surface area. However, not all these options are practically feasible and they actually interplay with one another. These correlations are useful for a prior prediction of CO2 adsorption capacities in to-be-synthesized adsorbents before experimental tests.
In conclusion, we have investigated CO2storage in 3D, 2D and 1D COFs using atomistic simulations. The 3D COFs possess larger free volume, porosity and surface area than the 2D and 1D COFs. COF-105 and COF-108 show exceptionally high storage capacity and even surpass the experimentally reported highest capacity in MOF-177. The compact atomic packing in COF-102 and COF-103 enhances isosteric heat and subsequently strong adsorption at low pressures. COF-6 exhibits the largest isosteric heat and Henry constant due to the constricted pores within the layered framework. Adsorption in 1D COF_NT is similar to that in a carbon nanotube. The complex factors like framework density, free volume, porosity and accessible surface area come into play in determining the adsorption capacity, and correlate well with the gravimetric and volumetric capacities of CO2 at 300 K and 30 bar. Using these structure–function correlations, COFs can be rationally screened for high CO2storage with minimal information on the structure.
Acknowledgements
We are grateful to Mário S. C. Mazzoni for kindly providing the structure of COF_NT and to the National University of Singapore for support (Grants R-279-000-198-112/133 and R-279-000-243-123).References
- A. Yamasaki, J. Chem. Eng. (Japan), 2003, 36, 361 Search PubMed.
- O. M. Yaghi, M. O'Keeffe, N. W. Ockwig, H. K. Chae, M. Eddaoudi and J. Kim, Nature, 2003, 423, 705 CrossRef CAS.
- A. R. Millward and O. M. Yaghi, J. Am. Chem. Soc., 2005, 127, 17998 CrossRef CAS.
- G. Garberoglio, A. I. Skoulidas and J. K. Johnson, J. Phys. Chem. B, 2005, 109, 13094 CrossRef CAS.
- R. Babarao, Z. Q. Hu, J. W. Jiang, S. Chempath and S. I. Sandler, Langmuir, 2007, 23, 659 CrossRef CAS.
- R. Babarao and J. W. Jiang, Langmuir, 2008, 24, 6270 CrossRef CAS.
- K. S. Walton, A. R. Millward, D. Dubbeldam, H. Frost, J. J. Low, O. M. Yaghi and R. Q. Snurr, J. Am. Chem. Soc., 2008, 130, 406 CrossRef CAS.
- A. P. Côté, A. I. Benin, N. W. Ockwig, M. O'Keeffe, A. J. Matzger and O. M. Yaghi, Science, 2005, 310, 1166 CrossRef CAS.
- A. P. Côté, H. M. El-Kaderi, H. Furukawa, J. R. Hunt and O. M. Yaghi, J. Am. Chem. Soc., 2007, 129, 12914 CrossRef CAS.
- H. M. El-Kaderi, J. R. Hunt, J. L. Mendoza-Cortes, A. P. Côté, R. E. Taylor, M. O'Keeffe and O. M. Yaghi, Science, 2007, 316, 268 CrossRef CAS.
- M. A. Lino, A. A. Lino and M. S. C. Mazzoni, Chem. Phys. Lett., 2007, 449, 171 CrossRef CAS.
- Y. W. Li and R. T. Yang, AIChE J., 2008, 54, 269 CrossRef CAS.
- G. Garberoglio, Langmuir, 2007, 23, 12154 CrossRef CAS.
- J. G. Harris and K. H. Yung, J. Phys. Chem., 1995, 99, 12021 CrossRef CAS.
- A. K. Rappe, C. J. Casewit, K. S. Colwell, W. A. Goddard III and W. M. Skiff, J. Am. Chem. Soc., 1992, 114, 10024 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, V. G. Zakrzewski, J. A. Montgomery, R. E. Stratmann, J. C. Burant, S. Dapprich, J. M. Millam, A. D. Daniels, K. N. Kudin, M. C. Strain, O. Farkas, J. Tomasi, V. Barone, M. Cossi, R. Cammi, B. Mennucci, C. Pomelli, C. Adamo, S. Clifford, J. Ochterski, G. A. Petersson, P. Y. Ayala, Q. Cui, K. Morokuma, D. K. Malick, A. D. Rabuck, K. Raghavachari, J. B. Foresman, J. Cioslowski, J. V. Ortiz, B. B. Stefanov, G. Liu, A. Liashenko, P. Piskorz, I. Komaromi, R. Gomperts, R. L. Martin, D. J. Fox, T. Keith, M. A. Al-Laham, C. Y. Peng, A. Nanayakkara, C. Gonzalez, M. Challacombe, P. M. W. Gill, B. G. Johnson, W. Chen, M. W. Wong, J. L. Andres, M. Head-Gordon, E. S. Replogle and J. A. Pople, Gaussian 03, Revision D.01 ed. Gaussian, Inc., Wallingford CT, 2004 Search PubMed.
- J. W. Jiang and S. I. Sandler, Langmuir, 2003, 19, 5936 CrossRef CAS.
- J. W. Jiang and S. I. Sandler, Phys. Rev. B, 2003, 68, 245412 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Atomic charges in COF-102, COF-103, COF-105, COF-108, COF-6, COF-8, COF-10 and COF_NT. See DOI: 10.1039/b805473h |
| This journal is © The Royal Society of Chemistry 2008 |