Organogels as scaffolds for excitation energy transfer and light harvesting
Ayyappanpillai
Ajayaghosh
*,
Vakayil K.
Praveen
and
Chakkooth
Vijayakumar
Photosciences and Photonics Group, Chemical Sciences and Technology Division, National Institute for Interdisciplinary Science and Technology, (NIST), CSIR, Trivandrum 695 019, India. E-mail: ajayaghosh62@gmail.com; Fax: (+91) 471-249-0186
First published on 10th September 2007
Abstract
The elegance and efficiency by which Nature harvests solar energy has been a source of inspiration for chemists to mimic such process with synthetic molecular and supramolecular systems. The insights gained over the years from these studies have contributed immensely to the development of advanced materials useful for organic based electronic and photonic devices. Energy transfer, being a key process in many of these devices, has been extensively studied in recent years. A major requirement for efficient energy transfer process is the proper arrangement of donors and acceptors in a few nanometers in length scale. A practical approach to this is the controlled self-assembly and gelation of chromophore based molecular systems. The present tutorial review describes the recent developments in the design of chromophore based organogels and their use as supramolecular scaffolds for excitation energy transfer studies.
 Ayyappanpillai Ajayaghosh | Ayyappanpillai Ajayaghosh is a native of Kerala, the south west coastal state of India. After his PhD in Chemistry from Calicut University (1988, from the group of Prof. V. N. R. Pillai), he joined the Regional Research Laboratory (presently NIST) as a scientist. Subsequently he was an Alexander von Humboldt fellow at the Max-Planck-Institut für Strahlenchemie, Muelheim an der Ruhr, Germany (1994–1996) and a visiting scientist to many universities in Germany, Japan and The Netherlands. Currently, he is a Senior Scientist of the National Institute for Interdisciplinary Science and Technology, Trivandrum and an Adjunct Professor of the Material Science Programme of the Indian Institute of Technology, Kanpur. He is a Fellow of the Indian Academy of Sciences and a member of the International Advisory Board, Chemistry: An Asian Journal. His research interests are in the area of photonically and electronically active organic and macromolecular materials, particularly the supramolecular chemistry of functional dyes and π-conjugated systems, molecular self-assemblies, organogels, light harvesting assemblies and molecular probes. |
 Vakayil K. Praveen | Vakayil K. Praveen obtained his MSc degree in Applied Chemistry (2001) from Calicut University, Kerala. Currently he is working as a senior research fellow and has completed his studies for a PhD under the guidance of Dr A. Ajayaghosh. His research is focused on π-organogels and their application as excitation energy donor scaffolds. |
 Chakkooth Vijayakumar | Chakkooth Vijayakumar obtained his MSc degree in Chemistry from Calicut University, Kerala (2002). Currently he is working as a PhD student under the guidance of Dr A. Ajayaghosh. His thesis is focused on chromophore self-assemblies and their application as donor scaffolds for resonance energy transfer. |
1 Introduction
Nature has the unique capability to tap the resources available for the benefit of living organisms. One such classic example is natural photosynthesis which harvests solar energy. Photosynthetic organisms including plants, algae and photosynthetic bacteria are considered as the great masters of solar energy harvesting. Pigments (chlorophylls and carotenoids) are the primary components of natural light harvesting systems, responsible for converting the energy of an absorbed photon into an electronic excitation. The mesoscale organization of these pigment molecules in the photosynthetic membrane allows the unidirectional excitation energy transfer towards a reaction center, which is the final destination of the collected energy.1 Inspired by Nature, scientists have been attempting to mimic such process with the help of synthetic molecular architectures. The understanding of the mechanism involved in the natural light harvesting processes is not only helped to the design of artificial systems but also made substantial contributions to the development of nanoscale optoelectronic and photonic devices.1–7 A challenging task to the design of artificial light harvesting assemblies is the organization of chromophores with precise control on distance and orientation, which is an indispensable criterion for efficient energy transfer. Several strategies have adopted for this purpose. Among them, self-organization of chromophores owing to its similarity with natural light harvesting assemblies has attracted considerable interest. Apart from this, self-assembly of chromophores, especially that of linearly π-conjugated systems have paramount importance in the emerging field of molecular and supramolecular electronics.8–11Recently, excitation energy transfer in supramolecular aggregates derived from the self-assembly of π-conjugated molecules have been reported.12–18 For example, in the excited state, self-assembled oligo(p-phenylenevinylene)s (OPVs) are known to be excellent energy donors to suitable acceptors.12–16 The majority of these studies have been demonstrated with aggregates which are present in solution as dynamic self-assemblies, though there are a few exceptions. In order to improve the efficiency of energy transfer and also from the view point of device applications, it is important to understand the energy transfer processes in extended self-assemblies of donor–acceptor systems. In this context, energy transfer in gel scaffolds has attracted significant attention. The purpose of the present tutorial review is to put into perspective the recent developments in the design of supramolecular light harvesting assemblies, particularly the use of chromophore based organogels as scaffolds for excitation energy transfer. Emphasis has been given to OPV based organogels as excitation energy transfer donors. Since molecular self-assembly and gel chemistry are at the centre stage of contemporary research, both from fundamental and technological significance, it is appropriate and timely to provide an overview of the recent developments and future potentials of this area to the scientific community. In the subsequent discussions, the basic requirements of donor–acceptor energy transfer systems, importance of chromophore organization, role of organogelators as media for energy transfer, and chromophore based organogelators as energy donor scaffolds to the design of light harvesting assemblies are included.
2 Excitation energy transfer in donor–acceptor systems
The transfer of excited-state energy from initially excited donor molecules to an acceptor in the ground state is often referred to as excitation energy transfer. The general requirements for excitation energy transfer between a donor and an acceptor are: (i) the energy of donor excited state should be higher than that of acceptor excited state and (ii) the rate of energy transfer should be more rapid than the decay rate of the donor excited state. Excitation energy transfer from donors to acceptors separated by a distance ‘R’ occurs through Dexter (collisional) or Förster (Coulombic) mechanisms.19 Dexter transfer is a wave-function overlap that has exponential distance dependence within ∼1 nm range whereas Förster transfer is a near-field resonant dipole–dipole interaction with a distance dependence of ‘R–6’ and cover a greater range of ∼10 nm. For efficient energy transfer from donors to acceptors at distances of >10 nm, fast energy migration is required, especially in the presence of very low concentrations of the acceptor molecules. In natural light harvesting assembly, these crucial criteria of energy transfer are satisfied by organizing the pigment molecules in a fixed spatial relationship by the surrounding polypeptides with the help of non-covalent interactions, which ultimately results in an efficient and directional energy transfer process.3 Importance of chromophore organization in energy transfer
As indicated above, the distance between the energy donor and the acceptor and their relative orientation is extremely important in facilitating the energy transfer process. However, in artificial systems, a precise arrangement of chromophores for optimum performance continues to be challenging. Several strategies have been developed to the organization of donor–acceptor molecules which involves covalent and noncovalent approaches. The use of dendrimers1–3,20,21 and fixation of chromophores on polymer backbones22,23 are examples of the covalent approaches (Fig. 1). Apart from these, attempts to use self-assembled monolayers,24 blends of organic dyes in conjugated molecules as thin films,25,26 encapsulation of molecules in zeolite crystals27 or cyclodextrin28,29 and the use of hydrogen-bonded supramolecular assemblies14–16,30,31 have been found efficient to the arrangement of chromophores (Fig. 1). Nevertheless, the current trend in the design of light harvesting assembly is the creation of hydrogen-bond or π-stack driven assembly of donor–acceptor molecules that form 1D nanoarchitectures.14–18 An alternative approach to this direction is the use of hierarchically self-assembled organogels as scaffolds for excitation energy transfer to an entrapped acceptor.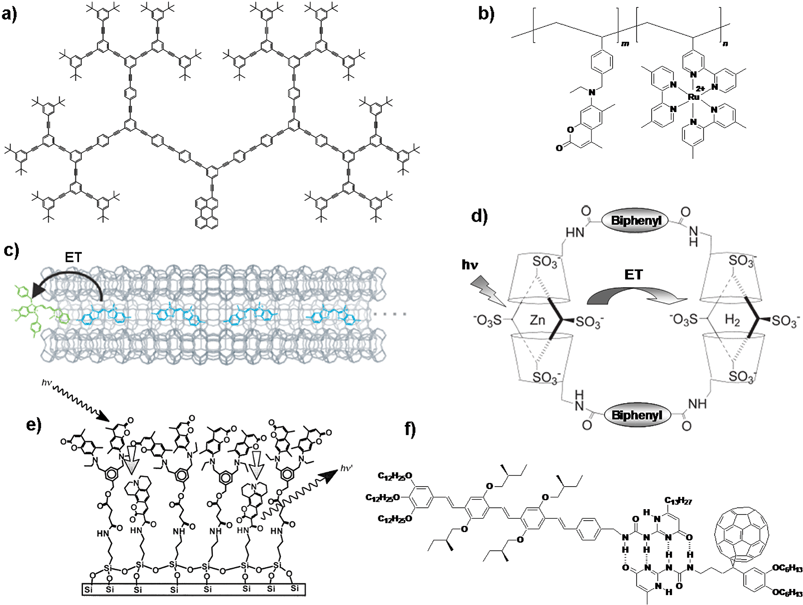 | ||
| Fig. 1 Different approaches to the design of organized donor–acceptor systems: (a) light harvesting dendrimer,21 (b) chromophore linked polymer,23 (c) dye encapsulated zeolite L crystals,27 (d) porphyrin cyclodextrin assembly,28,29 (e) self-assembled monolayer24 and (f) hydrogen bonded assembly.31 ET = energy transfer. Fig. 1(c) reprinted with permission from ref. 27. Copyright 2006, Wiley-VCH; Fig. 1(d) reprinted with permission from ref. 28. Copyright 2005, The Royal Society of Chemistry and Fig. 1(e) reprinted with permission from ref. 24. Copyright 2000, Wiley-VCH. | ||
4 Low molecular weight organogels: the world of soft materials
Certain organic molecules spontaneously self-assemble to form a soft solid-like mass called a ‘gel’ by entrapping a large volume of the solvent between the self-assembled structures.32–37 Gels can be defined as self-assembled materials that are composed of extended three-dimensional networks whose interstitial spaces are filled with solvents and hence behave like soft solids. Historically, most gels are composed of covalently crosslinked polymers having very high molecular weights. These gels can be swollen or shrunk by the addition or removal of a solvent, and by temperature. Another class of gelator comprises of small molecules. Recently, there has been an enormous increase of interest to the design of such low molecular weight gelators, which immobilize various organic fluids as a result of three-dimensional supramolecular network formation.In contrast to macromolecular gels, low molecular weight organogel networks are held together solely by noncovalent interactions such as hydrogen bonding, π-stacking, solvophobic effects and donor–acceptor interactions, rendering the gelation process completely reversible. The directional nature of intermolecular interactions allows gelator molecules to self-organize in one-dimensional arrays producing elongated fibrous structures. Entanglement of the fibers subsequently produces a three-dimensional network capable of trapping the solvent and yielding the gel. Usually, low molecular weight organogels are formed by dissolving a small amount of the gelator in hot solvent followed by cooling below the gel transition temperature (Tgel). The gelation of organic molecules can be visualized by the formation of a thick non-flowing mass, even when turned upside down. Organogels are characterized using a variety of analytical tools, particularly spectroscopic and microscopic techniques. A number of excellent reviews are available for the details of gel chemistry. Readers may go through these for a better understanding of the recent developments in this field.32–37
Molecular gels exhibit diverse supramolecular architectures with striking properties. The diversity of nanostructures provided by organogels make them promising candidates for several potential applications ranging from food, cosmetics, medicine, tissue engineering, biomineralization, catalysis, controlled release, and to advanced materials.32–37 In recent years, organogelators based on functional chromophores and dyes have attracted immense interest as novel functional materials for optoelectronic applications.16,35 Supramolecular alignment of chromophoric assemblies achieved through gelation provides functional materials with tunable optoelectronic properties which have potential applications in molecular and supramolecular electronic devices. For example, chromophore based organogels are excellent model systems for the active layers in plastic electronic devices.35,38,39 Therefore, a large variety of chromophore based organogelators have been synthesized and studied which include chromophores such as phthalocyanines, porphyrins, [n]acenes, pyrene, cyanine, squaraine, fluorene, tetrathiafulvene, phenanthroline, azobenzene, stilbene, butadiene, hexabenzocoronene, thiophene, phenylene, phenylacetylene, phenylenevinyleneetc.16,35 Among these, chromophores such as naphthalene, anthracene, pyrene, perylenes, phenylenevinylenes etc. are useful systems for energy transfer processes. In recent years, a large number of reports have appeared on organogelators based on these chromophores as scaffolds for energy transfer and light harvesting applications.
5 Organogelators for energy transfer and light harvesting
One of the early examples of energy transfer in a gel medium is reported by Sagawa et al.40Pyrene and porphyrin functionalized L-glutamate derivatives 1 and 2 have been synthesized and used for this purpose. Detailed gelation studies revealed that the pyrene derivative 1 forms physical gels in benzene and cyclohexane whereas the porphyrin derivative 2 failed to gelate any of the solvents tried. Spectroscopic studies suggest that amide hydrogen bonding leads these molecules to form ordered co-facial chiral aggregates even in the solution state. Energy transfer studies in the mixed assemblies of 1 and 2 revealed singlet–singlet energy migration from pyrene excimers to the free base porphyrin (Fig. 2).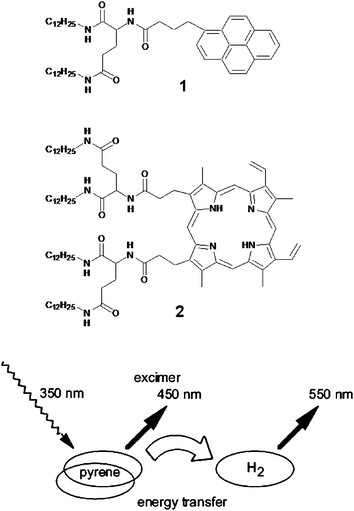 | ||
| Fig. 2 Didodecyl L-glutamic acid substituted pyrene (1) and porphyrin (2) form mixed assemblies in benzene. The energy transfer process from the self-assembled pyrene excimer to porphyrin is schematically depicted. Reprinted with permission from ref. 40. Copyright 2002, American Chemical Society. | ||
Nakashima and Kimizuka have used the electrostatic interaction of the anionic naphthalene 3 (energy donor) and the anthracene derivative 4 (energy acceptor) within the fibrous assemblies of the cationic L-glutamate derivatives 5 or 6 to the design of a light harvesting gel.41 The regular packing of the glutamate chains into the bilayer assemblies ensured an ordered arrangement of the naphthalene chromophores (Fig. 3). Such a supramolecular assembly helped the singlet energy hopping between the naphthalene chromophores. When a small concentration of the anthrancene derivative 4 is added to an aqueous gel of 3/5, efficient energy transfer from the naphthalene donors to the anthracene acceptors is observed as indicated by the quenching of the naphthalene fluorescence and the concomitant rise of the anthracene emission. Moreover, the efficiency of the energy transfer process in the gel state is high when compared to that in the solution state, demonstrating the importance of organizing the donor–acceptor chromophores using the gel matrix.
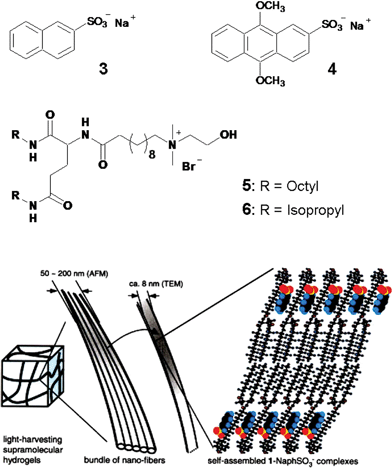 | ||
| Fig. 3 A cartoon representation of the self-assembly of the anionic naphthalene (3) and anthracene (4) within the cationic glutamate (5 or 6) derivatives resulting in supramolecular light harvesting hydrogels. Reprinted with permission from ref. 41. Copyright 2002, Wiley-VCH. | ||
Shinkai and co-workers have reported an energy transfer cascade in co-assembled organogels of cholesterol functionalized perylene bisimide gelators 7a–d (Chart 1).42 The difference in the HOMO–LUMO gap between perylene derivatives is exploited for energy transfer in various binary, ternary and quaternary perylene gels. For example, selective excitation of 7a at 457 nm resulted in the quenching of its emission at λem = 544 nm with efficiencies of 68% for 7b, 53% for 7c and 34% for 7d, consistent with decreasing donor–acceptor spectral overlap (Fig. 4). The perylene derivative 7d acts as an energy sink because of its low emissive nature that arises from the twisted intramolecular charge transfer (TICT) state. These trends are visually shown in the inset in Fig. 4. It is interesting to note that the supramolecular organization of the chromophores in the gel state guarantees the success of this cascade transfer process, whereas in solution the mentioned effects are not observed.
 | ||
| Chart 1 | ||
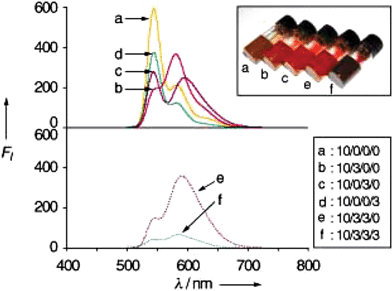 | ||
| Fig. 4 Fluorescence spectra of mixed perylene gels. The numbers in the inset denote the molar ratios for 7a/7b/7c/7d. The corresponding photographs of the mixed gels are also shown in the inset. Reprinted with permission from ref. 42. Copyright 2004, Wiley-VCH. | ||
The cholesterol appended 1,10-phenanthroline based gelator 8 is an example of a proton sensitive energy transfer system (Fig. 5).43 In the presence of two equivalents of trifluoroacetic acid (TFA), the purple emission (360 nm) of the gel phase of 8 is quenched completely, with the appearance of the yellow emission (530 nm) corresponding to the protonated form of 8 due to energy transfer from the neutral to the protonated form. However, when the gel was converted into the solution phase at 90 °C in the presence of TFA, a blue emission with a bimodal maximum was obtained which indicated the absence of energy transfer.
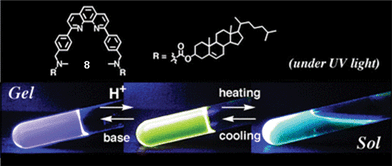 | ||
| Fig. 5 Emission changes in the cholesterol appended phenanthroline based gelator 8 upon protonation. Reprinted with permission from ref. 43. Copyright 2003, The Royal Society of Chemistry. | ||
The metallo-supramolecular gel of compound 9 which responds to metal–ligand interactions is an example of a stimuli responsive gel.44 The different binding modes of the tridentate ligand 2,6-bis(1′-methylbenzimidazolyl)-4-hydroxypyridine (BIP) with transition metal ions (2 : 1 complex) and lanthanide ions (3 : 1 complex) are advantageous to the formation of a supramolecular gel (Fig. 6(a)). Compound 9 spontaneously forms a gel upon addition of the lanthanoid(III) nitrate (3 mol%) followed by the transition metal ion perchlorate (97 mol%) to a solution of 9 in CHCl3–CH3CN. Excitation (λex = 340 nm) of the 9:Zn/Eu system results in intense emission from Eu(III) ions (Fig. 6(b)). The emission is due to efficient ligand to metal energy transfer process. While 9:Zn/La displayed emission from metal-bound ligand (λex = 397 nm), 9:Co/Eu did not emit at all, probably due to the presence of low energy metal centered levels which facilitate radiationless decay process. Addition of a small amount of formic acid to the luminescent gel of 9:Zn/Eu results in the disruption of the gel with strong quenching of Eu(III) emission. This could be due to the displacement of BIP ligand by formate ions, which eventually ‘switch off’ the antenna effect. This process could be reversed by the evaporation of the solvent followed by reswelling of the material with CH3CN, which will restore the Eu(III) emission.
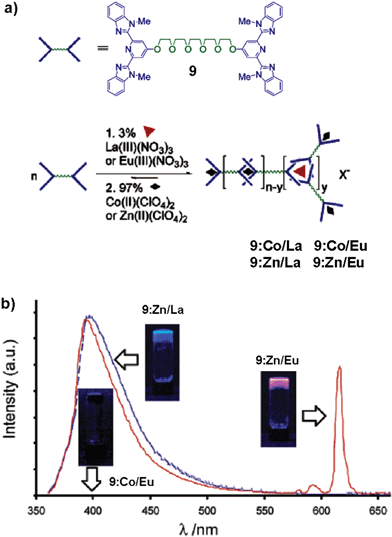 | ||
| Fig. 6 (a) Schematic representation of the formation of a metallo-supramolecular gel using a combination of lanthanoid and transition metal ions mixed with the monomer 9. (b) Photoluminescence spectra of metallo-supramolecular gels prepared in acetonitrile (λex = 340 nm). 9:Co/Eu shows no photoluminescence and lies essentially along the baseline. The insets show the gels under UV light (365 nm). Reprinted with permission from ref. 44. Copyright 2003, American Chemical Society. | ||
The versatility of the anthracene based organogel as a medium to study the energy transfer process has been demonstrated recently by Del Guerzo et al.45,46 They have used 2,3-n-didecyloxyanthracene (DDOA, 10) as the excitation energy donor and 5,12 or 2,3-n-dialkoxytetracene derivatives (DnOTs, 11–14) as the acceptors (Chart 2). These derivatives form gels in solvents such as alcohols, alkanes, aliphatic amines, dimethyl sulfoxide (DMSO), acetoneetc. The doping of DDOA gels with DnOTs facilitated energy transfer from excited anthracenes to the tetracene derivatives (Fig. 7(a)). Detailed energy transfer studies revealed that the efficiency of the process is highly influenced by the structural and chemical similarity between tetracene acceptors and DDOA. The high energy transfer efficiency of the present system with small amounts of the acceptor indicates the involvement of exciton migration in addition to the direct energy transfer between the closest donor and acceptor molecules (Fig. 7(b)).
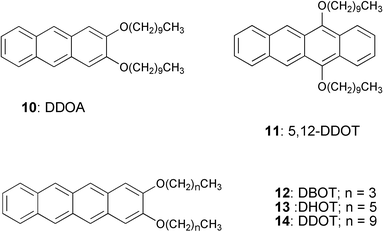 | ||
| Chart 2 | ||
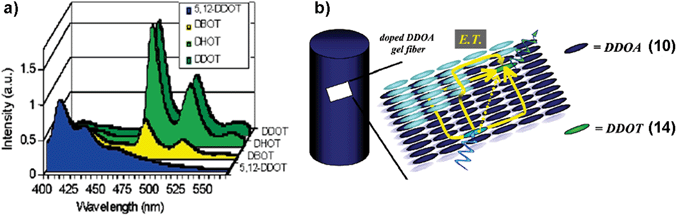 | ||
| Fig. 7 (a) Emission spectra of doped DDOA (10) gels in DMSO at 293 K, with 1 mol% of 5,12-DDOT (11), DBOT (12), DHOT (13) and DDOT (14), respectively. (b) Schematic representation of energy transfer process in DDOA gel fiber. Energy transfer pathways are depicted by yellow arrows. Reprinted with permission from ref. 45. Copyright 2005, American Chemical Society. | ||
Recently, energy transfer has been reported in the self-assembled cylindrical micellar structures of the amphiphilic triblock coil–rod–coil molecule 15 and the rod–coil–rod molecule 16 in aqueous solution (Fig. 8).47Addition of a small amount of 16 as a bridging agent to 15 resulted in the formation of a reversible nematic gel (Fig. 8(b)). Detailed morphological studies showed that these dynamic interconnections lead to stiff bundles composed of cylindrical micelles that are responsible for the formation of the nematic gel. More importantly, under this condition, excitation of the co-assembly at 290 nm, where most of the radiation is absorbed by 16, resulted in strong emission from 15 at 431 nm (Fig. 8(c)). These results indicate that energy transfer occurs from 16 to 15, which is an additional evidence for the formation of the co-assembly between these molecules.
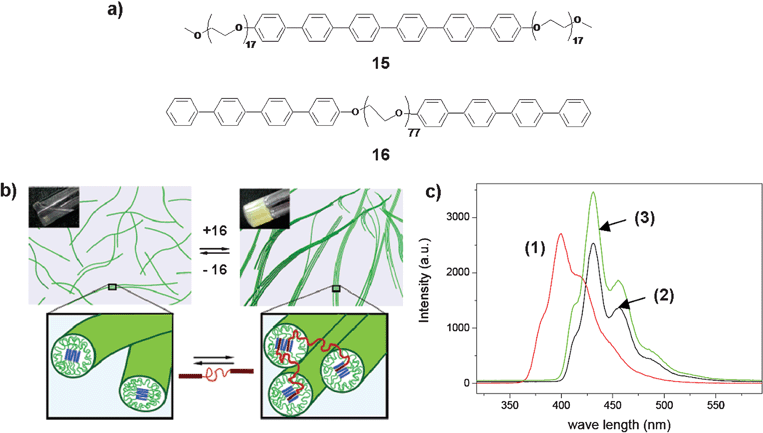 | ||
| Fig. 8 (a) Molecular structure of the coil–rod–coil molecule 15 and the rod–coil–rod molecule 16. (b) Cartoon representation of the reversible bridging between the isotropic fluid and the nematic gel of the supramolecular nanocylinders. (c) Fluorescence spectra (λex = 290 nm) of aqueous solutions of 16 (1), 15 (2) and 15 + 16 (3). Reprinted with permission from ref. 47. Copyright 2005, American Chemical Society. | ||
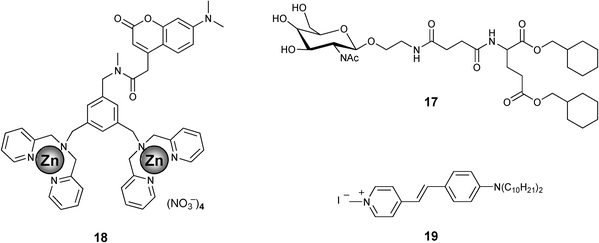
Energy transfer from artificial receptors to suitable acceptors which are noncovalently immobilized in a semi-wet supramolecular hydrogel is a unique system which is useful for the detection and discrimination of biologically important phosphate derivatives.48 A glycosylated amino acetate type hydrogelator 17 (Chart 3) is found to be effective for this purpose. The guest (phosphate derivatives) dependent redistribution of the artificial receptor in the hydrophobic and hydrophilic domain of the supramolecular hydrogel results in substantial difference in the FRET efficiency (Fig. 9(a)). For example, before guest addition, the coumarin-appended receptor 18 (Chart 3) is mainly localized in the bulk aqueous phase and the styryl dye 19 (Chart 3), which is the energy acceptor, is fixed in the hydrophobic domain of the hydrogelator 17. Upon phenyl phosphate (PhP) binding, the complexed receptor 18/PhP gets transferred to the more hydrophobic domain due to the increased hydrophobicity. Eventually, the average distance between the donor and the acceptor will be reduced which in turns facilitate efficient FRET. However, in the presence of adenosine triphosphate (ATP) the energy transfer process is found to be inefficient. This is attributed to the hydrophilic nature of the complexed receptor 18/ATP, which favors to localize predominately in the hydrophilic cavity of the gel. In the absence of the styryl dye 19, addition of PhP or ATP showed practically no change in the fluorescence of the receptor. Based on these observations a semi-wet molecular recognition (MR) chip for the rapid and high-throughput sensing has been developed (Fig. 9(b)).
 | ||
| Chart 3 | ||
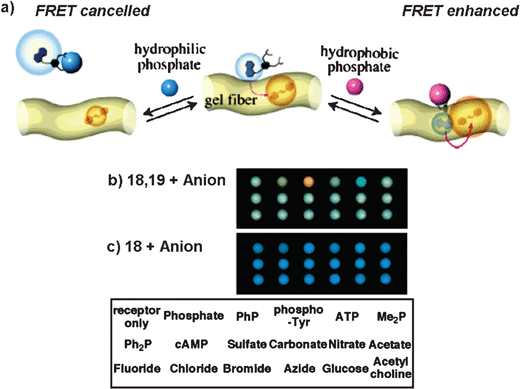 | ||
| Fig. 9 (a) Illustration of the guest-dependent FRET system using 18 and 19, by the addition of PhP or ATP in a hydrogel matrix. Digital camera photographs of the sensing patterns of the semi-wet molecular recognition (MR) chips of the hydrogel 17 containing (b) the coumarin-appended receptor 18 with the styryl dye 19 and (c) 18 without 19 in the presence of various anions. The spotted position of anions is shown in the bottom of the figure. Reprinted with permission from ref. 48. Copyright 2005, American Chemical Society. | ||
1,3,5-Cyclohexyltricarboxamide based hydrogelator 20, comprising of two hydrophilic moieties and one hydrophobic substituent containing a naphthalenic group is a good excitation energy donor to the dansyl fluorophore 21 (Fig. 10).49 The formation of H-type aggregates of the naphthalene moiety favors fast energy migration in the gel scaffold. Addition of 1% of the dansyl fluorophore 21, results in 50% quenching of the emission of the gel with strong emission at 490 nm. A five-fold higher concentration of 21 enhances the energy transfer efficiency to 75%. Under this condition both entrapped and free acceptors are present. The emission maximum of the entrapped 21 (λem = 490 nm, λex = 290 nm) after energy transfer indicates that the fluorophore is surrounded by a nonaqueous environment of the donor molecules within the water medium (Fig. 10(b)). On the other hand, direct excitation of 21 (λex = 365 nm) results in the shift of the emission maximum to 535 nm due to the combined emission from molecules existing in the gel fiber (λem = 490 nm) and in the solution (λem = 560 nm) (Fig. 10(c)). Detailed steady state and time resolved fluorescence spectroscopic studies revealed that around 30% of the total dansyl fluorophore (21) gets incorporated into the gel fiber and participated in the light harvesting process.
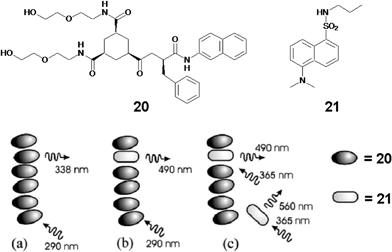 | ||
| Fig. 10 Representation of the energy transfer process in 1,3,5-cyclohexyltricarboxamide based hydrogelator 20 doped with dansyl fluorophore 21. Reprinted with permission from ref. 49. Copyright 2006, American Chemical Society. | ||
6 Oligo(p-phenylenevinylene) (OPV) derived light harvesting gels
Electron and energy transport properties of π-conjugated molecules are strongly influenced by the orientation of the chromophores within the self-assembly. A viable approach towards this end is the design of conjugated building blocks that form organogels in appropriate solvents through noncovalent interactions leading to entangled fibrous assemblies.16,35 Recently, we have shown the thermoreversible gelation of fluorescent OPV derivatives (Chart 4) and the control of their photophysical as well as morphological properties by structural modifications and noncovalent interactions.50–55 The most striking feature of the gelation of OPVs is the modulation in optical properties. Upon gelation, OPVs exhibit a large shift in the emission towards the long wavelength region (Fig. 11).50–52 | ||
| Chart 4 | ||
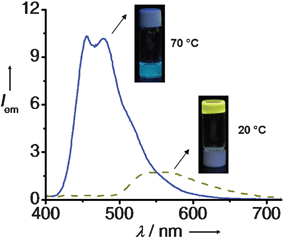 | ||
Fig. 11
Emission spectra of 21a gel (4 × 10–4 M) in cyclohexane at 20 °C (![[dash dash, graph caption]](https://www.rsc.org/images/entities/char_e091.gif) ) and 70 °C (—) (λex = 380 nm). Inset shows the corresponding emission color under illumination at 365 nm. ) and 70 °C (—) (λex = 380 nm). Inset shows the corresponding emission color under illumination at 365 nm. | ||
The appearance of the lower energy band in the absorption and a large red shift in the emission spectra during the self-assembly of OPVs indicate the delocalization of the excitons across more than one molecule.50–52,56 This could be due to efficient exciton migration within the aggregates of different HOMO–LUMO levels. This has been experimentally proved by time resolved emission studies.56 For example, the time resolved emission anisotropy measurements of a 21acyclohexane gel showed an extremely fast depolarization of emission corresponding to OPV aggregates, which is an indication of the fast interchromophore singlet exciton migration. Further evidence for energy migration is obtained from the time resolved emission spectra (TRES) and wavelength dependent emission decay profiles of the 21acyclohexane gel. With time, TRES of 21a showed a dynamic red shift in the emission spectra with a rapid decay of the lower wavelength shoulder band (Fig. 12(a)). Similarly, wavelength dependent decay profile of 21a revealed a rapid decay of emission in the short wavelength region and a delayed growth followed by decay in the higher wavelength region (Fig. 12(b)). Such observations point towards efficient exciton migration between the OPV aggregates. Such property of OPV gels is advantageous for their use as energy donor scaffolds to suitable acceptors. This has been demonstrated by entrapping rhodamine B dye within the self-assembled donor scaffolds of a variety of OPVs 21–23 (Chart 4) which facilitate resonance energy transfer (RET).57,58
![(a) Time-resolved emission spectra of 21a gel. (b) Emission decay curves of 21a gel monitored at different wavelengths. The inset shows initial growth in the emission decay of 21a gel within short time scales, monitored at different wavelengths after excitation. IRF (■), 478 nm (★), 504 nm (△), 536 nm (◆), 574 nm (□). In all experiments [21a] = 4 × 10–4 M in cyclohexane, l = 1 mm, λex = 375 nm. Reprinted with permission from ref. 56. Copyright 2007, Wiley-VCH.](/image/article/2008/CS/b704456a/b704456a-f12.gif) | ||
| Fig. 12 (a) Time-resolved emission spectra of 21a gel. (b) Emission decay curves of 21a gel monitored at different wavelengths. The inset shows initial growth in the emission decay of 21a gel within short time scales, monitored at different wavelengths after excitation. IRF (■), 478 nm (★), 504 nm (△), 536 nm (◆), 574 nm (□). In all experiments [21a] = 4 × 10–4 M in cyclohexane, l = 1 mm, λex = 375 nm. Reprinted with permission from ref. 56. Copyright 2007, Wiley-VCH. | ||
Energy transfer occurs exclusively from the gel phase and not from the corresponding single molecules. For example, when 21a in cyclohexane–chloroform (16 : 1) gel is excited at 380 nm in the presence of rhodamine B, the emission corresponding to the self-assembled species is selectively quenched (ca. 63%) with concomitant emission of the dye at 625 nm. Such an observation indicates the occurrence of RET from the self-assembled donors (Fig. 13(a)). Detailed energy transfer studies indicated that the efficiency of RET depends on the ability of the OPVs to form stable self-assemblies which is strongly influenced by the structure of the gelator as well as the solvent. Fig. 13(b) shows a comparison of the relative fluorescence intensities of rhodamine B with different OPVs against the respective molar ratios which revealed maximum RET efficiency for the OPV derivative 23 having carboxylic acid moieties as end functional groups. The improved efficiency of 23 with relatively less amount of the acceptor indicates a better interaction of the former with the latter when compared to that of 21 and 22. A unique feature of the present system is the thermal control on the RET process. The emission from the dyes is shut off in a thermoreversible fashion since the self-assembly breaks above the gel melting temperature.
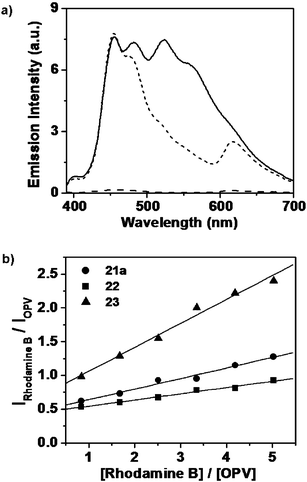 | ||
| Fig. 13 (a) Emission spectra of 21a in the absence (—) and in the presence () of rhodamine B in cyclohexane-chloroform (16 : 1) λex = 380 nm. Emission of rhodamine B (– – –) in the absence of 21a, on excitation at 380 nm is shown for a comparison. (b) Plots of relative fluorescence intensities against the molar ratio of the acceptor and the OPV donor in dodecane–chloroform (16 : 1). Reprinted with permission from ref. 58. Copyright 2006, American Chemical Society. | ||
In the case of the OPV–rhodamine gel system, the major drawback is the noncompatability of the cationic dye with the OPV self-assembly in nonpolar solvents, due to the aggregation and phase separation of the acceptor. A major portion of these aggregates may not be in contact with bulk of the gel matrix. Therefore, a large amount of the dye was required for the quenching of the gel emission. This could be solved to some extent in the case of a rhodamine B doped xerogel film. The RET efficiency was significantly enhanced (ca. 90%) in a xerogel film of 21a, which is dispersed with relatively less amount of the acceptor (33 mol%), when compared to that of the dodecane gel (Fig. 14).
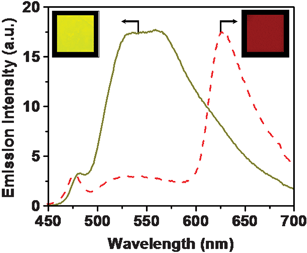 | ||
| Fig. 14 Emission from the xerogel film of 21a in the absence (—) and in the presence of rhodamine B () (33 mol%) upon excitation at 380 nm. Inset shows the emission colors of 21a and 21a + rhodamine B films under UV illumination (λex = 365 nm). Reprinted with permission from ref. 58. Copyright 2006, American Chemical Society. | ||
In order to design an efficient light harvesting system which can funnel excitation energy to a few acceptor molecules, it is necessary to have an acceptor which is compatible with the donor self-assembly. Recently we have addressed this issue with the help of the OPVs 24–27 having different HOMO–LUMO energy levels as donor and acceptor (Fig. 15(a)).59 The rational choice of OPVs with dipolar end functional groups allowed the tuning of the emission in the molecular level and further modulation of the optical properties in the supramolecular level (Fig. 15(b)). Detailed studies revealed that the optical properties of the gel-forming 24 (donor) and the nongelling 27 (acceptor) are suitable for energy transfer. Addition of 2.62 mol% of 27 to a n-decane gel of 24 showed a 90% quenching of the emission of the donor and the simultaneous emission from the acceptor (Fig. 15(c)). A three-fold increase in the luminescence of 27 after energy transfer (λex = 380 nm) when compared to that of the direct excitation (λex = 495 nm) is the indication of a significant light harvesting effect in the former case (Fig. 15(c) inset). However, RET is not efficient in polar solvents due to weak self-organization of the chromophores, which indicate the advantage of the gel scaffold in organizing the donor–acceptor chromophores.
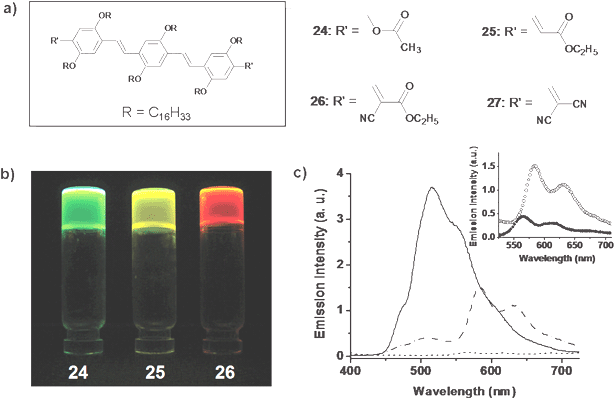 | ||
Fig. 15 (a) Molecular structure of OPV derivatives 24–27. (b) Photographs of the hexane gels under illumination at 365 nm. (c) Energy transfer between 24 and 27 in the gel state. Emission spectra of 24 in the absence (—) and in the presence (![[dash dot, graph caption]](https://www.rsc.org/images/entities/char_e090.gif) ) of 27 (2.62 mol%) in n-decane, and emission of acceptor in the absence of donor (). The inset shows the emission of the acceptor on indirect excitation at 380 nm (○) and direct excitation at 495 nm (●). Reprinted with permission from ref. 59. Copyright 2007, Wiley-VCH. ) of 27 (2.62 mol%) in n-decane, and emission of acceptor in the absence of donor (). The inset shows the emission of the acceptor on indirect excitation at 380 nm (○) and direct excitation at 495 nm (●). Reprinted with permission from ref. 59. Copyright 2007, Wiley-VCH. | ||
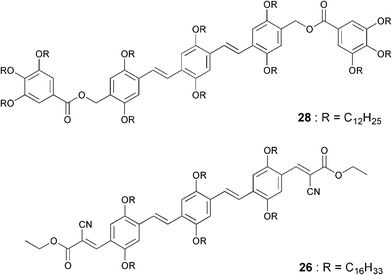
Efficient trapping of excitons by “isolated” or “aggregated” acceptors through a subtle control of the self-assembly and the photophysical properties of the donor–acceptor building blocks has been used to tune the emission of the acceptor. This could be successfully realized in a supramolecular light harvesting system consisting of OPVs 26 and 28 (Chart 5). These molecules were selected in view of their favorable optical and morphological features.60 For example, emission of the donor and the absorption of the acceptor have good spectral overlap. In addition, the acceptor has minimum aborption at the excitation wavelength of the donor thereby reducing the risk of self-excitation of the former. Excitation of a n-decane gel of 28 (donor) at 380 nm in the presence of 0–2 mol% acceptor molecules (26) resulted in the quenching of the donor emission at 509 nm with a concomitant formation of the monomer emission of the acceptor at 555 nm (Fig. 16(a)). This observation indicates that under this condition the acceptor molecules exist as an isolated energy trap. Upon further addition of 26 (2–20 mol%), the emission was continuously red-shifted to 610 nm which corresponds to the aggregate emission of 26. Consequently, 98% quenching of the donor emission at 509 nm was observed (Fig. 16(a)). Photographs (Fig. 16(b)) and fluorescence microscopic images (Fig. 16(c)–(e)) of the co-assembled gels of 28 and 26 under different compositions provided a visual evidence for the wavelength tunable FRET emission. Thus, efficient trapping of excitons by “isolated” or “aggregated” acceptors allowed a continuous shifting of the emission color anywhere between green and red (λmax = 509–610 nm).
 | ||
| Chart 5 | ||
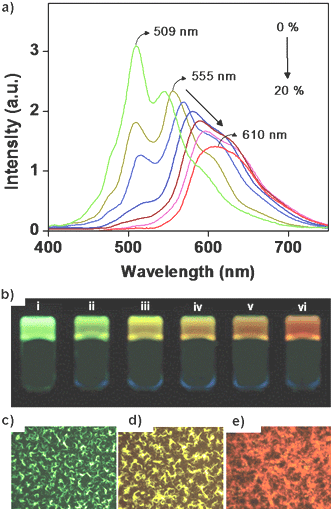 | ||
| Fig. 16 (a) Fluorescence emission (λex = 380 nm) of 28 on addition of different amounts of 26 (0–20 mol%). (b) Photographs of the gels of 28 and 26 at different compositions of 26: (i) 0 mol%, (ii) 1 mol%, (iii) 2 mol%, (iv) 6 mol%, (v) 12 mol%, (vi) 20 mol%, under illumination at 365 nm. (c, d and e) Fluorescence microscopy images of the drop-casted films from a decane solution of 28 in the presence of 0, 2, and 20 mol%, respectively, of 26. Reprinted with permission from ref. 60. Copyright 2006, American Chemical Society. | ||
In order to achieve the design of an efficient supramolecular gel based light harvesting antenna, it is necessary that the energy transfer should occur with a very low amount of the acceptor. Since energy migration is efficient in the gel state of OPVs, they are ideal excitation energy donors. However, the challenge is to identify a suitable acceptor that is compatible with the gel. Photoinduced energy migration and energy transfer are known in conjugated polymers and oligomers.61–63 Therefore, such systems may be good energy traps of excitons. Recently, a semiconducting molecular wire has been found suitable as an excellent acceptor when encapsulated within the self-assembled donor scaffold of a π-organogel.56 In this design, a gel forming oligo(p-phenylenevinylene) derivative 21a is used as the energy transfer donor and a copolymer of phenylenevinylene and pyrolylenevinylene (PYPV) 29 (average molecular weight (Mn) of ∼4358 g mol–1) as the acceptor.
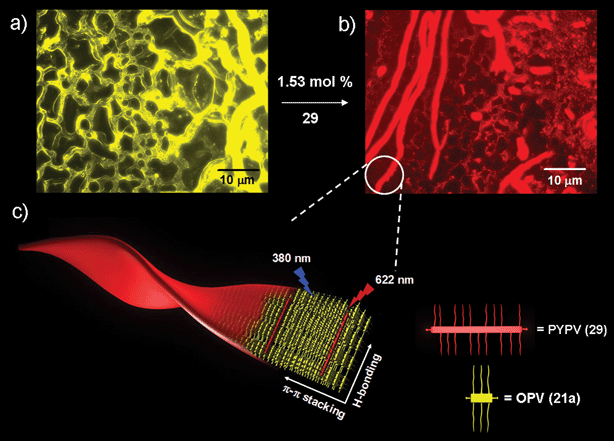 | ||
| Fig. 17 Fluorescence microscopy images of a drop-casted 21a-cyclohexane gel (1.12 mm): (a) in the absence and (b) presence of 29 (1.53 mol%). (c) A schematic representation of a PYPV (29)-encapsulated OPV (21a) supramolecular tape. Reprinted with permission from ref. 56. Copyright 2007, Wiley-VCH. | ||
Development of inorganic–organic hybrid materials consisting of gold nanoparticles and OPV organogelators has recently been reported which exhibited interesting optical properties.64 The OPV derivative 21b which forms a gel in toluene was selected as a template and another OPV derivative 30 which contains a disulfide moiety at one end was used to bind with gold nanoparticles. Mixing of 21b/30-Au in toluene at different ratios followed by heating and cooling of the solution resulted in clear gels with reduced thermal stability when compared to that of 21b alone. This observation indicated that 30-Au interacts with the 21b tapes. The formation of 21b/30-Au hybrid supramolecular tapes is visualized by TEM that showed arrays of 29-Au particles on both sides of the tape, indicating the formation of supramolecular hybrid tapes of 21b/30-Au (Fig. 18(b)). The results of cryo TEM and AFM studies support this observation. Fluorescence studies showed that incorporation of 30-Au into the tapes of 21b results in quenching of the OPV luminescence (Fig. 18(c)). In the case of the 100 : 1 21b/30-Au mixture, the intensity was reduced by a factor of 33 in comparison with the emission intensity of 21b alone. Time resolved photoluminescence studies revealed shortening of the lifetime of 21b upon addition of the OPV 30 attached nanoparticles, when compared to that of 21b alone. This observation indicates that at least part of quenching is due to a dynamic process that takes place in the nanosecond time scale and involves exciton energy migration towards docked gold nanoparticles. Further confirmation to this speculation is obtained from photoinduced absorption studies (PIA), which showed a clear difference in the population of the excited state for the 21b and 21b/30-Au hybrid gels, mainly in the nanosecond time scale. A very short lifetime observed for the 30/Au system in PIA studies points towards a rapid energy transfer from 30 to the gold core. The result of these studies demonstrate that the close proximity of gold nanoparticles to the OPV nanotapes facilitates strong electronic communication which makes them attractive candidates for the fabrication of electron and energy transport based nanodevices.
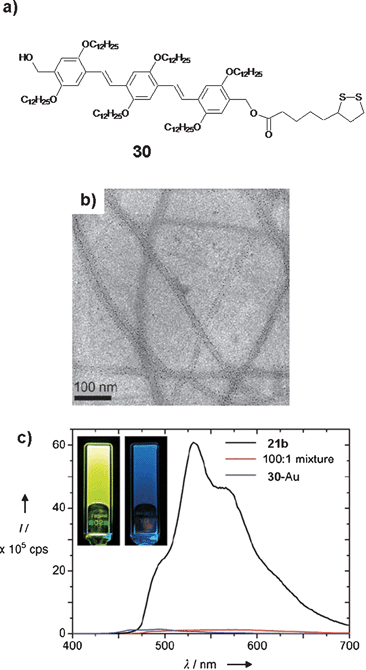 | ||
| Fig. 18 (a) Molecular structure of a thiol functionalized OPV derivative, 30. (b) TEM image of 21b/30-Au (100 : 1) tapes deposited from toluene. (c) Fluorescence spectrum of a 100 : 1 mixed gel of 21b and 30-Au and of the separate compounds in toluene. Inset: photographs of the luminescent 21b gel (left) and of the 100 : 1 21b/30-Au mixed gel (right). The latter displays a faint blue emission that can be ascribed to molecularly dissolved 21b. Reprinted with permission from ref. 64. Copyright 2007, Wiley-VCH. | ||
7 Conclusions
Excitation energy transfer in self-assembled donor–acceptor systems is important to the mimicry of natural processes and to the design of supramolecular electronic devices. Self-organization of chromophores is crucial in these cases. Gelation is found to be useful for the self-organization of chromophores which facilitate energy transfer processes. This approach has been successfully demonstrated in a variety of chromophore based gel forming molecules. OPV based organogelators are excellent excitation energy donors to suitable acceptors. As a result of fast exciton migration, efficient energy transfer is possible to encapsulated acceptors in the gel state. Thermoreversible transition of the sol–gel processes in OPV gels allows control on energy transfer process and facilitates tuning of the emission color from blue to red. Even though a significant amount of knowledge is available in the design of supramolecular light harvesting assemblies, control of the energy transfer processes in a few nanometers in length scale and their application in organic electronic devices remains challenging. Therefore, research in this area has tremendous opportunities particularly in the context of the emerging areas of supramolecular electronics and nanodevices.Acknowledgements
We thank the Department of Science and Technology (DST), New Delhi for financial support under the Nano Science and Technology Initiative. A. A. thanks DST for a Ramanna Fellowship. V. K. P. and C.V. acknowledge the Council of Scientific and Industrial Research (CSIR) for research fellowships. This is contribution No. NIST-PPG-253.References
- Energy Harvesting Materials, ed. D. L. Andrews, World Scientific, Singapore, 2005 Search PubMed.
- M.-S. Choi, T. Yamazaki, I. Yamazaki and T. Aida, Angew. Chem., Int. Ed., 2004, 43, 150 CrossRef CAS.
- S. Hecht and J. M. J. Fréchet, Angew. Chem., Int. Ed., 2001, 40, 74 CrossRef CAS.
- V. Balzani, M. Venturi and A. Credi, Molecular Devices and Machines, Wiley-VCH, Weinheim, Germany, 2003 Search PubMed.
- F. M. Raymo and M. Tomasulo, Chem. Soc. Rev., 2005, 34, 327 RSC.
- B. Liu and G. C. Bazan, Chem. Mater., 2004, 16, 4467 CrossRef CAS.
- K. E. Sapsford, L. Berti and I. L. Medintz, Angew. Chem., Int. Ed., 2006, 45, 4562 CrossRef CAS.
- R. L. Carroll and C. B. Gorman, Angew. Chem., Int. Ed., 2002, 41, 4378 CrossRef.
- J. M. Tour, Molecular Electronics: Commercial Insights, Chemistry, Devices, Architectures and Programming, World Scientific, River Edge, NJ, 2003 Search PubMed.
- A. P. H. J. Schenning and E. W. Meijer, Chem. Commun., 2005, 3245 RSC.
- A. C. Grimsdale and K. Müllen, Angew. Chem., Int. Ed., 2005, 44, 5592 CrossRef CAS.
- D. Oelkrug, A. Tompert, J. Gierschner, H.-J. Egelhaaf, M. Hanack, M. Hohloch and E. Steinhuber, J. Phys. Chem. B, 1998, 102, 1902 CrossRef CAS.
- F. J. M. Hoeben, I. O. Shklyarevskiy, M. J. Pouderoijen, H. Engelkamp, A. P. H. J. Schenning, P. C. M. Christianen, J. C. Maan and E. W. Meijer, Angew. Chem., Int. Ed., 2006, 45, 1232 CrossRef CAS.
- J. Zhang, F. J. M. Hoeben, M. J. Pouderoijen, A. P. H. J. Schenning, E. W. Meijer, F. C. De Schryver and S. De Feyter, Chem.–Eur. J., 2006, 12, 9046 CrossRef.
- F. J. M. Hoeben, M. Wolffs, J. Zhang, S. De Feyter, P. Leclère, A. P. H. J. Schenning and E. W. Meijer, J. Am. Chem. Soc., 2007, 129, 9819 CrossRef CAS.
- A. Ajayaghosh, S. J. George and A. P. H. J. Schenning, Top. Curr. Chem., 2005, 258, 83 CAS.
- X. Li, L. E. Sinks, B. Rybtchinski and M. R. Wasielewski, J. Am. Chem. Soc., 2004, 126, 10810 CrossRef CAS.
- T. Ishi-i, K.-i. Murakami, Y. Imai and S. Mataka, J. Org. Chem., 2006, 71, 5752 CrossRef CAS.
- N. J. Turro, Modern Molecular Photochemistry, University Science Books, Sausalito, CA, 1991 Search PubMed.
- A. Adronov and J. M. J. Fréchet, Chem. Commun., 2000, 1701 RSC.
- C. Devadoss, P. Bharathi and J. S. Moore, J. Am. Chem. Soc., 1996, 118, 9635 CrossRef CAS.
- S. E. Webber, Chem. Rev., 1990, 90, 1469 CrossRef CAS.
- X. Schultze, J. Serin, A. Adronov and J. M. J. Fréchet, Chem. Commun., 2001, 1160 RSC.
- L. A. J. Chrisstoffels, A. Adronov and J. M. J. Fréchet, Angew. Chem., Int. Ed., 2000, 39, 2163 CrossRef CAS.
- J. Cabanillas-Gonzalez, A. M. Fox, J. Hill and D. D. C. Bradley, Chem. Mater., 2004, 16, 4705 CrossRef CAS , and references cited therein.
- M. A. Wolak, J. S. Melinger, P. A. Lane, L. C. Palilis, C. A. Landis, J. Delcamp, J. E. Anthony and Z. H. Kafafi, J. Phys. Chem. B, 2006, 110, 7928 CrossRef CAS.
- C. Minkowski, R. Pansu, M. Takano and G. Calzaferri, Adv. Funct. Mater., 2006, 16, 273 CrossRef CAS.
- J. M. Haider and Z. Pikramenou, Chem. Soc. Rev., 2005, 34, 120 RSC.
- K. Sasaki, H. Nakagawa, X. Zhang, S. Sakurai, K. Kano and Y. Kuroda, Chem. Commun., 2004, 408 RSC.
- M. D. Ward, Chem. Soc. Rev., 1997, 26, 365 RSC.
- E. H. A. Beckers, P. A. van Hal, A. P. H. J. Schenning, A. El-ghayoury, E. Peeters, M. T. Rispens, J. C. Hummelen, E. W. Meijer and R. A. J. Janssen, J. Mater. Chem., 2002, 12, 2054 RSC.
- Molecular Gels, Materials with Self–Assembled Fibrillar Networks, ed. R. G. Weiss and P. Terech, Kluwer Press, Dordrecht, 2005 Search PubMed.
- L. A. Estroff and A. D. Hamilton, Chem. Rev., 2004, 104, 1201 CrossRef CAS.
- N. M. Sangeetha and U. Maitra, Chem. Soc. Rev., 2005, 34, 821 RSC.
- T. Ishi-i and S. Shinkai, Top. Curr. Chem., 2005, 258, 119 CAS.
- Z. Yang and B. Xu, J. Mater. Chem., 2007, 17, 2385 RSC.
- K. J. C. van Bommel, A. Friggeri and S. Shinkai, Angew. Chem., Int. Ed., 2003, 42, 980 CrossRef.
- J. Puigmartí-Luis, V. Laukhin, A. P. del Pino, J. Vidal-Gancedo, C. Rovira, E. Laukhina and D. B. Amabilino, Angew. Chem., Int. Ed., 2007, 46, 238 CrossRef CAS.
- Y. Yamamoto, T. Fukushima, Y. Suna, N. Ishii, A. Saeki, S. Seki, S. Tagawa, M. Taniguchi, T. Kawai and T. Aida, Science, 2006, 314, 1761 CrossRef CAS.
- T. Sagawa, S. Fukugawa, T. Yamada and H. Ihara, Langmuir, 2002, 18, 7223 CrossRef CAS.
- T. Nakashima and N. Kimizuka, Adv. Mater., 2002, 14, 1113 CrossRef CAS.
- K. Sugiyasu, N. Fujita and S. Shinkai, Angew. Chem., Int. Ed., 2004, 43, 1229 CrossRef CAS.
- K. Sugiyasu, N. Fujita, M. Takeuchi, S. Yamada and S. Shinkai, Org. Biomol. Chem., 2003, 1, 895 RSC.
- J. B. Beck and S. J. Rowan, J. Am. Chem. Soc., 2003, 125, 13922 CrossRef CAS.
- A. Del Guerzo, A. G. L. Olive, J. Reichwagen, H. Hopf and J.-P. Desvergne, J. Am. Chem. Soc., 2005, 127, 17984 CrossRef.
- J.-P. Desvergne, A. G. L. Olive, N. M. Sangeetha, J. Reichwagen, H. Hopf and A. Del Guerzo, Pure Appl. Chem., 2006, 78, 2333 CrossRef CAS.
- J. H. Ryu and M. Lee, J. Am. Chem. Soc., 2005, 127, 14170 CrossRef CAS.
- S. Yamaguchi, I. Yoshimura, T. Kohira, S.-i. Tamaru and I. Hamachi, J. Am. Chem. Soc., 2005, 127, 11835 CrossRef CAS.
- M. Montalti, L. S. Dolci, L. Prodi, N. Zaccheroni, M. C. A. Stuart, K. J. C. van Bommel and A. Friggeri, Langmuir, 2006, 22, 2299 CrossRef CAS.
- A. Ajayaghosh and S. J. George, J. Am. Chem. Soc., 2001, 123, 5148 CrossRef CAS.
- S. J. George and A. Ajayaghosh, Chem.–Eur. J., 2005, 11, 3217 CrossRef CAS.
- V. K. Praveen, S. J. George and A. Ajayaghosh, Macromol. Symp., 2006, 241, 1 CrossRef CAS.
- A. Ajayaghosh, C. Vijayakumar, R. Varghese and S. J. George, Angew. Chem., Int. Ed., 2006, 45, 456 CrossRef CAS.
- A. Ajayaghosh, R. Varghese, S. J. George and C. Vijayakumar, Angew. Chem., Int. Ed., 2006, 45, 1141 CrossRef CAS.
- A. Ajayaghosh and V. K. Praveen, Acc. Chem. Res., 2007, 40, 644 CrossRef CAS.
- A. Ajayaghosh, V. K. Praveen, C. Vijayakumar and S. J. George, Angew. Chem., Int. Ed., 2007, 46, 6260 CrossRef CAS.
- A. Ajayaghosh, S. J. George and V. K. Praveen, Angew. Chem., Int. Ed., 2003, 42, 332 CrossRef CAS.
- V. K. Praveen, S. J. George, R. Varghese, C. Vijayakumar and A. Ajayaghosh, J. Am. Chem. Soc., 2006, 128, 7542 CrossRef CAS.
- A. Ajayaghosh, V. K. Praveen, S. Srinivasan and R. Varghese, Adv. Mater., 2007, 19, 411 CrossRef CAS.
- A. Ajayaghosh, C. Vijayakumar, V. K. Praveen, S. S. Babu and R. Varghese, J. Am. Chem. Soc., 2006, 128, 7174 CrossRef CAS.
- J. Kim, D. T. McQuade, A. Rose, Z. Zhu and T. M. Swager, J. Am. Chem. Soc., 2001, 123, 11488 CrossRef CAS.
- C. Ego, D. Marsitzky, S. Becker, J. Zhang, A. C. Grimsdale, K. Müllen, J. D. MacKenzie, C. Silva and R. H. Friend, J. Am. Chem. Soc., 2003, 125, 437 CrossRef CAS.
- J. G. Müller, E. Atas, C. Tan, K. S. Schanze and V. D. Kleiman, J. Am. Chem. Soc., 2006, 128, 4007 CrossRef.
- J. van Herrikhuyzen, S. J. George, M. R. J. Vos, N. A. J. M. Sommerdijk, A. Ajayaghosh, S. C. J. Meskers and A. P. H. J. Schenning, Angew. Chem., Int. Ed., 2007, 46, 1825 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2008 |