Hybrid porous solids: past, present, future
Gérard Férey
Institut Lavoisier (UMR CNRS 8180), 45, Avenue des Etats-Unis, Université de Versailles St-Quentin en Yvelines 78035, Versailles, France. E-mail: ferey@chimie.uvsq.fr
First published on 19th September 2007
Abstract
This critical review will be of interest to the experts in porous solids (including catalysis), but also solid state chemists and physicists. It presents the state-of-the-art on hybrid porous solids, their advantages, their new routes of synthesis, the structural concepts useful for their ‘design’, aiming at reaching very large pores. Their dynamic properties and the possibility of predicting their structure are described. The large tunability of the pore size leads to unprecedented properties and applications. They concern adsorption of species, storage and delivery and the physical properties of the dense phases. (323 references)
 Gérard Férey | Gérard Férey received his PhD from Paris VI University in 1977. He was Professor of Inorganic Chemistry first in Le Mans University (1981–1996) and then in Versailles University where he created the Institut Lavoisier (1996–…). He is now Professor at the Institut universitaire de France and member of the French Académie des Sciences. After working on the magnetism of transition metal inorganic fluorides, his current interests concern the structural chemistry of inorganic and hybrid porous solids, their mechanisms of formation and their applications in gas storage, drug delivery and nanosciences. |
This new class of porous solids1 emerged as a new domain of research ca. fifteen years ago. More or less considered at the beginning as a curiosity, it has transformed into a fully qualified field of research with an explosion of papers (Fig. 1). As usual for a new domain, most of the papers currently refer to new phases with their crystal structure and sometimes some indications about the porosity. It was a necessary step for justifying the richness of the field, as it was previously for the mesoporous compounds.2,3 The latter impressed the community of zeolites and microporous phases by the huge gap in the dimensions of the pores. The interest towards hybrid porous solids concerns more the important increase in chemical versatility compared to ‘classical’ porous solids. The present contribution on hybrid solids will therefore be placed in the continuum of the story of porous solids, with their similarities with the other families, their advantages, their potentialities and the unprecedented properties they sometimes exhibit. It represents a global introduction to the domain of hybrid porous solids, not a complete review. Some of them have already been recently published: for a detailed inventory of what exists see ref. 4–12.
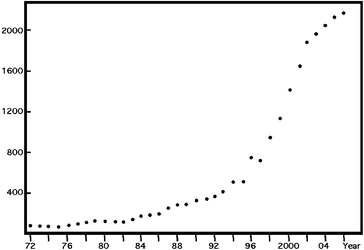 | ||
| Fig. 1 | ||
1. Some definitions
Hybrid porous solids result from the reaction between organic and inorganic species in order to build up three-dimensional frameworks whose skeleton contains both organic and inorganic moieties only linked by strong bonds, at variance to supramolecular chemistry. Conceptually, there is no difference between classical inorganic porous solids and hybrid ones (Fig. 2). Indeed, the three-dimensional skeleton can be described for both of them by the association of secondary building units (SBU).13 However, whereas the inorganic SBU contains only inorganic parts (tetrahedral species like SiO4, PO4, AsO4, SO4, associated with metallic cations in four-, five- or six-coordination), in the hybrid SBU, the anionic species are replaced by organic linkers, creating a contrast between the bonds within the framework: mainly covalent for the organic parts, ionocovalent for the inorganic. Moreover, as far as the porous character is concerned, organic ligands with multiple bonds must be preferred in order to ensure rigid topologies as for inorganic solids with an open framework.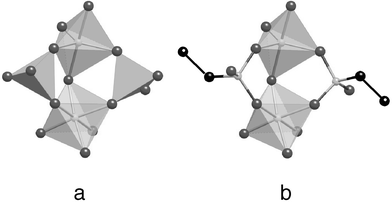 | ||
| Fig. 2 | ||
The tremendous development of this family generated a new vocabulary. In the first hybrid open frameworks14–19 the inorganic part contained either isolated polyhedra or small clusters, like in coordination chemistry. For this reason, these solids were first labelled as coordination polymers. However, very soon, it was shown that these hybrid solids could possess inorganic parts with a larger dimensionality, giving rise to chains (1D), layers (2D) and even inorganic frameworks (3D). The more general term Metal-Organic Frameworks20 (or MOFs) was then introduced with some derived acronyms [for instance IRMOFs (for IsoReticular MOFs), MMOFs (for microporous MOFs) PCP (for porous coordination polymers)…] to identify some specificities of the corresponding series. Some proposals have also been made in order to classify these solids according to the dimensionality of the inorganic subnetwork.21
MOFs provide also an ambiguity. It has become a generic term for the family. However, the old habit used by the community of zeolites to identify a given new solid by three letters (generally indicating the geographic origin of the new product) followed by a number, has been kept, and most of the published MOFs use this system. For example, our own compounds are given identifiers MIL-n [MIL for Materials of Institut Lavoisier].
2. Advantages and disadvantages of hybrids toward inorganic frameworks
At variance to zeolite-related inorganic solids, which require the use of inorganic or organic templates (amines, quaternary ammoniums…) beside the components of the skeleton and the solvent, the situation is much simpler for MOFs: the solvent itself acts as the main template. Such a feature presents a great advantage, the skeleton of most of the MOFs being therefore neutral. Indeed, many structures of zeolitic inorganic solids with a cationic skeleton often collapse during the extraction of the template owing to the strong electrostatic host–guest interactions, which energetically represent an important contribution to the lattice energy. In MOFs, the solvents have weaker interactions with the framework and therefore easily evolve the structure at low temperature, often keeping the framework intact and providing very quickly an important and readily accessible porosity. Moreover, the existence of inorganic and organic moieties in the structure allows hydrophilic and hydrophobic parts to coexist within the pores and may have some influence on the adsorption properties.Another interesting feature of MOFs concerns the great variety of cations which can participate in the framework. Indeed, compared to inorganic ones22 which are more based on a few cations [Si and Al for zeolites – eventually doped with some transition metals – with the exception of titanosilicates23–, Zr, Al, Ga, In phosphates and arsenates, sometimes fully substituted by transition metals (Ti,24 V,25 Fe,26,27 Co,28,29 Ni,30 Zn31,32)]–, MOFs can accept almost all the cations of the classification, at least those which are di-, tri- (including rare earth) or tetravalent. Keeping in mind the tremendous number of species previously isolated in coordination chemistry, this provides a huge number of possibilities for creating new MOFs.
This number is drastically increased considering the large choice of functionalized organic linkers which can be associated with the inorganic parts. The functions borne by the linker contain O or N donors. When O is concerned, they are mainly mono- or polycarboxylates, mono- or polyphosphonates, rarely sulfonates. All of them, even combined, can provide different possibilities of linkage with the inorganic cations (chelating, single bond…). The nitrogen derivatives (cyanides, pyridine, imidazoles...) are fixed directly to the cation. Moreover, the carbon subnetwork (rigid or not) of the linker can be itself functionalized, depending on the expected applications (halogeno-, amino groups…). This means that, potentially, the possibilities of combination within this new family of hybrids tend towards infinity. It is both the richness and the weakness of this family. The richness is clear, but the weakness comes from the quasi-infinite number of potential products. Among them, which are potentially interesting for applications? One cannot imagine testing all these products for eventual applications. A global and predictive approach to this family is therefore needed, in order to converge more easily towards potentially interesting compounds. This approach will be detailed below.
Fortunately, MOFs have a significant advantage for reducing the number of possible structure types. In the same way as inorganic zeolitic solids accept group substitutions while keeping the same topology (PO4 ⇒ AsO4 …), the functionalized linker can be substituted by larger ones, as soon as the connectivity with the inorganic moieties is preserved. When it is possible, this creates series of so-called ‘isoreticular’ solids (IRMOFs),20,33 decreasing the number of possibilities, while taking advantage of the increase of the pore size of the corresponding solid with length of the ligand. In other words, MOFs not only allow modularity for a given structural type but, within it, can create a new type of porous solid, as was recently shown by us.34–36 Indeed, beside the usual crystallized microporous solids, with small pores (ø < 20 Å) and mesoporous solids, with larger pores (ø > 20 Å) but amorphous frameworks, there is place for hybrid mesophases with crystallized frameworks, opening a new window in terms of applications. This will be discussed in Section 5.
During the syntheses of MOFs (see below), the coordination of the metallic species, the nuclearity and/or the dimensionality of the inorganic subnetwork strongly depend on the temperature of reaction. Within the same system, the other synthesis parameters being fixed, the increase of temperature favours first an enhanced condensation of supplementary metallic polyhedra on the starting cluster (increased nuclearity) and after, a change into chains then layers of inorganic polyhedra. The increase of the inorganic dimensionality creates the onset of long range interactions (for instance, magnetic ones) and therefore, the appearance of physical properties usually encountered in dense solids. This introduces a new field of potential applications beside the usual properties of porous solids (fluids separation and storage, catalysis…). On the other hand, such a variability increases once more the possibilities for the creation of new solids. In the face of this infinity, claiming that new solids are obtained by design is at least utopian.37 The community will have first to increase its knowledge about the mechanisms of formation of hybrid porous solids, to introduce more rationale in the syntheses before speaking seriously about ‘design’.
Have MOFs disadvantages? For the moment, only one is apparent: the weak thermal stability of MOFs [limited to 350–400 °C, rarely more (500 °C)38] which rules out any application at high temperatures. However, it allows specific ones, described at the end of this paper.
3. Syntheses
Usually, MOFs are synthesized at low temperature (<250 °C). Below 100 °C, the classical ways familiar to coordination chemistry are used. Above 100 °C, solvothermal syntheses are required. Beside water (the most frequently used), the main solvents are alcohols, dialkyl formamides, pyridine. For many syntheses, the recipes underline the addition of amines (ETA) which seem to favour the reaction without participating in the formula of the final compound. It must be recalled that MOFs can be obtained with almost all of the di-, tri- or tetravalent cations.The pertinent chemical parameters of the synthesis are pH (mostly acidic), concentrations (which can vary over a large range) and temperature. As mentioned above, temperature is a fundamental parameter. First, it defines two regimes, at least with water as a solvent: below and above 100 °C. In hydrothermal conditions, the dielectric properties of the solvent change, leading to weakened interactions between the solvent molecules and increased dissociation of the latter. For instance, the pH value of water, measured at 180 °C in hydrothermal conditions by an original in situ NMR method, is 5.5.39 This means that an extrapolation of the conditions applied at room temperature is not yet valid. Another chemistry begins.
A textbook example is provided by cobalt(II) succinates.40–44 Keeping the same starting mixture, this system was studied from room temperature to 250 °C. It provided seven different solids with first a gradual decrease of the number of coordinated water molecules per cobalt atom (T ≤ 100 °C), an increased edge-sharing connectivity for the Co2+ octahedra, higher coordination numbers for carboxylate groups and incorporation of hydroxide groups in the phases formed above 100 °C. The temperature increase also augments the dimensionality of both the whole structure and the inorganic subnetwork. First one-dimensional (with isolated octahedra for the inorganic part at 60 °C and trimers at 100 °C), it becomes two-dimensional at 150 °C with 14-membered rings of corner-shared Co octahedral, and finally three-dimensional above (190 and 250 °C), the inorganic subnetwork being always 2D, but with an increased connectivity of M–O–M bonds.
Anyhow, each cation or association of cations into clusters is a particular case, depending on its chemical nature and its acido-basic characteristics. For instance, the trimeric octahedral µ3-O chromium cluster45 which exists in the structure of chromium(III) acetate, is stable in solution from 25 °C to 200 °C without modification. It allows exchange reactions46 to be performed in this range of temperature and gives rise to very interesting structures.
For a given system, the nature of the starting metallic salt also greatly influences both the nature of the resulting products and their crystal growth.47 For example, in the Zn2+/1,4-benzenedicarboxylic acid/solvent reaction system, many different compounds, for example, Zn(BDC)·DMF·H2O, Zn3(BDC)3·6CH3OH and Zn4O(BDC)3 were discovered. Keeping the same 1 : 1 metal/ligand ratio of the original synthesis of MOF-5,20 but changing the counter anion of the salt (NO3–, Cl–, SO4–, CH3CO2–, O2–) in DMF medium leads to MOF-5 with NO3–, CH3CO2–, O2–, to an unknown phase with SO4, while reactions using Cl– only result in DEF soluble reaction products, with a profound influence on the crystal size and the morphology. Using slightly basic conditions leads to a new zinc terephthalate Zn3(OH)2(BDC)2·2DEF. The addition of small amounts of nitric acid to the synthesis mixture results in the new microporous zinc terephthalate (H2NEt2)2[Zn3(BDC)4]·3DEF.48
This underlines the role of precursors for the synthesis of MOFs. Counter ions of metal salts, owing to their own acido-basic or redox characteristics, can influence the conditions of reaction. This was also demonstrated for carboxyphosphonates in the system Co2+/(H2O3PCH2)2NCH2C6H4COOH (H5L3)/NaOH.47 in which unexpected results were obtained with the redox-active nitrate as counter ion. Under acidic conditions, NO3– ions induce an in situ oxidation of one of the P–C bonds of the ligand and a new phosphonic acid is formed which is stabilized by complexation of the Co2+ ions.
Beside these classical methods, four new routes are currently being developed for the synthesis of MOFs. The first49 uses a mixture of non miscible solvents for the hydrothermal synthesis (heavy alcohols and water, for instance). The solid forms at the interface of the biphasic mixture and, most of the time, provides single crystals of the desired phase.
The second represents the first trial for synthesizing MOFs using an electrochemical route.50 It concerns the previously known copper (1,3,5-benzenetricarboxylate).51 Bulk copper plates (thickness 5 mm) are arranged as the anodes in an electrochemical cell with the carboxylate linker dissolved in methanol as solvent and a copper cathode. After a period of 150 mn at a (voltage: 12–19 V, current: 3 A), the greenish blue precipitate is formed.
The third concerns the microwave synthesis, a method already applied to dense solids and inorganic porous compounds51–57 which seems very promising for MOFs. The microwave method already attracted growing attention for the synthesis of nanoporous inorganic materials which normally require several days for their hydrothermal crystallization. It provides an efficient way to synthesize them with short crystallization times, narrow particle size distributions, facile morphology control, and efficient evaluation of process parameters,58,59etc. However, the microwave method has rarely been applied to the synthesis of porous hybrid materials to date.60
Chang and co-workers have recently shown that the microwave syntheses of the latter offer several advantages such as fast crystallization as well as phase selective synthesis.61,62 Hybrid solids with giant pores, chromium trimesate and terephthalate (MIL-100 and -101), have been formed under microwave irradiation after less than 1 h at 220 °C, instead of 96 h using the conventional route.61 Moreover, at very short times (1 min) of synthesis, MIL-101 is obtained as quasi-monodisperse nanoparticles, a feature which could afford very quickly applications in nanosciences. In addition, the formation of the cubic nickel glutarate, [Ni20{(C5H6O4)20(H2O)8}]·40H2O (MIL-77) previously synthesized by conventional heating for several hours or days depending on the synthesis temperature, was greatly accelerated by microwave irradiation. The system results also in the formation of the more stable tetragonal nickel glutarate, Ni22(C5H6O4)20(OH)4(H2O)10]·38H2O only within a few minutes.62 The cubic phase was formed preferentially at low pH, low temperature and predominantly with conventional electrical heating. In contrast, the tetragonal phase was favored at high pH, high temperature and especially with microwave irradiation. These selective results suggest the efficiency of microwave technique in the synthesis of MOFs.
The exceptionally rapid crystallization of nickel glutarates was attributed to different formation pathways compared with conventional zeolites. While the formation of aluminosilicate zeolites appeared to involve complex crystallization pathways via hydrolysis, hydrophobic hydration, gelation, nucleation, and crystal growth,63 the porous nickel glutarates apparently grew directly from the reactants once the solution was raised to the appropriate reaction temperature. Consequently, the long induction periods required for zeolite formation are not necessary in the synthesis of nickel glutarates.
Morris and co-workers have reported that two new isostructural coordination polymers with anionic MOFs are synthesized classically and under microwave conditions using an ionic liquid, 1-ethyl-3-methylimidazolium bromide, as solvent and template.64 They conclude that the MOF samples prepared under microwave conditions are purer in phase and have higher crystallinity. All these results show that the extremely fast hydrothermal formation of metal–organic hybrid materials is not only a new route for synthesis of MOFs but also an important step towards developing commercially viable routes toward producing MOFs.
Finally, the richness of the possibilities of isolating new MOFs and the race for describing them has incited some authors to develop a dedicated application of high throughput synthesis to MOFs systems.65–69 This method was already employed for zeolites, inorganic frameworks and polymers for applications in catalysis and phosphors.70–72 High-throughput (HT) methods imply four major steps: design of experiment, synthesis, characterization, and data evaluation which have to be integrated in a workflow in order to reach a maximum of productivity and innovation. While HT-methods can produce a tremendous amount of data in a very short time, their success depends on proper application. Thus, the design of experiment is a step of paramount importance. Statistical methods in combination with data evaluation programs, genetic algorithms and neural networks have been shown to be powerful tools.73 The number of reactions must also be minimized by including chemical data and chemical knowledge into the synthesis set-up. In addition, investigations are most often limited to certain parameters, mainly composition, but can also process parameters such as temperature, time and pressure. In a typical experiment, 48 mini autoclaves are filled by various compositions within the same system.
For MOFs, the current focus of the applications of HT concerns the investigation of the parameter space of solvothermal reactions in solid state sciences, the discovery of new compounds,66,74 the optimization of reactions75 (including the influence of dilution on crystal growth66) as well as the identification of reaction trends, particularly the influence of pH and water content. In general, the large amount of data obtained in a short time leads to an improvement towards the understanding of the role that the chemical parameters play in the formation of materials. For example, with the cobalt succinates HT methods investigated the parameter space from 384 individual reactions in a few days. Beside the already known solids, two new materials were discovered.44
Current trends for new porous systems
The number of possibilities of combining inorganic and organic moieties is immense. It is out of the scope of this paper to give an extensive review which can be obtained in some other papers.4,7 The infinite variation on the nature of the linkers provides either series of isostructural compounds or new structures. With rigid linkers, a particular emphasis is put on MOFs which do not have a counterpart in inorganic porous solids (particularly those containing metallic cations like rare earths whose phosphates are so insoluble that they exclude any formation of templated porous equivalents76–101), on chiral ligands for their applications in enantioselective catalysis and separation, and more recently, on the use of amino acids for biological purposes.In the case of chiral ligands,102–108 some recent examples are particularly interesting. A chiral ligand derived from tartaric acid gives a layered zinc compound109 (POST-1) that has triangular chiral channels (side length of a triangle 13.4 Å) which enantioselectively adsorbs transition metal complexes. A 3-D cadmium carboxylate derived from quinine110 enantioselectively adsorbs (S)-2-butanol and (S)-2-methyl-1-butanol. Also, the three-dimensional coordination polymer zinc saccharate features two types of channels, hydrophilic and hydrophobic, which can adsorb molecules such as azobenzene.111 Finally, two recent nickel aspartates,112–115 one 1-D composed of helical chains (11.6 Å in diameter) with extended Ni–O–Ni bonding), and the other 3-D Ni2.5(OH)(L-Asp)2), 6.55 H2O composed of the same helical chains 11.6 Å in diameter with extended Ni–O–Ni bonding but connected by additional nickel octahedra to ensure the chiral open framework are very promising.
Related to chiral solids, the use of amino acids and peptides in general as building blocks for the construction of crystalline open-frameworks is emerging.116 Such systems are expected to exhibit rich metal–amino acid coordination chemistry that could serve as model systems for the study of important biocoordination compounds like metalloproteins. Open frameworks developed through the interaction of biomolecules and inorganic primary or secondary building units may allow for the creation of novel materials that combine the high chemical and thermal stability of inorganic oxides with the biological functionality of biomolecules. Furthermore, the direct incorporation of chiral amino acids may lead to the creation of noncentrosymmetric solids which could have applications in enantioselective catalysis and separation or non-linear optical applications.117,118 Because of these advantages, there has been an increasing interest in using amino acids as building blocks to create MOFs and a small number of amino acids have been explored already, including asparagines, histidine and aspartic and glutamic acids.119–123 A particularly interesting example is provided by the combination of histidine with zinc phosphite which lead to a layered MOF with four-ring units.116
Toward tailor-made porous solids?
Even recently, it was a dream to create by purely rational synthesis tailor-made structures fitting the requirements for dedicated applications. Some premises arise now in this connection. It requires the knowledge of the chemical conditions associated with the existence in the primitive solution of a given type of INORGANIC brick which will be preserved during the crystallization of the desired porous solid. This is the key point. The organic linker is topologically neutral during the synthesis since it remains invariant, but this is not the case for the inorganic brick whose nuclearity and dimensionality can vary greatly if the synthesis parameters are not controlled. This was seen above. The mastery of nuclearity and dimensionality then defines the active inorganic brick which will be responsible for the final structure. Its potential connectivity indeed orientates the topology, and its dimensionality will determine the physical properties and their intensity. A great effort must be made in two directions for such a knowledge: (i) more carefully to look at the relations between the chemical conditions of synthesis and the existence of bricks with a given geometry in the resulting structures45 and (ii), development of the use of in situ techniques (NMR, IR, Raman, EXAFs …) in respect of the conditions of synthesis in order to identify these bricks in solution, as has already been done for some porous inorganic solids systems (AlPOs, GaPOs).124–129Despite being long to perform and difficult to analyse, this knowledge of the mechanisms of formation is important for the future. For instance, an in situ EXAFs study of the formation of a porous Fe(III) muconate using Fe(III) acetate as a precursor recently showed46 that the trimeric unit, which already exists in the precursor, remains intact during the whole reaction, including the initial formation of an amorphous phase followed by crystallisation of the final muconate.
Finally, after the synthesis, a correct evacuation of the template or the solvent from the cages and tunnels is needed for obtaining a solid with accessible pores. Even if it is easier with MOFs, since they usually only necessitate evacuation of the solvents, the proper activation of the as-synthesized solid remains essential since upon it depends the surface area, the accessibility to the pores and the porous volume, which will drastically influence the adsorption and catalytic properties.
4. Structures and “design” of structures
In the broader sense of solid state chemistry, the emergence of a new field always corresponds first to a tremendous effort in chemical synthesis followed by a correlative research on the corresponding crystal structures. In a first step, this feeds the databases, but, very rapidly, a rationalization becomes necessary for organizing these structural data into classes. This was already done for dense phases (metals, inorganic solids) in the nineties by Anderson, O'Keeffe and Hyde.130,131 after the pioneer works of Wells,132 and of Smith133 for zeolites.In the new field of MOFs, basic principles for such a classification were needed owing to the already mentioned infinite number of possible combinations between organic and inorganic parts. Curiously, in 2000, in the same issue of the same journal, O'Keeffe134 and Férey135 paved the way for the development of the topological rules governing the structures of porous solids. Their starting point was the same. They both observed that some structures of MOFs corresponded to extended versions of simple structures (diamond, sodalites …). Even if their approaches were slightly different (nets for O'Keeffe, SBU for Férey), both concepts were concerned by the topology of structures and their invariance whatever the chemical associations. They aimed also at finding porous solids with larger and larger pores136 for specific applications which are allowed with mesoporous solids and not with micropores.
In continuation of his former way of describing the structure of the solids in terms of nets,131,132 O'Keeffe defined the geometrical design principles for frameworks of extended solids134 based on the concept of ‘augmented’ nets. Every solid can indeed be described by the geometric figure (net) obtained by the connection of the entities of the structure. The idea of these connecting nets is underlaid by the old concept of coordination. For instance, in an [N,M] connected net, some vertices are connected to N neighbors and some to M neighbors. With the idea of creating low density structures, and formalizing a previous idea of Hansen,137 O'Keeffe defined the concept of decoration138 which describes the process of replacing a vertex by a group of vertices, the augmentation134 corresponding to the special case where each vertex of a N-connected net is replaced by a group of N-vertices. In other words, this replacement does not affect the connectivity of the parent net.
An illustration of this concept is provided by the example of platinum oxide Pt3O4 taken as a parent structure (Fig. 3). In this solid, platinum ions are in square planar coordination by oxygen atoms, and the latter are connected to three platinum atoms (Fig. 3a), thus creating a (3,4) net and a structure based on the three-dimensional assembly of square planes (Fig. 3b).The augmented net will replace the platinum atom by a square (which provides the same connectivity as Pt) and the oxygen atom by a triangle (which provides the same connectivity as O). Once related by a line, the vertices of these two polygons create the augmented net (Fig. 3c). The squares and the triangles may be referred to as the topological SBU; that is, they represent species whose connectivity is four for the squares and three for the triangles, whatever their chemical nature. Moreover, the line joining the polygons can be a bond, but also a sequence of bonds, the latter case being called expansion by O'Keeffe.134 It is what I called ‘the simplicity of complexity’139 and such an approach allowed Chen et al. to describe a copper(II) 4,4′,4″-benzene-1,3,5-triyl-tribenzoic carboxylate as an augmented Pt3O4 net.140 In this solid, the squares are taken up by a binuclear Cu carboxylate moiety (Fig. 3d). and the triangles correspond to the three corners of the benzene ring acting as the vertices of the inner triangle, the linkers being phenyl groups (Fig. 3e).
 | ||
| Fig. 3 Principle of augmented nets (a) description of Pt3O4 in terms of connection of squares (b) balls and sticks representation of Pt3O4 (Pt: blue; O: red) showing the fourfold coordination of Pt and the threefold coordination of O (c) augmented version of Pt3O4; O is replaced by a triangle and Pt by a square. Both polygons are related by linkers; keeping the same topology in copper(II) 4,4′,4″-benzene-1,3,5-triyl-tribenzoic carboxylate, the triangles correspond to the connecting points of the central phenyl ring of benzene-1,3,5-triyl-tribenzoic carboxylate (d) and the square to the Cu dimer linked to four carbons of the carboxylate functions. | ||
The O'Keeffe's principle of “augmented nets” was not only a tool for describing complex structures in a simple manner, but was also very inspiring for the synthesis of new MOFs with large pores showing very simple basic topologies dictated by the shape and connectivity of the building units. O'Keeffe anticipated that a few simple high symmetry topologies would be of paramount importance in the future for such a purpose. In a further work based on this principle, his group detailed the rules for reticular synthesis141 (defined as the authors as ‘the process of assembling judiciously designed rigid molecular building blocks into predetermined ordered structures which are held together by strong bonding’) and the design of new materials and proposed a classification of the known structure types of MOFs,142 labelled, as for inorganic porous solids13 by three small letters with oblique characters, often referring to an abbreviation of the formula of the parent structure.
The approach of Férey, known as the ‘scale chemistry’ concept,135 started from his analysis of solids in terms of Secondary Building Units (SBU). Instead of describing structures by the connection of single polyhedra, he showed that it was possible to analyze them using larger units (SBU or ‘bricks’) which, by translation and/or rotation and further sharing of vertices, built up the final solid (Fig. 4). SBU remained however only a tool of description.
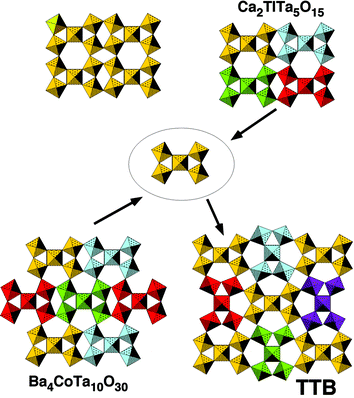 | ||
| Fig. 4 Some complex oxides described in terms of connection of pentameric SBUs. | ||
Playing on the size of the SBU, he illustrated his concept by numerous examples, for instance the following (Fig. 5). If the brick is an atom, for example metallic, the topology of the resulting solid is generally face centered cubic (fcc); if the brick is a cube of atoms, the resulting structure is the fluorite type, always fcc; if the brick is a C60, the fullerene structure is obtained, which also exhibits a fcc arrangement. This means that, whatever the size of the SBU, the topology of the resulting structure remains invariant and, in terms of porosity, ‘the larger the brick, the larger the pores’.
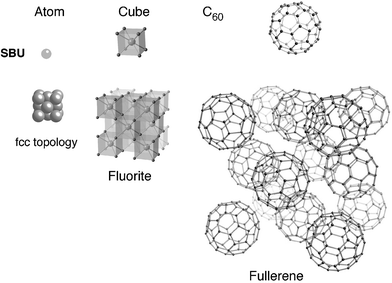 | ||
| Fig. 5 Principle of scale chemistry: whatever the size of the brick, the three-dimensional arrangement keeps the same topology (here fcc). | ||
This defined one of his strategies for a possible access to giant pores and to new applications related to their large sizes.
His in situ studies on the mechanisms of formation of inorganic porous solids124–129 changed his approach. He indeed proved the existence within the solution of SBU identical to those which existed in the final solid. These SBU, initially a tool of description, became a reality and then could act as ‘bricks’ for the construction of the solid. Moreover, as he proved that the increase of the size of the inorganic moiety (and therefore the extent of oligomeric condensation of inorganic species in the solution) was strongly dependent on the weakening of the charge density of the template, the validity of the concept was proved with the rational synthesis of MIL-74,143 an aluminophosphate using TREN as a template. MIL-74 has a supersodalite structure (Fig. 6). The square brick of four tetrahedra which exists in the pure sodalite is replaced by a square of nine tetrahedra which create a cage eight times larger than that of the sodalite.
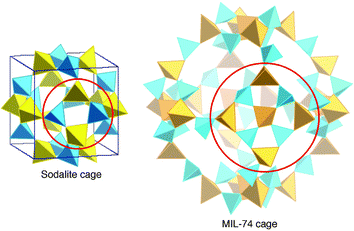 | ||
| Fig. 6 Comparison of the sodalite and MIL-74 cages with the same topology. The latter is eight times larger than the former. | ||
This concept of SBU applies to MOFs with however more difficulties for playing on the size of the inorganic bricks. Indeed, the rare inorganic bricks evidenced up to now are stable ones which exist in a large range of temperature and are known since a long time with their associated chemical conditions of existence. In situ studies are currently performed in his group for finding new inorganic SBU able to increase the number of possibilities and new connectivities to enrich once more the possibilities of getting tailor-made solids. The two first examples are the discovery of the hexameric cluster of six octahedra144 and the invariance of the trimeric cluster of three octahedral during the reaction with carboxylic acids.46 Another example, explained in detail below for MIL-100 and MIL-101 solids, will show that the search of new and unexpected inorganic bricks appears to be at its very beginning.
Both concepts are complementary for the creation of new topologies. Both are simple, inspiring for the search of solids with dedicated applications. Even if they do not pretend to exhaustivity, they provide elements of thought which stimulate the imagination of researchers without neglecting the unexpected results of serendipity which will refine the concepts. Indeed, both principles have limitations. In the case of MOFs (but not in the case of inorganic porous solids), the O'Keeffe's principle seems to be restricted, at least for most of the cases, to the creation of coordination polymers in which the inorganic part is a cluster because the augmentation of the primitive net always respects the alternation of the cation and the anion of the original simple structure and therefore the alternation of organic and inorganic moieties. In terms of predictability, the principle does not consider the variability of the inorganic SBU with the chemical conditions. It is predicted for already known bricks with a variation on the organic linker. By contrast, the Férey's principle takes into account this variability of the bricks, but considers the same linkage in a whole series. At least at the beginning, the extension concerned only the inorganic brick, the connection being invariant but recent examples145–147 have proved that scale chemistry can apply as well to the shared vertices. Moreover, it is not restricted to organic–inorganic connections, but accepts also inorganic–inorganic ones for explaining, for instance, the existence of MOFs with 1D or 2D inorganic subnetworks.
However, these two principles correspond to geometrical rules and, as noticed by O'Keeffe, ‘In such considerations, the structure directing role of guests and counterions is of paramount importance and careful attention will have to be placed on guest–host interactions. These will depend strongly on the specific chemistry, as opposed to the simple geometry which has been our concern in this paper’. It is true that both concepts do not take into account the thermodynamics of the systems for the isolation of new series, for instance by using reticular chemistry.141 The series are observed only because the thermodynamics (and sometimes kinetics) of the studied systems allows their existence. In other terms, one must never forget that the structures of MOFs are governed by thermodynamics and not by geometry. At variance to the opinion of some authors, the above rules can only suggest some possibilities of arrangements of the SBU. They cannot predict them. This point will be discussed further in this paper.
This important remark brings into question so-called ‘design’. Indeed, in many papers concerning MOFs, one can read ‘this solid was obtained by design’. This term, despite being fashionable, becomes more and more controversial.148 The etymology of this term comes from the old Italian ‘designo’ which has two senses: on one hand: drawing, sketch and on the other hand: idea, aim, objective… Its evolution with time, mainly during the XXth century, concerned architecture, fashion, new shapes (aesthetic or not) and became indistinguishable from a certain idea of beauty…Moreover, ‘design’ is inherent to the macroscopic scale and often associated with the notion of pleasant use. An architect can design a building, its shape, its height, its arrangement of spaces because he has the masteryat the macroscopic scale of the materials he uses and of the shaping he wants to give to them. Indeed, at the macroscopic scale, materials can easily take a desired form by smithing (metals), moulding (plastics)… A car designer can imagine the lines of the coachbuilding of a Ferrari because he can choose the proper material which allies rigidity and lightness, he can optimize the lines for giving to the car a good penetration in air, and therefore a higher speed… A great dressmaker will choose a material, its colours and texture, will cut the different pieces and assemble them in a certain way, to give the final dress that he imagined from his sketches. Design is the realization of what one imagined. The result may be bad or good, depending on the quality of the designer, but it is design, and from the design, one recognizes a style. Frank Lloyd Wright, Pei (architecture), Pininfarrina (cars), Y. Saint-Laurent (fashion) were famous designers because they worked on macroscopic matter.
Can design exist at the atomic scale? If there is design in chemistry, only organic chemists can insist on this talent because they play on single molecules. Experts in total synthesis of natural products do so because they know the recipes for a possible grafting of some groups in special places of the molecule, when the thermodynamic conditions are satisfied. As soon as the dimensionality of the compound increases, it becomes almost impossible. If it was possible, this would imply that, in the domain of hybrid solids, chemists are able to provide the structures they imagined. This would imply too that only one structure is possible, which is clearly not the case because it would exclude the possibility of polymorphism. For instance vanadium(III) terephthalate, VIII(OH)[OOC–C6H4–COO], exists in at least two forms MIL-47149 and MIL-68.150 Both are built up from the same SBU associating one vanadium octahedron with one terephthalate (Fig. 7) but they correspond to a 44 and 6.3.6.3 topologies respectively. This is a first argument against design.
![Perspective view of MIL-47 and MIL-68, two polymorphs of VIII(OH)[OOC–C6H4–COO].](/image/article/2008/CS/b618320b/b618320b-f7.gif) | ||
| Fig. 7 Perspective view of MIL-47 and MIL-68, two polymorphs of VIII(OH)[OOC–C6H4–COO]. | ||
The other is much more general and takes into account the landscape of energies for the formation of solids. Schoen and Jansen showed by simulation148 that the number of possibilities (which each corresponds to a local minimum of energy) is so large, with very small differences in the minima that it is almost impossible to ‘design’ or predict anything. With these authors, one can conclude that design is probably an ‘illusion’, even if I have to confess to have used this word at the very beginning.135,139
Anyhow, the rules emerging from these two principles are a good guideline for the search for new structures. Both concepts are based on the invariance of the connectivity between the building bricks, but with a huge possibility of variations around both the inorganic moiety and the linker. This can lead to almost unlimited possible networks. MOFs represent the microscopic version of Legoland™.
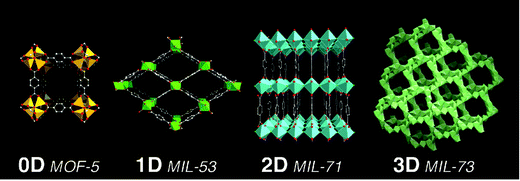
When the inorganic subnetwork is concerned, several possibilities are offered to the chemist. As already noticed, playing on the chemical conditions, he can increase progressively the dimensionality of the inorganic subnetwork, from the original 0D coordination polymers to 3D (Fig. 8). and introduce for the highest dimensionalities long range physical properties usually encountered in dense solids as will be seen in the paragraph of applications.
Keeping the 0D character of the inorganic subnetwork and its connectivity, he can play either on the nuclearity of the cluster (for instance using polyoxometalates ions155,156 or new large hexameric units157) or the characteristics of the ligand either for increasing the distance between the clusters, for providing them a convex curvature158 (Fig. 9) which enhances the accessible dimension of the cage, for introducing chirality in the MOF (see aspartates112–115), for introducing in the linker large functionalized substitutes whose steric hindrance will modify the orientation of the phenyl rings and therefore the opening of the windows.
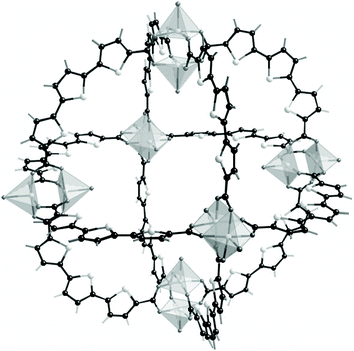 | ||
| Fig. 9 The curved ligands in MOP-28.158 | ||
The three latter strategies apply also to MOFs with higher dimensionalities of the inorganic subnetwork. A special case worth noting concerns decorated inorganic porous solids when the oxygens shared between two polyhedra are replaced by ligands which have the same twofold connectivity as oxide ions. It is for example the case of imidazole which create by M–N bonds the same topology as the corresponding inorganic porous solids. With such a strategy, the upper homologues of sodalite, analcime and zeolite-rho topologies145–147 were isolated and provide pore volumes eight times those of the parent structures. Such a variety of parameters on which the chemist can play shows once more that the immense number of possible combinations is to come. The game is just beginning.
5. The structural originality of MOFs: dynamic frameworks and breathing
Beside all these topologies, a strange feature arises with some MOFs, which relates to their flexibility. It is a general problem which concerns not only 0D coordination polymers, but all the dimensionalities of the inorganic subnetwork.As early as 1997, Kitagawa159 suggested classifying the hybrid porous frameworks in three categories which he called ‘generations’. The first concerns frameworks which are sustained only with guest molecules and collapse on removal of the guest, most of the time irreversibly. The second generation corresponds to stable and robust porous frameworks which exhibit permanent porosity without any guest in the pores. The last category refers to flexible frameworks which change – most of the time reversibly – their structure to respond to external stimuli. The stimulus can be temperature, pressure, light, electric or magnetic field, guests… Depending on the structure itself, the input is associated with either an expansion or a contraction of the cell volume and can generate induced movements larger than 10 Å during the transformation.
Such a phenomenon, illustrated by several examples, has lead Kitagawa160 to distinguish six classes of dynamic frameworks (Fig. 10) in relation with the dimensionality of the inorganic subnetwork.
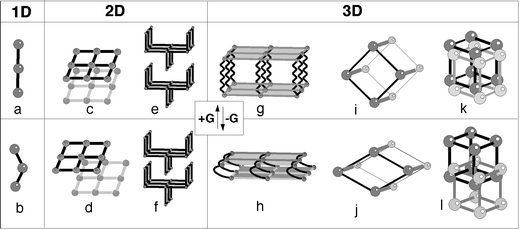 | ||
| Fig. 10 The six classes of Kitagawa. | ||
In the 1D class (Fig. 10a,b), the voids between the chains are occupied by small molecules and can exhibit ion exchange160. In the first case of 2D class (Fig. 10c,d), the manner of stacking of the layers (superimposed or shifted) is strongly dependent on the nature of the guest and the weak interactions they have with the layers. In the second case (Fig. 10e,f), the interdigitated layers are superimposed and form 1D channels.161–163 Closed without guests, they open with some of them, resulting in an elongation of the stacking parameter.
In the 3D cases, three situations occur. When pillared layers are concerned (Fig. 10g,h), the reversible phenomenon of interlayer elongation and shortening is realized by non-rigid pillars.164 The expanding and shrinking frameworks (Fig. 10i,j) act as sponges. Keeping the same topology, the drastic volume change is induced by strong host–guest interactions. Depending on the structure, the volume increase is associated with either the evacuation149 or the inclusion of the guests.165–169 Finally, in the case of interpenetrated grids, they are densely packed in the absence of guests and the introduction of molecules generates a sliding of one network (Fig. 10k,l).170,171
Most of the cases imply significant atomic movements during the transition, typically in the range 2–4 Å. However, these displacements can reach values in the range 5–10 Å whereas the topology of the framework is maintained. This is observed in class 5 (expanded and shrinked grids) and concern two structure types (MIL-53 and MIL-88) isolated in our laboratory.
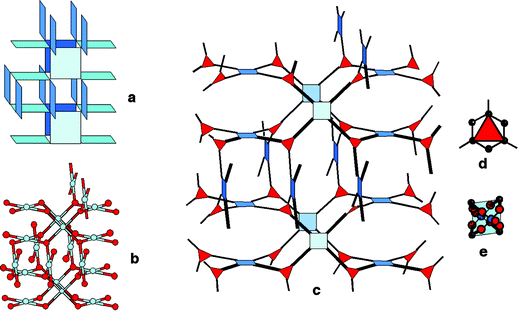
The MIL-53 type149,152 [(MIII(OH)L, Guest) with M = Al, V, Cr, Fe; L = terephthalate, naphthalene dicarboxylate, G: guest] is built up from chains of octahedra sharing OH vertices, which are linked in the two other directions by linkers (Fig. 11) in order to create 1D lozenge-shaped tunnels.
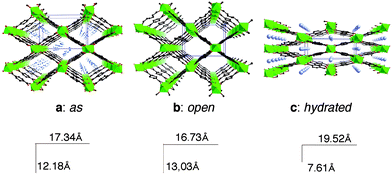 | ||
| Fig. 11 The different forms of MIL-53: (a) as synthesized (as); disordered terephthalic acid molecules lie within the tunnels; (b) high temperature (open); (c) room temperature hydrated form (hydr.). Note the changes in the cell parameters during the thermal treatments. | ||
The expanded empty form occurs at high temperature. By cooling, a water molecule is trapped in the tunnels and induces a drastic shrinkage of the framework with a decrease of the cell volume of ca. 40% associated with atomic displacements of –5.2 Å in one direction and of +3 Å in the other one. The transition is fully reversible. It is worth noting that, in the case of MIL-53, the input (water) provokes a contraction of the framework. An in situ solid state NMR study proved38 that two types of strong hydrogen bonds between water and the skeleton were responsible for the shrinkage. This very large breathing induces some selectivity during the exchange of water by other solvents. Acetone and ethanol are not exchangeable, whereas dmf is, owing to the sufficiently strong hydrogen bonds it forms with the skeleton. This large breathing effect induces new applications, described in the last chapter.
The MIL-88 type, with an hexagonal symmetry,45,46,172,173 corresponds to a coordination polymer based on trimeric units with three octahedra sharing a µ3-oxygen (Fig. 12). Within the 3D structure, the carboxylates linking the trimers create both tunnels and triangular bipyramidal cages in such a way that, in the latter, there is no connection between the trimers in the equatorial plane of the bipyramid.
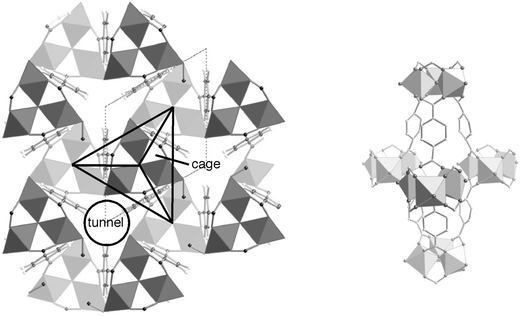 | ||
| Fig. 12 (001) projection (left) of MIL-88B and perspective view of its cage (right) | ||
This peculiarity induces unprecedented very large breathing effects, but at variance to MIL-53, the input (solvent) is associated with an expansion of the framework (Fig. 13). The extent of these swellings (most of the time reversible) strongly depends on the nature of the dicarboxylate linking the trimers in the solids (MIL-88A for muconate, B for terephthalate, C and D for naphthalene and biphenyl dicarboxylates respectively).
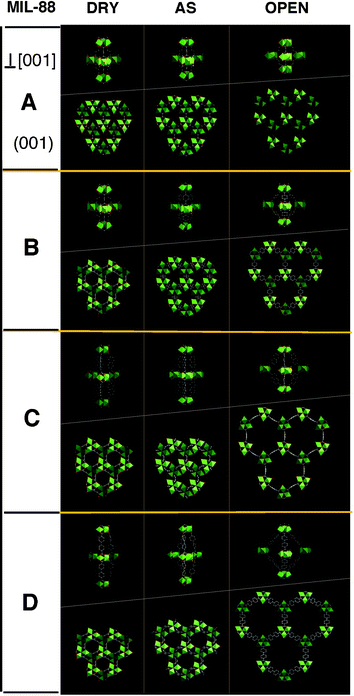 | ||
| Fig. 13 Evolution of the breathing in the MIL-88 family. For each solid, the (001) projections and the view of the cages are represented (from left to right, the dry closed form, the as synthesized structure (as) and the open extended one (open). | ||
The as-synthesized solids always contain a few solvent molecules and drastically increase their volume by solvent exchange while keeping the same topology. The evacuation of the solvent molecules by heating provides the dry forms, with the same symmetry, but with a strong decrease of the cell volume. For example, in MIL-88D (chromium(III) biphenyl dicarboxylate), the ratio Vopen/Vdry is larger than 3, which means a difference of more than 300% between the two states.174 Correlatively, this expansion/shrinkage implies very large reversible atomic displacements in the structures (>10 Å for MIL-88D), the topology remaining invariant with apparently no bond breaking.
Structural reasons must exist for explaining such a behaviour.174 They concern two types of situation: (i) the host–guest interactions (hydrogen bonds, VDW forces, π–π interactions) and (ii) the intrinsic flexibility of the framework itself, induced by the existence of ‘weak points’ within the skeleton, which allow the deformation of the network under the action of the stimulus.
On the first point, the interactions created by the guest must be sufficiently energetic to induce the structural changes described by Kitagawa, and once this condition is fulfilled, the extent of breathing will depend on the strength of the host–guest interactions. For instance, with MIL-88C, and as far as hydrogen bonds are concerned, the volume expansion when the dry form is put in contact with a solvent is ten times more important for DMF than for H2O.174 This could find applications in separation.
The second point is more specific and relates to the structural characteristics of each structure. For MOFs with non rigid ligands, the weak point of the structure concerns the carbon chain of the ligand itself which can change its conformation when stimulated. A nice example is provided by [Cu2(pzdc)2(dpyg)]164,165 which exhibits contraction–expansion during the desorption–adsorption process, with a magnitude of breathing of 3.6 Å and 27% in volume variation. This compound adsorbs MeOH and H2O but neither N2 nor CH4. For MOFs with rigid ligands, and in the limit of the rare known examples, two weak points of the structure (Fig. 14) seem to be responsible for the breathing:174 (i) the connection of the carboxylate functions with the metals of the inorganic subnetwork; the O–O axis of the carboxylate which acts as a kneecap between the inorganic part and the carbon chain during the transition, and (ii) the free rotation of the phenyl rings around the axis of the linker, which can relax the constraints during the structure change and minimize the lattice energy, as in MIL-53. When the inorganic subnetwork is made of clusters, the latter can also play the above role of the phenyl rings. It occurs for MIL-88 (Fig. 14) where the trimers also rotate by 30° during the transition.
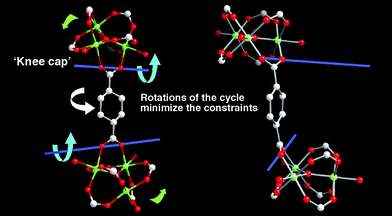 | ||
| Fig. 14 Part of the structure of the cage in MIL-88B, which corresponds to one edge of the trigonal bipyramid. It explains the framework displacements during breathing, occurring around the ‘knee cap’ O–O axis (blue line and sense of rotation (blue arrow)) of the carboxylates. This allows the rotation around this axis of the whole trimeric units (green arrows). The free rotations of the phenyl ring and of the trimers around the OOC–COO axis occur only for relaxing the constraints and minimize the lattice energy during the transformation. | ||
There is also a topological restriction for breathing. As exemplified by MIL-68,150 a structure cannot breathe if odd cycles exist in the structure. Indeed, MIL-68 is a polymorph of MIL-53. Despite the same formula and the same inorganic chain, the structures are different. Instead of lozenge-shaped tunnels in MIL-53, MIL-68 exhibits large hexagonal and small triangular tunnels. The latter confers a strong rigidity to the structure, which was verified as a function of temperature, excluding any breathing. The same rule applies for MOF-5.
6. Are MOF structures predictable?
Regarding topological aspects, the inorganic subnetwork may be formally described as clusters (0D), chains (1D), layers (2D) or frameworks. Its combinations with organic ligands (carboxylates, phosphonates, crown ethers, polyamines...) on which one can play with the size, shape, rigidity together with the number and relative positions of the complexing N-donors or O-donors functions leads to virtually infinite possibilities of topologies. But, are all the associations possible?The predictability of hybrid architectures and the control of their dimensionality are therefore essential, however confronted with the underlying issue of polymorphism.6 The concept of rational design developed by O'Keeffe et al. was rooted in the fact that topochemically-selected reactions govern the construction process of the metal–organic framework in hydrothermal conditions. Although metal-containing SBUs may not be isolated, their repeated occurrence in a significantly large number of structures suggested that the targeted inorganic sub-unit pre-exists in the solution and may be obtained with adequate synthetic conditions for participating in a systematic way in the construction of frameworks.45,46 As soon as a prototypic structure is known, the possible modulation of the pore sizes is directly achieved through the length of the ligand or the nature of the inorganic SBU. The isoreticular synthesis of IRMOFs 1–16 derived from the prototypic MOF-533,175,176 is a good example of this approach.
A real structural prediction of new MOFs required the development of a global optimization approach for predicting libraries of viable MOFs in the same way as our group and others used for the prediction of inorganic structures.177–187 Indeed, MOFs offer ideal features and concepts for the efficient computational developments already applied by our AASBU method177 for inorganic structures: the bricks exist or are known, the global optimizations techniques for identifying the local minima of the “energy “ landscape and the candidate structures are valid. My group therefore made an adaptation of the AASBU method applied to MOFs.188 The automated assembly of SBU is performed in 3D space with minimal input, aiming at computationally exploring the possibilities of connection. The simulations provide a list of hybrid candidates (existing or not-yet-synthesized structures), with their space group, cell parameters and atomic positions, while tackling the issue of polymorphism by limiting the domain of structures that are possible for a given metal–organic ligand pair. The inorganic (modelled by a rigid body) and organic (treated as a flexible body) counterparts may be either treated independently or encapsulated in a single hybrid building-block. The computational assembly, is further controlled through the use of pre-defined “sticky-atoms”. The rules that control the possible assembly are encapsulated in a forcefield that includes “sticky-atoms” pairs, parameterized on an atom–atom basis by a simple Lennard–Jones potential.
This simulation was successfully validated on the best known MOFs (MOF-5, HKUST-1, MIL-53) before testing it with other bricks and ligands. Our first choice focused on the trimeric cluster of metallic octahedral sharing a µ3-oxygen, for which the chemical conditions (including hydrothermal) of existence were mastered in the group.45,46 Combined with the two simplest carboxylates: terephthalate (1,4-benzene dicarboxylate or BDC) and trimesate (1,3,5-benzene tricarboxylate or BTC), this cluster gave two solids, MIL-10034 and MIL-101,36 exclusively in powdered form, with complex XRD patterns and Bragg peaks at very low angle, indicating a huge cell. As preliminary attempts to solve the structures ab initio failed, the AASBU programme was tested. After calculating all the steps, three candidate polymorphic structures with reasonable energies were found, with different symmetries and cell parameters. Among the calculated XRP patterns, only one perfectly fits with experimental results After refinement of the data, it appears that MIL-100 and-101, which both exhibit an augmented MTN zeolitic topology, (scale chemistry!) are ‘the two largest non proteinic structures ever evidenced’,189 with cell volumes of 380,000 and 706,000 Å3 respectively, with unprecedented cage volumes (from 10,000 to 20,000 Å3) (Fig. 15) and Langmuir surface [for MIL-101 (5900 m2 g–1)]. In the usual classification of porous solids, they represent the first example of perfectly crystallized mesoporous solids.
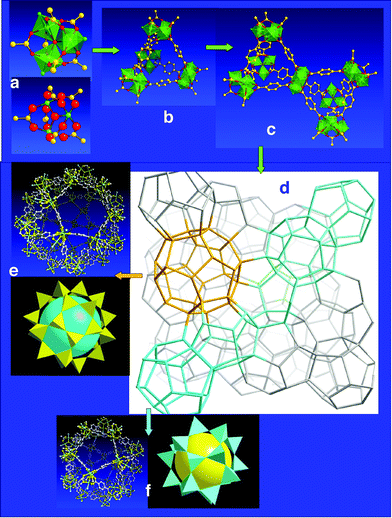 | ||
| Fig. 15 Structure and building of MIL-101: (a) the trimeric inorganic brick; (b) the supertetrahedral SBU; (c) their connection; (d) the framework of MIL-101; the lines join the centers of the supertetrahedra and show two types of cages (yellow and blue); (e) ball and sticks and polyhedral representations of the large cages, (f) ball and sticks and polyhedral representations of the ‘small’ cages. | ||
Beside their spectacular character, the results show that prediction, exclusively based on the combination of mastered chemistry and computer simulations based on energy concepts, allows the structure of not-yet-synthesized solids to be anticipated. This renders computer simulation extremely promising in the field of MOFs. Not only does it provide structural solutions, but it stimulates the synthesis activity in the search for new systems without the need of single crystals. Beyond that, and just by comparison with X-ray powder patterns, it provides structural solutions which could even not be reachable in the absence of single crystals (the upper limit of possibility of solving a structure in a cubic F lattice was calculated to be 288,000 Å3190). As it can be anticipated that the discovery of new solids in the future will probably lead to more and more complex structures and difficulties in getting single crystals, the simulation facet in the research of new MOFs opens a new window for their knowledge, their applications and aesthetics (Fig. 16).
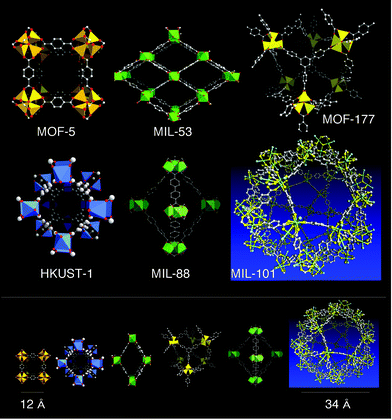 | ||
| Fig. 16 The currently most cited MOFs (up). In the lower part of the figure, their cages are represented at the same scale. | ||
7. Hybrids as efficient materials: some physical properties and applications
Porous solids have been for a long time strategic materials and some authors191 claim that they represent more than 20% of the Gross Domestic Product of the industrial countries for the applications they imply, directly or indirectly. These applications concern mainly petrochemistry, catalysis and selective separation using the porous character, the high thermal stability and the interesting surface areas of inorganic porous solids. The principal limitation, up to the discovery of mesoporous compounds, was the relatively small size of the pores in the crystallized solids , which were further shown to be rather disappointing regarding applications. MOFs provided a breakthrough, as shown in Section 2, since they can combine all the desired possibilities of the above classical porous solids together this time with potentially unlimited pore sizes and surface areas, with the physical properties of dense solids which were quasi-inexistent for zeolites and related compounds.This gap opens a number of windows for new potential applications, at a moment of our civilisation when energy problems become crucial and sustainable development a way for surviving. MOFs can provide many solutions in these areas owing to their already mentioned infinity of possibilities, the increasingly rational approach for their synthesis, and the ability to play on the tunability of all the characteristics of porous solids (skeleton, surface, cages and/or tunnels) for dedicated applications. Most of them are inspired by previous orientations (catalysis, gas separation/storage), but with highly improved performances. Some of them are unprecedented.
Catalysis by porous hybrid materials
Although catalysis is potentially one of the most important applications of metal–organic porous materials, as was the case in microporous zeolites and mesoporous materials, only a handful of examples have been so far reported.192–194 Fujita's group first achieved195 shape-specific catalytic activity for the cyanosilylation of aldehydes over [Cd(NO3)2(4,4′-bpy)2]n in 1994. For catalytic applications using metal–organic open-framework materials, apparently five types of catalyst systems or active sites have been utilized: (a) homochiral metal–organic frameworks, (b) metal ions or ligands in the metal–organic frameworks, (c) coordinatively unsaturated metal (CUM) centers in metal–organic porous materials, (d) metal complexes in supramolecular porous frameworks, (e) highly dispersed metal or metal oxide nanoparticles loaded onto porous MOF host lattices.Homochiral, porous MOFs that look like heterogeneous enzymatic catalysts are particularly attractive candidates as heterogenenous asymmetric catalysts for the production of optically active organic compounds due to the lack of chiral, inorganic zeolites. However, despite considerable efforts, attempts to synthesize homochiral metal–organic porous materials capable of enantioselective catalysis have met with only limited success. Only a few groups have recently provided preliminary evidence for the potential utility of homochiral porous MOFs in enantioselective separation and catalysis.196–199 Among the related works, Lin and co-workers have designed a homochiral porous Cd–MOF ([Cd3Cl6L3]. X) which, after chemisorption of titanium isopropoxide onto the hydroxyl units, catalyses ZnEt2 additions to aromatic aldehydes for highly enantioselective heterogeneous asymmetric catalysis rivalling its homogeneous counterparts.197 Recently, Dybtsev et al.199 have isolated a Zn-based MOF with bdc and lactate ligands, intrinsically homochiral, with size- and enantioselective guest sorption properties and a remarkable catalytic activity with size and chemoselectivity, and high conversion in the oxidation of thioethers to sulfoxides.
Most popular examples for catalytic applications belong to framework catalysis by metal ions in the metal–organic frameworks even though the metal ion and the ligand are usually selected as the building blocks rather than as catalysts. After the pionneer works of Clearfield on phosphonates200–202 framework catalysis by MOFs includes now cyanosilylation,203 the Diels–Alder reaction,204 the hydrogenation,205 esterification,44 CO oxidation,206etc.
The introduction of CUM centers into porous MOFs can offer a promising tool in catalysis because a regular arrangement of metal centers in the pore channels induces regioselectivity or shape- or size-selectivity towards guest molecules or reaction intermediates.207 For example, Kitagawa and co-workers have shown that pore surface engineering using a metaloligand as a building unit could provide the introduction of CUM centers.208 Some examples in framework catalysis may have been achieved by CUM centers in MOFs although they have not clearly mentioned.
Given that inorganic porous materials that contain metal complexes encapsulated in their porous cavities take advantage of heterogeneous catalysts, it might be a good approach to encapsulate metal complexes into MOFs through supramolecular self-assembly. A few authors recently illustrated this strategy.209,210 For instance, Qiu et al.209 have encapsulated the metal complex [Mn(phen)2(H2O)2]2+ into supramolecular frameworks through hydrogen bonding and π–π interactions. The resulting supramolecular frameworks showed size- and shape-selective catalytic activity in the oxidation of phenols with H2O2 to form dihydroxybenzenes.
The use of highly dispersed metal or metal oxide nanoparticles inside porous MOF host lattices is very rare. However, Thompson et al. showed that Pd- and Pt phosphonates were active catalysts for the photochemical production of H2211 and the production of hydrogen peroxide from streams of H2 and O2.212,213 More recently, Fischer and co-workers have shown that metal–organic chemical vapor deposition gave inorganic nanoparticles (Cu and Pd) in MOF-5 that were moderately active for methanol synthesis (Cu@MOF-5) and hydrogenation of cyclooctene (Pd@MOF-5), respectively.210 However, the surface functionalization of pores for catalytic applications remains still unexplored in porous MOFs in spite of a promising research area, previously exemplified in mesoporous materials.
Whereas catalytic applications concern the surface of the pores, many others use either the pores and their possibilities to be filled by inserted species, or the skeleton as soon as properties close to those of dense solids (magnetism, conductivity, optical properties) are required.
Insertion of species and their applications
Solvents are easily evacuated from the pores of MOFs. The tunability of pore sizes render them particularly attractive for insertion of species, including gases, liquids, molecules, inorganic nanoparticles and metals. On the point of tunability, a false debate is emerging: what is better, large pores or small pores? Such a question is not reasonable. The choice will only depend on the required applications and on the size of the species to be inserted. It is clear that if a selectivity between small species is sought, there is no need for large pores. In contrast, if the aim is to insert drugs in the pores, the larger the cage, the better the storage. In other words, instead of opposing the two ways, it is better to use all the possibilities of dimensions provided by the literature to fit with a given application and optimize it. This does not prevent us from searching for larger and larger pores. Beside the idea of a world record (but every record is to be beaten), more important is to enlarge the possibilities of insertion for dedicated applications.The first success,159 due to Kitagawa in 1997, was the introduction of large amounts of methane in a coordination polymer. This opened the way for a tremendous search for materials able to store these gases, due both to their high specific surface areas (SSA) and large pore sizes. For the moment, the main efforts concentrate on H2, CH4 and CO2 with however a striking difference between the first and the others. Indeed, MOFs adsorb large amounts of hydrogen only at 77 K; at room temperature, adsorption is negligeable, at variance to CH4 and CO2 which exhibit interesting performances at 300 K and above. This low temperature adsorption of H2 prevented, for a long time, applications for its use in cars until the recent discovery of technical solutions.215
At this point, two general remarks must be made. The first concerns the performances of MOFs and their reproducibility. It originates from the works of Panella and Hirscher216 who showed that the claimed performances of a MOF are strongly dependent on the method of synthesis, on the scale of production (laboratory or large-scale preparations) and, as already mentioned, on the efficiency of activation of the MOF. For example, for MOF-5, the first measurements on small-scale laboratory synthesis claimed an uptake at 77 K of 4.5 wt.% at 0.8 bar,217 further corrected to 1.6218 and 1.3219 wt.% before reaching 4.7 wt.% at 50 bars (5.1 wt.% at saturation)216 when the sample was prepared by a large scale fast synthesis. This means that every published value, even unprecedented, must be taken with care and needs to be verified by other groups before becoming credible, using for instance round robin procedures, as is done in other disciplines prior to acceptance. Moreover, it is currently difficult to compare the real performances because the data (for instance wt.%) refer to a given P/P0 ratio (often at 1 bar) whereas the true capacity must be measured at high pressure (60–70 bars). For the future, there is an urgent need of normalization, with a complete set data including isotherms of adsorption (classical) and desorption (currently rare), gas capacity at high pressure (wt.%; cm3 g–1; cm3 cm–3) as well as surface areas (BET, Langmuir). At the laboratory scale, to the best of my knowledge, the performances of only three MOFs have been validated: MOF-5 (see above), MIL-53220 and HKUST-1.221 Moreover, a material will be efficient for industrial developments50 if the performances, measured at the laboratory scale on a few milligrams, are still valid at a large scale.
The second remark concerns an emerging trend. The improvement of the performances in gas adsorption will go through a better understanding of mechanism and thermodynamics of adsorption, and of a better knowledge of the adsorption sites. This was done for zeolites. Measurements of heats of adsorption are currently very scarce,50,222 as is the localization of adsorbed molecules, experimentally (using X-ray and neutron diffraction),223–225 or theoretically using computer simulations.226–234 This will be a major requirement for the future. The identification of the active sites, either on the inorganic or the organic moieties, will be of paramount importance for elaborating new syntheses.
Up to now, the three best verified MOFs for hydrogen storage at 77 K are MOF-5 (5.1 wt.% at saturation, SSABET: 2296 m2 g–1, SSALangmuir: 3840 m2 g–1), HKUST-1 (3.6 wt.% at saturation; SSABET: 1154 m2 g–1, SSALangmuir: 1958 m2 g–1)), and MIL-53(Al) (3.8 and 4.5 wt.% at 15 bars and at saturation, SSABET: 1100 m2 g–1, SSALangmuir: 1540 m2 g–1).50Fig. 17 shows their isotherms of adsorption. The heats of adsorption were calculated to be –3.8 and –4.5 kJ mol–1 for MOF-5 and HKUST-1 respectively.
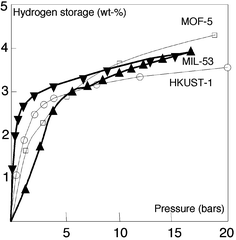 | ||
| Fig. 17 Adsorption isotherms of a) MOF-5 (□), b) HKUST-1 (○), and MIL-53 (▲) at 77 K (σ adsorption; τ desorption). | ||
The adsorption–desorption curve of MIL-53 presents an hysteresis which is due to the breathing effect in this solid. 90% of its total capacity is reached in less then one minute; 2.2 wt.% of hydrogen is recovered at 0.1 bar.
Recently, a capacity of 7.5 wt.% at 70 bar was mentioned for MOF-177242 but needs to be verified according to the above remarks. Anyhow, all these MOFs show H2 capacities much larger than those reported for zeolites A, X, Y and RHO (1.8 wt.%).243 Currently the above MOFs compare favourably to high-grade activated carbon with an SSA close to 2500 m2 g–1.244,245
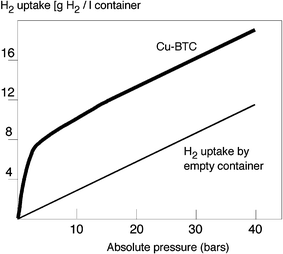
Müller et al,50 from BASF, recently showed the real efficiency of MOFs for industrial applications. Indeed, they noted that, compared to pressurizing an empty container with hydrogen, MOF-5, IRMOF-8 and Cu-BTC-MOF (electrochemically-prepared HKUST-1) increasingly take up higher amounts of hydrogen, all of them exceeding the standard pressure–volume–temperature (PVT) uptake curve of the empty container and with the steepest incline below 10 bar. At 40 bar, the PVT-relationship of hydrogen in an empty canister is registered as 12.8 g H2 l–1,244–246 whereas containers filled with Cu-BTC-MOF reach a plus 44% capacity of up to 18.5 g H2 l–1. For comparison, the volume-specific density of liquid hydrogen at its boiling point (20 K) is 70 g H2 l–1. Above 10 bar, the curves run mostly parallel to the conventional H2-pressure–volume relationship. On a per weight calculation, it becomes clear that the saturation of MOFs with hydrogen is already achieved at pressures of less than 15 bar (Fig. 18). For electrochemically-prepared Cu-BTC-MOF this attributes to about 3.3 wt% H2-uptake. Some dedicated reviews appeared on the subject in 2005.235–237
For a few experimental determinations of the localization of H2 molecules in MOFs,223–225 several computer simulations have been performed, which either anticipated experience or agreed with it.226–234 Whatever the case, experience and simulation show that it is molecular dihydrogen which is adsorbed, and that the metal–oxygen clusters are the preferential adsorption sites for H2 in MOFs; the effect of the organic linkers becomes evident with increasing pressure. For example, Yildirim and Hartman performed a nice neutron diffraction study of MOF-5 under D2 increasing pressure. Their results (confirmed by simulation) show first that the amount of adsorbed D2 can reach 10 wt.% at high pressure at 4 K (which corresponds to 46 D2 per Zn4 cluster), moreover, they identified five types of sites Fig. 19. When the pressure is increased, the three first sites, progressively occupy up to 26 D2 per Zn4 cluster; all are situated around the clusters at distances in the range 3.1–3.6 Å of the oxygens of the carboxylates. It is only above that interactions with the phenyl rings begin to occur.
 | ||
| Fig. 19 The different adsorption sites for H2 in MOF-5 (data from224). | ||
The H2 storage capacity of MOFs is definitively larger than that of carbon nanotubes, which is much higher than zeolites. Furthermore, diffusion of H2 in MOFs is an activated process, similar to diffusion in zeolites. This domain is currently a great challenge, combining targeted storage capacities, thermodynamics and kinetics of exchange. It will become more and more interdisciplinary, using chemistry, physics and engineering science.
At variance with hydrogen, they are both adsorbed at room temperature. However, studies on their adsorption are by far less numerous than for hydrogen,222,249–256 even if it is with CH4 that the story of gas adsorption began for MOFs.159 In a general way, methane is less adsorbed than CO2. Even if it is not yet clearly understood, it seems that the difference in adsorption is due to the existence of large quadrupolar moment for CO2 (–1.4 × 10–35 C m2)222 which does not exist with CH4. This moment induces specific interactions with adsorbents (molecular orientation, hydrogen bonding…) which, depending on the host structure, will give different behaviours for the adsorption isotherms.
From their adsorption isotherms, the currently best adsorbers are MOF-177255 (SSA: 4500 m2 g–1, 11 × 17 Å pores) and MIL-10136,257 (SSA: 5900 m2 g–1, 29 and 34 Å diameters) which correspond to the solids having the largest pores ever evidenced (Fig. 20). They adsorb more than 30 mmol g–1 (33.5 at 16 bar and 33.5 at 40 bar for MOF-177 and 16 at 16 bar and 40 at 70 bar for MIL-101), which means ca. ten times the amount of pure CO2 in a container at the same pressure. Whereas MOF-177 presents a sigmoid shape, MIL-101 does not. This particular shape, which also occurs for other MOFs in the same study, was attributed by Millward to larger effective pore sizes, which lead to a behavior closely related to that of the bulk fluid.258 However, no structural information can confirm this hypothesis.
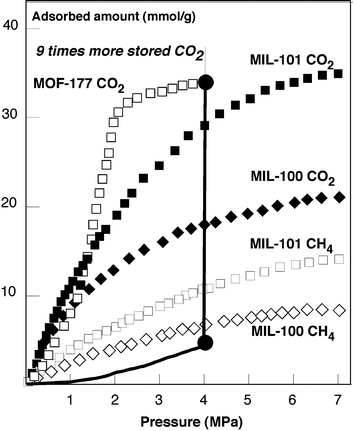 | ||
| Fig. 20 CO2 and CH4 adsorption isotherms of MIL-101 and MOF-177 at 300 K. Comparison with pure CO2 | ||
A possible explanation can be provided by the only two structural studies on CO2 adsorption, due to the groups of Takamizawa223 and Férey.259 Both show that flexibility might have an influence. The rhodium(II) benzoate pyrazine evidenced by Takamizawa223 did not exhibit large pores (9 × 4 × 3 Å) but evidenced a small breathing effect that Takamizawa explained from single crystal studies by a phase transition, implying the host–guest interactions which produced a cell contraction by 3%. The enthalpy of adsorption was close to 35 kJ mol–1. Anhydrous MIL-53 presented a much pronounced behaviour, related to its already mentioned large breathing effect (ca. 40%). Whereas the isotherm is classical with CH4, the CO2 one presents a two-step behaviour (Fig. 21a). After a first increase of CO2 uptake, it marks a clear plateau at 3 mmol g–1 up to 5 bar before a second increase up to 9 mmol g–1 at 20 bar.222 The enthalpy of adsorption is found once more close to 35 kJ mol–1. In situ diffraction studies provide the explanation.259 When dehydrated under vacuum before CO2 adsorption, MIL-53 adopts its expanded structure, but at low pressures of CO2 it structure re-adopts the shrunken form up to 5–6 bar. At higher pressure, the structure reopens to give the expanded form.
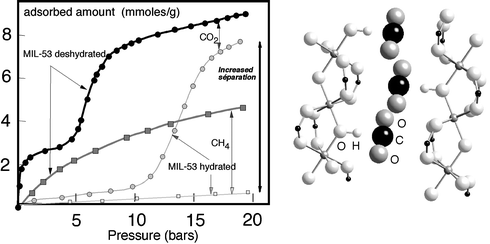 | ||
| Fig. 21 CO2 and CH4 adsorption isotherms of anhydrous (a) and hydrated (b) MIL-53 and location of CO2 molecules within the tunnels. | ||
A complete structural study at 1 bar shows that the CO2 molecules lie at the center of the tunnels, as did water molecules,38 but the interactions are different, even if the OH groups shared between the octahedra of the skeleton are concerned in both cases. Whereas with H2O, the interactions occurred between the OH groups and the oxygens of the water molecules of the tunnels, with CO2 it is the carbon and not the oxygen of CO2 which interacts with OH. This induces a strong but continuous modification of the IR spectrum, which was followed by in situ measurements,259,260 and correlatively, a clear bending of the CO2 molecule by 6°.
This tends to demonstrate an influence of the polar character of the adsorbed molecules on the host–guest interactions and on the shape of the isotherms. This could have applications in selective adsorption of polar molecules. It is the reason why our group recently studied the adsorption of CO2 and CH4 by the hydrated form of MIL-53256 in order to see the behaviour of a non-polar and polar probe in the presence of a second polar molecule (here water). The isotherms are drastically changed (Fig. 21b). Whereas CH4 is almost not adsorbed up to 20 bar (0.2 mmol g–1), there is a very little uptake of CO2 up to 10 bar and a sudden increase (8 mmol g–1) after. XRPD patterns show that, up to 10 bar, the shrunken form of MIL-53 prevails and in situ IR spectroscopy indicates a progressive displacement of the water. This could find applications in the recovery of CO2 in mixed gas streams, particularly natural gas.
The tunability of the pore size in MOFs presents also a big advantage for the separation of alcanes. This application, well known for zeolites,22 is just emerging for MOFs. A recent structural paper by J. Li et al.275 proves that the pore size of one of their coordination polymers is sufficiently large for trapping methane, ethane, propane, while butane is not inserted. Moreover, in the petroleum industry, the separation of alkane isomers is a very important process. Narrow pore zeolites sieve linear from branched alkanes, to boost octane ratings in gasoline.22 Recently, a solid described simultaneously by different groups,276–278 exhibits a similar behaviour and is used for gas-chromatographic separation of linear and branched alkanes.278 Whereas the first example described real trapping of small alkanes, the second one, owing to the small dimensions of the cage (4 × 4 Å), concerns only the size of the windows allowing an accessibility to the interior of the pores. They can accommodate only the linear part of the branched isomer and the retention of alkanes on the column mainly depends on the length of the linear part of the alkane, and its van der Waals interactions with the microporous MOF walls.
The immense possibilities of adsorption of MOFs begin also to apply to liquids. Recently, Jacobson et al.279 introduced aniline, thiophene and acetone in crystals of MIL-47 (V4+) by impregnation and localized the species within the tunnels. The intercalated aniline molecules show substantial ring–ring π–π interactions between them and with the BDC ligand through short distances. The π–π interactions are complemented by weak C–H⋯π and N–H⋯π interactions between the aniline molecules and the BDC ligands. The packing of thiophene molecules is similar to that of aniline. A clear C–H⋯π interaction between the thiophene molecules and the framework BDC seems to play a major role in determining the thiophene orientation. The acetone molecules are stacked with an antiparallel packing pattern with weak dipolar carbonyl–carbonyl interactions. The packing density calculated for the guest molecule is 122.2 Å3 per acetone molecule which is almost identical to that of liquid acetone. In contrast, the guest packing densities calculated for aniline and thiophene are both ca. 21% lower than the corresponding liquid densities of the guest molecules, probably because the oriented interactions between the guest molecules and the framework BDC ligands determine the packing.
As already observed with this topology, breathing occurs during insertion, but with a lowering of the space group symmetry from centrosymmetric Pnma to non-centrosymmetric P212121. This was confirmed by second harmonic generation (SHG) measurements. The SHG efficiency of intercalated solids is comparable to that of quartz. This shows the importance of non-covalent orientated weak interactions in the packing of organic molecules within nanopores of MOFs. As mentioned by Jacobson, such interactions, although relatively weak, may readily cause remarkable deformation and symmetry change of the framework, which point to effective ways of manipulating known materials or designing new materials with targeted properties through intercalation chemistry. As recently shown by De Vos et al.,280 the same solid MIL-47 exhibits also remarkable properties of separation between the different isomers of xylene. These features open new opportunities of application for MOFs.
A decisive gap has been reached with the discovery of the mesoporous MOFs MIL-10034 and -101.36 Their augmented MTN zeolite topology exhibits two types of cages with 20 and 28 vertices, the first with exclusively pentagonal windows and the other with pentagonal and hexagonal ones, with large aperture (up to 16 Å). These windows allow the introduction of large molecular species, particularly drugs. The analgesic Iboprofen was used as a probe for validation.281
MIL-100 and MIL-101 show remarkable Ibuprofen uptake (Fig. 22) compared to what was known before, but they adsorb drastically different amounts of drug (0.35 and 1.4 g g–1of dehydrated MILs, respectively) as a result of their different pore sizes (25 and 29 Å for MIL-100; 25 and 29 Å for MIL-101). These findings are very important as only very small amounts of material are required for the administration of high dosages. Structural reasons may explain this discrepancy, particularly the free apertures of the windows of the cages [4.8 Å (MIL-100) and 12 Å (MIL-101) for the pentagonal; 8.6 Å (MIL-100) and 16 Å (MIL-101) for the hexagonal] compared to those of Ibuprofen (6 × 10.3 Å). Therefore, ibuprofen fills only the large cages in MIL-100, and all of them in MIL-101. According to the weight increase, each small and large cage of MIL-101 hosts approximately 56 and 92 Ibuprofen molecules which represent four times the capacities of MCM-41 toward Ibuprofen282 Moreover, the release of the drug at physiological pH occurs in 3 days with MIL-100 and six days with MIL-101, in two steps. The faster release involves the molecules which fill the cages. Afterwards it involves the molecules in noticeable interaction with the framework.
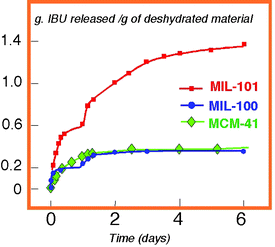 | ||
| Fig. 22 Compared adsorptions of ibuprofen by MCM-41, MIL-100 and MIL-101. | ||
This unprecedented behavior illustrates three facts: (i) the ever-growing need for very large pores,136 (ii) when tunable, the hierarchy of mesopores can act as an internal molecular sieve for a given guest of important dimensions with a selective occupation of the cages, the empty cages remain able to host a different species; and (iii) such matrices may provide tools for the study of nanoassemblies of organic compounds and help the development of nano-organic chemistry. Moreover, it adds a new route for drug storage and release. Two routes have been previously set up: the “organic route”, which uses either biocompatible dendritic macromolecules or polymers283,284 and the “inorganic route”, in which the hosts are inorganic porous solids, such as zeolites285,286 or mesoporous silicate materials.287 In the first case, a wide range of drugs can be encapsulated but a controlled release is difficult to achieve in the absence of a well-defined porosity.283,284 In the second case, this release is performed by grafting organic molecules on the pore walls but implies a decrease in the drug-loading capacity.288 Crystallized mesoporous MIL-100 and -101 introduce a third way: the “hybrid” route. Indeed, the combination of a high and regular porosity with the presence of organic groups within the framework may cumulate the advantages to achieve both a high drug loading and a controlled release.
These mesopores allow also the introduction of large molecular inorganic species within the cages. For instance, MIL-101 incorporates36 the Keggin polyanion PW11O407–. Owing to the large dimension of this anion (ca. 13 Å), only the large cages can host it. From XRPD, TGA, specific surfaces and solid state NMR measurements, it was proved that each cage can accept five Keggin ions, representing 50% of the volume of the cage. This successful incorporation of large amounts of Keggin anions strongly suggests that MIL-101 is an ideal candidate for the introduction of other nano-objects with chemical, physical, biological or medicinal properties.
In a general way, a fit must exist, mainly between the size of the guest and the dimensions of the windows. A recent example289 illustrates this point. Fischer looked at the absorption of volatile or very soluble metal organic CVD precursors [(η5-C5H5)Pd(η3C3H5), [(η5-C5H5)Cu(PMe3), (CH3)Au(PMe3) and Cu(OR)2 (R = CH(CH3)CH2NMe2)]. Only the latter does not incorporate the structure because its dimensions exceed those of the pore window of MOF-5 (8 Å). Reduction of the intercalated compounds by H2 leads to the formation of metallic nanoparticles. In the case of Pd, the framework of MOF-5 is strongly affected by the treatment whereas, with Cu and Au, it remains intact. However, the range of observed particle sizes (13–15 Å for Pd, 30–40 Å for Cu, 50–200 Å for Au) are far above the dimensions of the cage, indicating a biphasic metal–MOF mixture instead of incorporation at the nanometric scale at variance to what is observed with MCM.290 However, the corresponding solids exhibit some catalytic properties.
Except in one case,323 inclusion of metallic nanoparticles in MOFs remains a challenge. Suh et al.291 recently tried to generate in situ nanoparticles of Ag and Au within a flexible two-dimensional MOF [NiII(cyclam)2][BPTC] through the reduction of noble metals by the Ni2+ of the cyclam complex. Reduction effectively occurs but a thorough study including many types of characterization proves that, the MOF network remaining intact, the nanoparticles (40 Å for Ag, 20 Å for Au) are not incorporated between the layers but form a metal–MOF composite. The authors suggest that Ag(I) metal ions are introduced between the host layers and react with the Ni(II) species incorporated in the host to form Ag(0) atoms, which diffuse to the surface of the solid to grow into nanoparticles. Since the Ag nanoparticles grow on the surface of the solid, the host structure can be maintained even after the nanoparticles of ca. 4 nm are formed. Such an explanation seems valid also for the above metalocenes.
MOFs as nanoreactors and nanomoulds for nanosciences and applied physics
The work of Suh was an elegant but unsuccessful trial to use MOFs as reactors within which nanospecies could be synthesized in situ and confined in the pores. The high reactivity and diffusion of metallic aggregates was probably responsible for the failure. However, as soon as such a strategy becomes applicable, a myriad of new possibilities will arise, in relation to the physical properties which are usually encountered in dense solids (magnetism, conductivity, optical properties…), but this time at the nanoscale. The unique advantage of MOFs is to provide large, tunable, and well defined crystallized pores. Beside their now classical role of storage and sieving, they can act as nanoreactors and perform chemical reactions and the formation of known (or unknown) products within the cages, with the possibility to see the effect of confinement on the structure of the included species. Once this nanoreactor role is realized, the cages would act as nanomoulds for the synthesized species because the size of the latter is fixed by the dimensions of the cage, thereby leading to calibrated monodisperse nanoparticles, a feature which is scarcely reached by the usual methods of obtaining nano-objects. This opens the way to a new step in the knowledge of nanophysics with the study of strictly monodisperse assemblies, their size being tunable as a function of the size of the cage.The first example of such a strategy is provided by MIL-101.36 Once desolvated and impregnated in a Zn2+ solution, it provides nanoparticles of the semiconductor ZnS when the carefully washed Zn-impregnated solid is treated by an H2S stream at 300 K. High resolution electron microscopy with associated chemical nanoanalysis on small crystals unambiguously proves that the semiconductor particles reside only within the pores and not on the surface. Both types of cages are partially occupied with 4 ZnS per trimer, which means 80 and 40 ZnS assemblies within each type of cage, corresponding to a 50% occupancy of the volume of the pores. From preliminary studies, it seems that the sphalerite form of ZnS predominates in the aggregates, whereas, for 60 ZnS, computer simulation of the nucleation and growth of ZnS nanoparticles predicted the existence of bubbles close to a sodalite arrangement.292 The evolution of the semiconducting properties is currently in progress. Such a result opens large perspectives for nanosciences: it becomes therefore possible to imagine the introduction of known dense materials in mesoporous MOFs, such as semiconductors (or a mixture of them), photocatalysts (TiO2), oxide conductors (YBaCuO), ferro- and ferrimagnets (CrO2, spinels…) as soon as the dedicated chemistry is discovered for such a purpose. For that, the accumulated knowledge on the preparation of these nanophases, but in a non-confined form, will be useful.
Conductivity of porous MOFS seemed impossible for a long time because conductivity implies either mobility of species (ionic conductivity) or delocalization (electronic conductivity, isotropic or anisotropic). For MOFs, if ionic conduction is theoretically possible when dedicated inserted species are chosen, electronic conduction seems to be almost inaccessible for at least two reasons: (i) there is no clear mention of MOFs containing mixed-valence species, a necessary condition for eventual electronic delocalization and (ii) most of the MOFs are coordination polymers (0D for the inorganic framework), whereas, except in a few cases,293,294 at least a one dimensional inorganic subnetwork is required.
A recent breakthrough circumvents these limitations in terms of mixed-valence and at least ionic conductivity.295 The problem was solved by the electrochemical insertion of lithium in MIL-53(Fe3+) or [Fe(OH)0.8F0.2(BDC)] which presents one dimensional chains of corner-linked octahedra connected by terephthalate ions, and is known to present large breathing effects. The progressive introduction of Li within the tunnels induces the onset of Fe2+ within the chains. Structural, Mössbauer, and electrochemical studies and DFT calculations unambiguously establish the mixed valence of the electrochemically synthesized phase [LixFeIIxFeIII1–x(OH)0.8F0.2[O2C–C6H4–CO2] (x ≤ 0.6). Used as an electrode in Li-half cell, this material shows a reversible redox process around 3.0 V vs. Li+/Li° exchanging 0.6 Li per formula unit with excellent capacity retention and rate capability, even if the induced electronic conductivity is rather low. Moreover, the introduction of mixed-valence for Fe changes the magnetic properties from antiferromagnetic for MIL-53(Fe3+) to ferrimagnetic for Li-MIL-53(Fe3+). It reveals also an interesting property of MOFs with large galleries, related to the breathing effect: the uptake of electrolyte molecules within their channels facilitating ionic transport in MOFs. Mixed valence in MOFs opens a wealth of opportunities towards the elaboration of materials with tuneable properties for various applications.
Magnetic properties of MOFs are an emerging field.5,21 As for inorganic porous solids, the results are rather scarce.27 Despite their academic interest, it seems that they will not give rise to applications, owing to their current very low magnetic ordering temperatures. Anyhow, magnetic MOFs present interesting features, some of them being specific to the hybrid nature of the related structures, for instance the use of ligands with phenyl rings which, through their delocalized π electrons, can transmit the magnetic information between several inorganic moieties and create long-range interactions even for 0D coordination polymers.
Depending on the dimensionality of the inorganic subnetwork, transition metal-containing MOFs (d and f) satisfy the rules of molecular magnetism for 0D coordination polymers and, for one- to three-dimensional subnetworks, exhibit long-range magnetic interactions governed, as for dense solids, by the Kanamori–Goodenough laws.296–298 The sign and the strength of the magnetic superexchange interactions depend on both the nature of the magnetic carriers (which can give either isotropic (Mn2+, Fe3+) or anisotropic (Co2+) couplings) and on geometrical criteria conerning the M–X–M superexchange angles. Depending on these angles, the coupling can change from antiferromagnetic to ferromagnetic, and the strength from strong (when M–X–M angle is close to 180°) to weak (when M–X–M angle is close to 90°). For 2D solids, dipolar magnetic interactions must also be taken into account for the long range interactions. This means that all the magnetic behaviours encountered in dense solids are also encountered in MOFs, including frustration problems when odd cycles of cations in antiferromagnetic interactions are involved in the structure.299,300 An increasing number of papers refer to this new trend. The current state-of-the-art was recently reviewed by Veciana et al.301 for coordination polymers.
The same authors302–305 recently introduced a new strategy for enhancing the magnetic properties of MOFs. They used the purely organic radical (polychlorinated triphenylmethyl tricarboxylate or PTMTC) as ‘spacer’ paramagnetic ligands which interact magnetically with the transition metal. The so-called MOROFs series exhibit interesting properties. The layered Cu-MOROF-1 is a ferrimagnet below 2 K and exhibits a spectacular reversible breathing behaviour upon solvent uptake and release with a change by 30% of the volume, the desolvated phase exhibiting turbostratic disorder. In the 3D Co-MOROF-3, ferro-and antiferromagnetic interactions coexist.
Spin crossover (SCO) phenomena, which relate to changes under stimuli of the electronic configuration of some 3d ions between high- and low spin forms, also occur in MOFs. The spin crossover is there guest dependent. For example, FeII2(azpy)4–(NCS)4.EtOH306 exhibits a SCO below 150 K in which only a fraction of Fe(II) ions is concerned in the solvated solid whereas the desolvated solid remains high spin. Other examples exist with imidazolates.307,308
The guest dependence is also nicely illustrated by the porous manganese(II) formate309 [Mn3(HCOO)6]MeOH.H2O with a diamond-type framework built up from MnMn4 supertetrahedra, and which exhibits a guest-modulated ferrimagnetic behaviour, owing to the steric effects of the guest which induce subtle changes in the framework. The Curie temperature, originally measured as 8 K can vary from 5 to 10 K according to the nature of the guests re-inserted in the porous framework. Such effects could also explain the complex 4f–3d interactions in the trimetallic Cu–Gd–Fe coordination polymer.310
Finally, one could expect an increase of the critical magnetic temperatures for higher dimensionalities but, they currently do not exceed 100 K, even if a remnant magnetization has been mentioned at room temperature for [Cu2(py)2(BDC)2.311 Up to now, only Ni2+ carboxylates exhibit 3D inorganic subnetworks. Higher Tc values could therefore be expected but, despite interesting ferri- and ferromagnetic properties, they occur only at T < 10 K312–314 because the polyhedra in these structures are only linked by edges and faces, a characteristic which is not favourable for high critical temperatures.
The optical properties of MOFs mainly concern luminescence. Here also, the possibility for MOFs to accept many metals, including rare-earths, the tunability of the chromophors as ligands offer great opportunities for new phosphors and probes. A number of lanthanide carboxylates have been isolated76–101 and some of them were submitted to spectroscopic investigations, but most of these are qualitative and restricted to the presentation of the emission spectra. This quasi-absence of deep interest is motivated by the fact that, in Ln–MOFs, the rare-earth polyhedra have water ligands which are well known to quench fluorescence due to the loss of excited-state energy to vibrational energy of an OH oscillator of close proximity, although it has been shown that the evolution of the Eu3+ lifetimes can serve as an indicator of the degree of hydration of some Ln-glutarates.315
However, a characteristic of lanthanide fluorescence is low absorbance coefficients, and a coordinated ligand with π delocalized system can stimulate energy transfer to the metal center via an intramolecular pathway. This effect, named the ‘antenna effect’316,317 can realize efficient UV light conversion devices. This goal incites research in many fields and new concepts have emerged.318–319 MOFs could be good candidates for such a purpose but serious and quantitative studies are currently extremely rare.320,321 For example, the rare earth trimesate Ln[(C6H3)(COO)3]321 has demonstrated such an ‘antenna effect’. The ligand triplet state is located 200 cm–1 higher than the 5D0 (Eu3+) emitting level, and the migration of the excitation occurs along the chains of rare-earth polyhedra with an activation energy quite similar to the energy separation between the triplet state and the 5D0 (Eu3+) level.
8. Conclusion
There are few examples of topics which, in fifteen years, have known such a tremendous development. Beside the current explosion of new products, due to the quasi-infinity of possible combinations between inorganic and organic moieties, and the understanding of their formation, the current trends, mainly evidenced and developed during the last three years, offer now a myriad of potential applications. In my opinion, the only limitation is the imagination of researchers. A good example is that in a few years, hybrid solids passed from curiosities to strategic materials. Not only do they amplify most of the performances of the usual porous solids (sieving, adsorption, storage…) but they reach domains usually reserved to other disciplines: solid state chemistry and physics with the introduction of physical properties, usually devoted to dense phases; life sciences with their ability to store and deliver drugs; the emerging field of nanoscience and the possibility to provide monodisperse nanoparticles of many kinds of solids… Even polymer science is now concerned322 with the polymerization of the monomers influenced by the restricted space of the pores which induces confinement effects. These solids represent a new world. We just discover it. So, using the title of one of my recent papers,8 the last sentence of this paper could be: ‘Hybrid porous solids: are there limits to the possible?’References
- B. F. Hoskins and R. Robson, J. Am. Chem. Soc., 1989, 111, 5962 CrossRef CAS.
- C. T. Kresge, M. E. Leonowicz, W. J. Roth, J. C. Vartuli and J. S. Beck, Nature (London), 1992, 359, 710 CrossRef CAS.
- J. S. Beck, J. C. Vartuli, W. J. Roth, M. E. Leonowicz, C. T. Kresge, K. D. Schmitt, C. T. W. Chu, D. H. Olson, E. W. Sheppard, S. B. McCullen, J. B. Higgins and J. L. Schlenker, J. Am. Chem. Soc., 1992, 114, 10835.
- S. Kitagawa, R. Kitaura and S. Noro, Angew. Chem., Int. Ed., 2004, 43, 2334 CrossRef CAS.
- G. Férey, Chem. Mater., 2001, 13, 3084 CrossRef CAS.
- O. M. Yaghi, Acc. Chem. Res., 2005, 38, 176 CrossRef CAS.
- C. N. R. Rao, S. Natarajan and R. Vaidhyanathan, Angew. Chem., Int. Ed., 2004, 43, 1466 CrossRef CAS.
- G. Férey, C. Mellot-Draznieks, C. Serre and F. Millange, Acc. Chem. Res., 2005, 38, 317.
- D. Bradshaw, J. B. Claridge, E. J. Cussen, T. J. Prior and M. M. Rosseinsky, Acc. Chem. Res., 2005, 38, 273 CrossRef CAS.
- C. Janiak, Dalton Trans., 2003, 2781 RSC.
- C. J. Kepert, Chem. Commun., 2006, 695–700 RSC.
- O. M. Yaghi, H. L. Li, C. Davis, D. Richardson and T. L. Groy, Acc. Chem. Res., 1998, 31, 474 CrossRef CAS.
- W. M. Meier, D. H. Olson and Ch. Baerlocher, Atlas of zeolite structure types, International Zeolite Association, 4th edn, 1996 [see also http://www-iza-ethz.ch/IZA-SC/Atlas/AtlasHome.html] Search PubMed.
- E. Kokot and R. L. Martin, J. Chem. Soc., 1965, 187 Search PubMed.
- C. Biondi, M. Bonamico, L. Torelli and A. Vaciago, J. Chem. Soc., 1965, 191 RSC.
- E. A. Tomic, J. Appl. Polym. Sci., 1965, 9, 3745–3752 CrossRef CAS.
- A. Weiss, E. Riegler, I. Alt, H. Bohme and C. Robl, Z. Naturforsch., B: Anorg. Chem., Org. Chem., 1986, B41, 18; C. Robl, Mater. Res. Bull., 1992, 27, 99 CrossRef CAS.
- K. A. Hirsch, D. Venketaraman, S. R. Wilson, J. S. Moore and S. Lee, J. Chem. Soc., Chem. Commun., 1995, 2199 RSC.
- D. Venketaraman, G. B. Gardner, S. Lee and J. S. Moore, J. Am. Chem. Soc., 1995, 117, 11600 CrossRef CAS.
- H. Li, M. Eddaoudi, M. O'Keeffe and O. M. Yaghi, Nature (London), 1999, 402, 276 CrossRef CAS.
- D. Maspoch, D. Ruiz-Molina, K. Wurst, N. Domingo, M. Cavallini, F. Biscarini, J. Tejada, C. Rovira and J. Veciana, Nat. Mater., 2003, 2, 190 CrossRef CAS.
- Handbook of porous solids, ed. F. Schüth, K. S. W. Sing and J. Weitkamp, J. Wiley-VCH, Weinheim, 2002 Search PubMed.
- J. Rocha and M. W. Anderson, Eur. J. Inorg. Chem., 2000, 801 CrossRef CAS.
- C. Serre, F. Taulelle and G. Férey, Chem. Commun., 2003, 2755 RSC.
- M. I. Khan, L. M. Meyer, R. C. Haushalter, A. L. Schweitzer, J. Zubieta and J. L. Dye, Chem. Mater., 1996, 8, 43 CrossRef.
- M. Cavellec, D. Riou and G. Férey, J. Solid State Chem., 1994, 112, 441 CrossRef CAS.
- M. Cavellec, D. Riou and G. Férey, Inorg. Chim. Acta, 1999, 291, 317 CrossRef CAS and refs therein.
- J. Chen, R. H. Jones, S. Natarajan, M. B. Hursthouse and J. M. Thomas, Angew. Chem., Int. Ed. Engl., 1994, 33, 639 CrossRef.
- S. Natarajan, S. Neeraj, A. Choudhury and C. N. R. Rao, Inorg. Chem., 2000, 39, 1426–33 CrossRef CAS.
- N. Guillou, Q. Gao, M. Nogues, R. E. Morris, M. Hervieu, G. Férey and A. K. Cheetham, C. R. Acad. Sci., Ser. IIc: Chim., 1999, 2, 387 CrossRef CAS.
- P. Feng, X. Bu and G. Stucky, Nature (London), 1997, 388, 735 CrossRef CAS.
- P. Feng, X. Bu and G. Stucky, Science, 1997, 278, 2080 CrossRef CAS.
- M. Eddaoudi, J. Kim, N. Rosi, D. Vodak, J. Wachter, M. O'Keeffe and O. M. Yaghi, Science, 2002, 295, 469 CrossRef.
- G. Férey, C. Serre, C. Mellot-Draznieks, F. Millange, J. Dutour, S. Surblé and I. Margiolaki, Angew. Chem., Int. Ed., 2004, 43, 6296 CrossRef CAS.
- G. Férey, C. Mellot-Draznieks, C. Serre and F. Millange, Acc. Chem. Res., 2005, 317.
- G. Férey, C. Mellot-Draznieks, C. Serre, F. Millange, J. Dutour, S. Surblé and I. Margiolaki, Science, 2005, 309, 2040 CrossRef CAS.
- M. Jansen and J. C. Schön, Angew. Chem., Int. Ed., 2006, 45, 3406 CrossRef CAS.
- T. Loiseau, C. Serre, C. Huguenard, G. Fink, F. Taulelle, M. Henry, T. Bataille and G. Férey, Chem.–Eur. J., 2004, 10, 1373 CrossRef CAS.
- C. Gérardin, M. In, L. Allouche, M. Haouas and F. Taulelle, Chem. Mater., 1999, 11, 1285 CrossRef CAS.
- C. Livage, C. Egger, M. Nogues and G. Férey, J. Mater. Chem., 1998, 8, 2743 RSC.
- C. Livage, C. Egger and G. Férey, Chem. Mater., 1999, 11, 1546 CrossRef CAS.
- C. Livage, C. Egger and G. Férey, Chem. Mater., 2001, 13, 410 CrossRef CAS.
- P. M. Forster, A. R. Burbank, C. Livage, G. Férey and A. K. Cheetham, Chem. Commun., 2004, 368 RSC.
- P. M. Forster, N. Stock and A. K. Cheetham, Angew. Chem., Int. Ed., 2005, 44, 7608–7611 CrossRef CAS.
- C. Serre, F. Millange, S. Surblé and G. Férey, Angew. Chem., Int. Ed., 2004, 43, 6285 CrossRef.
- S. Surblé, F. Millange, C. Serre, G. Férey and R. I. Walton, Chem. Commun., 2006, 1518–1520 RSC.
- S. Bauer, T. Bein and N. Stock, Inorg. Chem., 2005, 44, 5882 CrossRef CAS.
- E. Biemmi, T. Bein and N. Stock, Solid State Sci., 2006, 8, 363 CrossRef CAS.
- P. M. Forster, P. M. Thomas and A. K. Cheetham, Chem. Mater., 2002, 14, 17 CrossRef CAS.
- U. Mueller, M. Schubert, F. Teich, H. Puetter, K. Schierle-Arndt and J. Pastré, J. Mater. Chem., 2006, 16, 626 RSC.
- S. S.-Y. Chui, S. M.-F. Los, J. P. H. Charmant, A. G. Open and I. D. Williams, Science, 1999, 238, 1148 CrossRef.
- D. M. P. Mingos and D. R. Baghurst, Chem. Soc. Rev., 1991, 20, 14 Search PubMed.
- G. G. Tompsett, W. C. Conner and K. S. Yngvesson, ChemPhysChem, 2006, 7, 296 CrossRef CAS.
- S.-E. Park, J.-S. Chang, Y. K. Hwang, D. S. Kim, S. H. Jhung and J.-S. Hwang, Catal. Surv. Asia, 2004, 8, 91 CrossRef CAS.
- Y.-K. Hwang, U.-H. Lee, J.-S. Chang, Y.-U. Kwang and S.-E. Park, Chem. Lett., 2005, 34, 1596 CrossRef CAS.
- S.-H. Jhung, J. W. Yoon, J.-S. Hwang, A. K. Cheetham and J.-S. Chang, Chem. Mater., 2005, 17, 4455 CrossRef CAS.
- S.-H. Jhung, J.-H. Lee, J. W. Yoon, J.-S. Hwang, S.-E. Park and J.-S. Chang, Microporous Mesoporous Mater., 2005, 80, 147 CrossRef CAS.
- Y. K. Hwang, J.-S. Chang, S.-E. Park, D. S. Kim, Y.-U. Kwon, S. H. Jhung, J.-S. Hwang and M.-S. Park, Angew. Chem., Int. Ed., 2005, 44, 557.
- S. H. Jhung, J.-S. Chang, D. S. Kim and S.-E. Park, Microporous Mesoporous Mater., 2004, 71, 135 CrossRef CAS.
- S. Kitagawa, T. Okubo, S. Kawata, M. Kondo, M. Katada and H. Kobayashi, Inorg. Chem., 1995, 34, 4790 CrossRef CAS.
- S. H. Jhung, J.-H. Lee and J.-S. Chang, Bull. Korean Chem. Soc., 2005, 26, 880 CAS.
- S. H. Jhung, J.-H. Lee, P. M. Forster, G. Férey, A. K. Cheetham and J.-S. Chang, Chem.–Eur. J., 2006, 12, 7899 CrossRef CAS.
- S. Mintova, N. H. Olson, J. Senker and T. Bein, Angew. Chem., Int. Ed., 2002, 41, 2558 CrossRef CAS.
- Z. Lin, D. S. Wragg and R. E. Morris, Chem. Commun., 2006, 2021 RSC.
- N. Stock and T. Bein, Solid State Sci., 2003, 5, 1207–1210 CrossRef CAS.
- N. Stock and T. Bein, Angew. Chem., Int. Ed., 2004, 43, 749 CrossRef CAS.
- S. Bauer, T. Bein and N. Stock, Inorg. Chem., 2005, 44, 5882–5889 CrossRef CAS.
- C. Serre, J. A. Groves, P. Lightfoot, A. M. Z. Slawin, P. A. Wright, N. Stock, T. Bein, M. Haouas, F. Taulelle and G. Férey, Chem. Mater., 2006, 18, 1451–1457 CrossRef CAS.
- P. M. Forster, A. R. Burbank, M. C. O'Sullivan, N. Guillou, C. Livage, G. Férey, N. Stock and A. K. Cheetham, Solid State Sci., 2005, 7, 1549–1555 CrossRef CAS.
- D. Akporyaye, I. M. Dahl, A. Karlsson, M. Plassen, R. Wendelbo, D. S. Bem, R. W. Broach, G. J. Lewis, M. Miller and J. Moscoso, Microporous Mesoporous Mater., 2001, 48, 367 CrossRef CAS.
- B. Jandeleit, D. J. Schaefer, T. S. Powers, H. W. Turner and W. H. Weinberg, Angew. Chem., Int. Ed., 1999, 38, 2494–2532 CrossRef CAS.
- B. Archibald, O. Brümmer, M. Devenney, D. M. Giaquinta, B. Jandeleit, W. H. Weinberg and T. Weskamp, in Handbook of Combinatorial Chemistry, ed. K. C. Nicolaou, R. Hanko and W. Hartwig, Wiley-VCH, Weinheim, 2002, pp. 1017–1057 Search PubMed.
- A. Corma, M. Moliner, J. M. Serra, P. Serna, M. J. Diaz-Cabanas and L. A. Baumes, Chem. Mater., 2006, 18, 3287 CrossRef CAS.
- N. Stock, M. Rauscher and T. Bein, J. Solid State Chem., 2004, 177, 642 CrossRef CAS.
- N. Stock, N. Hilbrandt, K. Choi and T. Bein, Stud. Surf. Sci. Catal., 2001, 135, 550 CAS.
- F. Serpaggi and G. Férey, J. Mater. Chem., 1998, 8, 2749 RSC.
- F. Serpaggi and G. Férey, Inorg. Chem., 1999, 38, 4741 CrossRef CAS.
- F. Serpaggi and G. Férey, J. Mater. Chem., 1998, 8, 2737 RSC.
- F. Serpaggi and G. Férey, Microporous Mesoporous Mater., 1999, 32, 311 CrossRef CAS.
- C. Serre, F. Millange, J. Marrot and G. Férey, Chem. Mater., 2002, 14, 2409 CrossRef CAS.
- C. Serre and G. Férey, J. Mater. Chem., 2002, 12, 3053 RSC.
- F. Millange, C. Serre, J. Marrot, N. Gardant, F. Pelé and G. Férey, J. Mater. Chem., 2004, 14, 642 RSC.
- F. Millange, C. Serre, J. Marrot, N. Gardant, F. Pelé and G. Férey, J. Am. Chem. Soc., 2005, 127, 12788 CrossRef CAS.
- F. Millange, C. Serre, J. Marrot, N. Gardant, F. Pelé and G. Férey, Chem. Mater., 2004, 16, 1177 CrossRef CAS.
- C. Serre, J. Marrot and G. Férey, Inorg. Chem., 2005, 44, 654 CrossRef CAS.
- T. Devic, C. Serre, N. Audebrand, J. Marrot and G. Férey, J. Am. Chem. Soc., 2005, 127, 12788 CrossRef CAS.
- T. M. Reineke, M. Eddaoudi, M. Fehr, D. Kelley and O. M. Yaghi, J. Am. Chem. Soc., 1999, 121, 1651 CrossRef CAS.
- T. M. Reineke, M. Eddaoudi, M. O'Keeffe, D. Kelley and O. M. Yaghi, Angew. Chem., Int. Ed., 1999, 38, 2590 CrossRef CAS.
- N. L. Rosi, J. KIm, M. Eddaoudi, B. L. Chen, M. O'Keeffe and O. M. Yaghi, J. Am. Chem. Soc., 2005, 127, 1504 CrossRef CAS.
- B. D. Alleyne, A. R. Wiliams and L. A. Hall, Inorg. Chem., 2001, 40, 1045 CrossRef CAS.
- S. S.-Y. Chui, A. Siu, X. Feng, Z. Y. Zhang, T. C.-W. Mak and I. D. Williams, Inorg. Chem. Commun., 2001, 4, 467 CrossRef.
- L. P. Wu, J. Coord. Chem., 1996, 37, 361 CAS.
- L. P. Wu, M. Munataka, T. Kuroda-Sowa, M. Maekawa and Y. Suenaga, Inorg. Chim. Acta, 1996, 249, 183 CrossRef CAS.
- L. Pan, X. Huang, J. Li, Y. Wu and N. Zheng, Angew. Chem., Int. Ed., 2000, 39, 527 CrossRef CAS.
- Y. Wu, N. Zheng, R. Yang, H. Xu and E. Ye, J. Mol. Struct., 2002, 610, 181 CrossRef CAS.
- R. Cao, D. Sun, Y. Liang, M. Hong, K. Tatsumi and Q. Shi, Inorg. Chem., 2002, 41, 2087 CrossRef CAS.
- L. Pan, E. B. Woodlock and X. Wang, Inorg. Chem., 2000, 39, 4174 CrossRef.
- D.-L. Long, A. J. Blake, N. R. Champness, C. Wilson and M. Schröder, J. Am. Chem. Soc., 2001, 123, 3401 CrossRef CAS.
- B.-Q. Ma, D.-S. Zhang, S. Gao, T.-Z. Jin, C.-H. Yang and G.-X. Xu, Angew. Chem., Int. Ed., 2000, 39, 3644 CrossRef CAS.
- Y.-Q. Sun, J. Zhang, Y.-M. Chen and G.-Y. Yang, Angew. Chem., Int. Ed., 2005, 44, 5814 CrossRef CAS.
- D. T. de Lill, N. S. Gunning and C. L. Cahill, Inorg. Chem., 2005, 44, 258 CrossRef CAS.
- N. M. Maier, P. Franco and W. J. Lindner, J. Chromatogr., A, 2001, 906, 3 CrossRef CAS.
- A. Baiker, Curr. Opin. Solid State Mater. Sci., 1998, 3, 86 CrossRef CAS.
- C. E. Song and S. G. Lee, Chem. Rev., 2000, 102, 3495.
- Chirality in Natural and Applied Science, ed. W. J. Lough and I. W. Wainer, CRC Press, Boca Raton, 2002 Search PubMed.
- B. Kesanli and W. Lin, Coord. Chem. Rev., 2003, 246, 305 CrossRef CAS.
- Y. Cui, H. L. Ngo and W. Lin, Chem. Commun., 2003, 1338 RSC.
- N. G. Pschirer, D. Ciurtin, M. D. Smith, U. H. F. Bunz and H. C. zur Loye, Angew. Chem., Int. Ed., 2002, 41, 583 CrossRef CAS.
- J. S. Seo, D. Whang, H. Lee, S. I. Jun, Y. J. Jeon and K. Kim, Nature (London), 2000, 404, 982 CrossRef CAS.
- R.-G. Xiong, X.-Z. You, B. F. Abrahams, Z. Xue and M. C. Che, Angew. Chem., Int. Ed., 2001, 42, 4422 CrossRef.
- B. F. Abrahams, M. Moylan, S. D. Orchard and R. Robson, Angew. Chem., Int. Ed., 2003, 42, 1848 CrossRef CAS.
- E. V. Anokhina and A. J. Jacobson, J. Am. Chem. Soc., 2004, 126, 3044 CrossRef CAS.
- E. V. Anokhina, Y. B. Go, Y. Lee, T. Vogt and A. J. Jacobson, J. Am. Chem. Soc., 2006, 128, 9957 CrossRef CAS.
- X. Q. Wang, M.-L. Liu and A. J. Jacobson, Angew. Chem., Int. Ed., 2006, 45, 6499 CrossRef CAS.
- R. Vaidhyanathan, D. Bradshaw, J. N. Rebilly, J. P. Barrio, J. A. Gould, N. G. Berry and M. J. Rosseinsky, Angew. Chem., Int. Ed., 2006, 45, 6495 CrossRef CAS.
- L. Chen and X. Bu, Chem. Mater., 2006, 18, 1857 CrossRef CAS.
- K. Nomiya, S. Takahashi, R. Nogughi, S. Nemoto, T. Takayama and M. Oda, Inorg. Chem., 2000, 39, 3301 CrossRef CAS.
- M. E. Davis, Top. Catal., 2003, 25, 3 CrossRef CAS.
- H. Ratajczak, J. Barycki, A. Pietraszko, J. Baran, S. Debrus, M. May and J. Venturini, J. Mol. Struct., 2000, 526, 269 CrossRef CAS.
- L. E. Gordon and W. T. A. Harrison, Inorg. Chem., 2004, 43, 1808 CrossRef CAS.
- J. Fan, C. Slebodnick, R. Angel and B. E. Hanson, Inorg. Chem., 2005, 44, 552 CrossRef CAS.
- Y. Zhang, M. K. Saha and I. Bernal, CrystEngComm, 2003, 5, 34 RSC.
- M. Mizutani, N. Maejima, H. Masuda and H. Einaga, Inorg. Chim. Acta, 1998, 283, 105 CrossRef CAS.
- M. Haouas, C. Gérardin, F. Taulelle, C. Estournes, T. Loiseau and G. Férey, J. Chim. Phys. Phys.–Chim. Biol., 1998, 95, 302 CrossRef CAS.
- F. Taulelle, M. Haouas, C. Gérardin, C. Estournes, T. Loiseau and G. Férey, Colloids Surf., A, 1999, 158, 299 CrossRef CAS.
- F. Taulelle, M. Pruski, J. P. Amoureux, D. Lang, A. Bailly, C. Huguenard, M. Haouas, T. Loiseau and G. Férey, J. Am. Chem. Soc., 1999, 121, 12148 CrossRef CAS.
- R. J. Francis, S. O'Brien, A. M. Fogg, P. S. Halasyamani, D. O'Hare, T. Loiseau and G. Férey, J. Am. Chem. Soc., 1999, 121, 1002 CrossRef CAS.
- R. Walton, T. Loiseau, D. O'Hare and G. Férey, Chem. Mater., 1999, 11, 3201 CrossRef CAS.
- T. Loiseau, L. Beitone, F. Millange, F. Taulelle, R. D. O'Hare and G. Férey, J. Phys. Chem. B, 2004, 108, 20020 CrossRef CAS.
- B. G. Hyde and S. Anderson, Inorganic Crystal Structures, Wiley-Interscience, New York, 1988 Search PubMed.
- M. O'Keeffe and B. G. Hyde, Crystal Structures I: Patterns and Symmetry, Am. Mineral. Assoc., Washington, DC, 1996 Search PubMed.
- A. F. Wells, Further Studies of Three-Dimensional Nets, Am. Crystallogr. Assoc., Monograph 8, Mineralogical Society of America, Washington, 1979 Search PubMed.
- S. Han and J. V. Smith, Acta Crystallogr., Sect. A: Found. Crystallogr., 1999, A55, 332 CrossRef CAS and references therein.
- M. O'Keeffe, M. Eddaoudi, H. Li, T. Reineke and O. M. Yaghi, J. Solid State Chem., 2000, 152, 3–20 CrossRef CAS.
- G. Férey, J. Solid State Chem., 2000, 152, 37–53 CrossRef CAS.
- G. Férey and A. K. Cheetham, Science, 1999, 283, 1125 CrossRef CAS.
- S. Hansen, Nature (London), 1990, 346, 799 CrossRef.
- M. O'Keeffe, Z. Kristallogr., 1991, 196, 21–37.
- G. Férey, Science, 2001, 291, 994–995 CrossRef CAS.
- B. Chen, M. Eddaoudi, S. T. Hyde, M. O'Keeffe and O. M. Yaghi, Science, 2001, 291, 1021 CrossRef CAS.
- O. M. Yaghi, M. O'Keeffe, N. W. Ockwig, H. K. Chae, M. Eddaoudi and J. Kim, Nature (London), 2003, 423, 705–714 CrossRef CAS.
- N. W. Ockwig, O. Delgado-Friedrichs, M. O'Keeffe and O. M. Yaghi, Acc. Chem. Res., 2005, 38, 176–182 CrossRef CAS.
- L. Beitone, C. Huguenard, A. Gansmuller, M. Henry, F. Taulelle, T. Loiseau and G. Férey, J. Am. Chem. Soc., 2003, 125, 9102 CrossRef CAS.
- N. Guillou, C. Livage and G. Férey, Angew. Chem., Int. Ed., 2005, 44, 6488 CrossRef CAS.
- X.-C. Huang, Y.-Y. Lin, J.-P. Zhang and X.-M. Chen, Angew. Chem., Int. Ed., 2006, 45, 1557 CrossRef CAS.
- J.-P. Zhang and X.-M. Chen, Chem. Commun., 2006, 1689 RSC.
- Y. Liu, V. C. Kravtsov, R. Laren and M. Eddaoudi, Chem. Commun., 2006, 1488 RSC.
- J. C. Schoen and M. Jansen, Angew. Chem., Int. Ed., 2006, 45, 3406 CrossRef CAS.
- K. Barthelet, J. Marrot, D. Riou and G. Férey, Angew. Chem., Int. Ed., 2002, 41, 281 CrossRef CAS.
- K. Barthelet, J. Marrot, D. Riou and G. Férey, Chem. Commun., 2004, 520 RSC.
- H. Li, M. Eddaoudi, M. O'Keeffe and O. M. Yaghi, Nature (London), 1999, 402, 276 CrossRef CAS.
- C. Serre, F. Millange, C. Thouvenot, M. Nogues, G. Marsolier, D. Louer and G. Férey, J. Am. Chem. Soc., 2002, 124, 13519 CrossRef CAS.
- K. Barthelet, K. Adil, F. Millange, C. Serre, D. Riou and G. Férey, J. Mater. Chem., 2003, 13, 2208 RSC.
- N. Guillou, C. Livage, M. Drillon and G. Férey, Angew. Chem., Int. Ed., 2003, 42, 5314 CrossRef.
- S. Xiang, X. Wu, J. Zhang, R. Fu, S. Hu and X. Zhang, J. Am. Chem. Soc., 2005, 127, 16352 CrossRef CAS.
- A. Dolbecq, C. Mellot-Draznieks, P. Mialane, J. Marrot, G. Férey and F. Sécheresse, Eur. J. Inorg. Chem., 2005, 3009 CrossRef.
- C. Livage, N. Guillou, J. Chaigneau, P. Rabu, M. Drillon and G. Férey, Angew. Chem., Int. Ed., 2005, 44, 6488 CrossRef CAS.
- Z. Ni, A. Yassar, T. Antoun and O. M. Yaghi, J. Am. Chem. Soc., 2005, 127, 12752 CrossRef CAS.
- S. Kitagawa and M. Kondo, Angew. Chem., Int. Ed. Engl., 1997, 36, 1725 CrossRef CAS.
- S. Kitagawa and K. Uemura, Chem. Soc. Rev., 2006, 34, 109 Search PubMed and refs. therein.
- K. Uemura, S. Kitagawa, M. Kondo, K. Fukui, R. Kitaura, H.-C. Chang and T. Mizutani, Chem.–Eur. J., 2002, 8, 3586 CrossRef CAS.
- K. Uemura, S. Kitagawa, K. Fukui and K. Saito, J. Am. Chem. Soc., 2004, 126, 3817 CrossRef CAS.
- K. Birhada, Y. Hongo and M. Fujita, Angew. Chem., Int. Ed., 2002, 41, 3395 CrossRef CAS.
- R. Kitaura, K. Seki, G. Akiyama and S. Kitagawa, Angew. Chem., Int. Ed., 2003, 42, 428 CrossRef CAS.
- R. Kitaura, K. Fujimoto, S. I. Noro, M. Kondo and S. Kitagawa, Angew. Chem., Int. Ed., 2002, 41, 133 CrossRef CAS.
- G. Alberti, S. Murcia-Mascaros and R. Vivani, J. Am. Chem. Soc., 1998, 120, 9291 CrossRef.
- L. C. Tabares, J. A. R. Navarro and J. M. Salas, J. Am. Chem. Soc., 2001, 123, 383 CrossRef CAS.
- S. K. Makinen, J. Melcer, M. Parvez and G. K. H. Shimizu, Chem.–Eur. J., 2001, 7, 5176 CrossRef CAS.
- J.-Y. Lu and A. M. Babb, Chem. Commun., 2002, 1340 RSC.
- L. Carlucci, G. Ciani, M. Moret, D. M. Proserpio and S. Rizzato, Angew. Chem., Int. Ed., 2000, 39, 1506 CrossRef CAS.
- C. J. Kepert, T. J. Prior and M. J. Rosseinsky, J. Am. Chem. Soc., 2000, 122, 5158 CrossRef CAS.
- C. Mellot-Draznieks, C. Serre, S. Surblé and G. Férey, J. Am. Chem. Soc., 2005, 127, 16273 CrossRef CAS.
- S. Surblé, C. Serre, C. Mellot-Draznieks, F. Millange and G. Férey, Chem. Commun., 2006, 284 RSC.
- C. Serre, C. Mellot-Draznieks, S. Surblé, N. Audebrand, Y. Filinchuk and G. Férey, Science, 2007, 315, 1828 CrossRef CAS.
- M. Eddaoudi, J. Kim, N. Rosi, D. Vodak, J. Wachter, M. O'Keeffe and O. M. Yaghi, Science, 2002, 295, 469 CrossRef.
- H. K. Chae, D. Y. Siberio-Pérez, J. Kim, Y. Go, M. Eddaoudi, A. J. Matzger, M. O'Keeffe and O. M. Yaghi, Nature (London), 2004, 427, 523–527 CrossRef CAS.
- C. Mellot-Draznieks, J. M. Newsam, A. M. Gorman, C. M. Freeman and G. Férey, Angew. Chem., Int. Ed., 2000, 39, 2270–2275 CrossRef CAS.
- C. Mellot-Draznieks, S. Girard and G. Férey, J. Am. Chem. Soc., 2002, 124, 15326–15335 CrossRef CAS.
- S. Girard, P. Pullumbi, C. Mellot-Draznieks and G. Férey, Stud. Surf. Sci. Catal., 2001, 135, 254 Search PubMed.
- C. Mellot-Draznieks, G. Férey, C. Schön, Z. Cancarevic and M. Jansen, Chem.–Eur. J., 2002, 8, 4102 CrossRef CAS.
- C. Mellot-Draznieks and G. Férey, Curr. Opin. Solid State Mater. Sci., 2003, 7, 13 CrossRef CAS.
- C. M. Freeman, A. M. Gorman and J. M. Newsam, in Computer Modelling in inorganic crystallography, ed. C. R. A. Catlow, Academic Press, London, 1997, pp. 177–150 Search PubMed.
- J. M. Newsam, C. M. Freeman and F. J. J. Leusen, Curr. Opin. Solid State Mater. Sci., 1999, 4, 515 CrossRef CAS and references therein.
- J. C. Schön and M. Jansen, Acta Crystallogr., Sect. A: Found. Crystallogr., 1999, 55 Search PubMed , Supplement.
- J. C. Schön and M. Jansen, in Inorganic Chemistry Highlights, ed. G. Meyer, D. Naumann and L. Wesemann, Wiley-VCH, Weinheim, 2002 Search PubMed.
- M. D. Foster, O. Delgado Friedrichs, R. G. Bell, F. A. Alameida Paz and J. Klinowski, Angew. Chem., Int. Ed., 2003, 42, 3896–3999 CrossRef CAS.
- Y. Li, J. Yu, J. Jiang, Z. Wang, J. Zhang and R. Xu, Chem. Mater., 2005, 17, 6086 CrossRef CAS.
- C. Mellot-Draznieks, J. Dutour and G. Férey, Angew. Chem., Int. Ed., 2004, 43, 6291.
- M. O'Keeffe, Mater. Res. Bull., 2006, 41, 911 CrossRef CAS.
- A. Le Bail, Standard and special strategies in structure from powder data, presented at ACA'97, Saint-Louis (Missouri), 1997 Search PubMed.
- M. E. Davis and I. E. Maxwell, Curr. Opin. Solid State Mater. Sci., 1996, 1, p.35.
- W. Mori, S. Takamizawa, C. N. Kato, T. Ohmura and T. Sato, Microporous Mesoporous Mater., 2004, 73, 31–46 CrossRef CAS.
- P. M. Forster and A. K. Cheetham, Top. Catal., 2003, 24, 79 CrossRef CAS.
- W. Lin, J. Solid State Chem., 2005, 178, 2486 CrossRef CAS.
- M. Fujita, Y. J. Kwon, S. Washizu and K. Ogura, J. Am. Chem. Soc., 1994, 116, 1151–1152 CrossRef CAS.
- J. S. Seo, D. Whang, H. Lee, S. I. Jun, J. Oh, Y. J. Jeon and K. Kim, Nature (London), 2000, 404, 982–986 CrossRef CAS.
- B. Kesanli and W. Lin, Coord. Chem. Rev., 2003, 246, 305–326 CrossRef CAS.
- D.-D. Wu, A. Hu, L. Zhang and W. Lin, J. Am. Chem. Soc., 2005, 127, 8940–8941 CrossRef CAS.
- D. N. Dybtsev, A. L. Nuzhdin, H. Chun, K. P. Bryliakov, E. P. Talsi, V. P. Fedin and K. Kim, Angew. Chem., Int. Ed., 2006, 45, 916–920 CrossRef CAS.
- C. Y. Yang and A. Clearfield, React. Polym., 1987, 5, 13 CAS.
- A. Clearfield and Z. K. Wang, J. Chem. Soc., Dalton Trans., 2002, 2937 RSC.
- A. Clearfield, Z. K. Wang and P. Bellinghausen, J. Solid State Chem., 2002, 167, 376 CAS.
- O. R. Evans, H. L. Ngo and W. Lin, J. Am. Chem. Soc., 2001, 123, 10395–10396 CrossRef CAS.
- T. Sawaki and Y. Aoyama, J. Am. Chem. Soc., 1999, 121, 4793–4798 CrossRef CAS.
- B. Gomez-Lor, E. Gutierrez-Puebla, M. Iglesias, M. A. Monge, C. Ruiz-Valero and N. Snejko, Inorg. Chem., 2002, 41, 2429–2432 CrossRef CAS.
- Q. Zou, H. Sakurai and Q. Xu, Angew. Chem., Int. Ed., 2006, 45, 2542–2546 CrossRef CAS.
- S. Kitagawa, S.-I. Noro and T. Nakamura, Chem. Commun., 2006, 701–707 RSC.
- R. Kitaura, G. Onoyama, H. Sakamoto, R. Matsuda, S.-I. Noro and S. Kitagawa, Angew. Chem., Int. Ed., 2004, 43, 2684 CrossRef CAS.
- L.-G. Qiu, A.-J. Xie and L.-D. Zhang, Angew. Chem., Int. Ed., 2004, 43, 2684–2687 CrossRef CAS.
- S. Hermes, M.-K. Schröter, R. Schmid, L. Jhodeir, M. Muhler, A. Tissler, R. W. Fischer and R. A. Fischer, Angew. Chem., Int. Ed., 2005, 44, 6237 CrossRef CAS.
- H. Byrd, A. Clearfield, D. Poojary, K. P. Reis and M. E. Thompson, Chem. Mater., 1996, 8, 2239 CrossRef CAS.
- A. Dokoutchaev, V. V. Krishnan and M. E. Thompson, J. Mol. Struct., 1998, 470, 191 CrossRef CAS.
- K. P. Reis, V. K. Joshi and M. E. Thompson, J. Catal., 1996, 161, 62 CrossRef CAS.
- Hydrogen, Fuel Cells & Infrastructure Technologies Program: Multi-yrear research, Development and Demonstration Plan, U.S. Department of Energy, February 2005, Chapter 3, http://www.eere.energy.gov/hydrogenandfuelcell/mypp/; Basic Reearch Needs for the Hydrogen Economy, report of the Basic Energy Sciences workshop on Hydrogen, Production, Storage and Use, U. S. Department of Energy, May 13–15, 2005, http://www.sc.doe.gov/bes/. Search PubMed.
- U. Eberle, personal communication.
- B. Panella, M. Hirscher, H. Pütter and U. Müller, Adv. Funct. Mater., 2006, 16, 520 CrossRef CAS.
- N. L. Rosi, J. Eckert, M. Eddaoudi, D. T. Vodak, J. Kim and O. M. Yaghi, Science, 2003, 300, 1127 CrossRef CAS.
- B. Panella and M. Hirscher, Adv. Mater., 2005, 17, 538 CrossRef.
- J. L. C. Rowsell, A. R. Milward, K. S. Park and O. M. Yaghi, J. Am. Chem. Soc., 2004, 125, 5666 CrossRef.
- G. Férey, M. Latroche, C. Serre, F. Millange, T. Loiseau and A. Percheron-Guégan, Chem. Commun., 2003, 2976 RSC.
- P. Krawiec, M. Kramer, M. Sabo, R. Lunschke, H. Fröde and S. Kaskel, Adv. Eng. Mater., 2006, 8, 293 CrossRef.
- S. Bourrelly, P. L. Lewellyn, C. Serre, T. Loiseau, F. Millange and G. Férey, J. Am. Chem. Soc., 2005, 126, 13519 CrossRef.
- S. Takamizawa and E.-I. Nakata, CrystEngComm, 2005, 7, 476 RSC.
- T. Yildirim and M. R. Hartman, Phys. Rev. Lett., 2005, 95, 215504 CrossRef CAS.
- E. C. Spencer, J. A. K. Howard, G. J. McIntyre, J. L. C. Rowsell and O. M. Yaghi, Chem. Commun., 2006, 278 RSC.
- Q. Yang and C. Zhong, J. Phys. Chem. B, 2006, 110, 655 CrossRef CAS.
- Q. Yang and C. Zhong, J. Phys. Chem. B, 2005, 109, 11862 CrossRef CAS.
- T. Düren, L. Sarkizov, O. M. Yaghi and R. Q. Snurr, Langmuir, 2004, 20, 2683 CrossRef CAS.
- T. Mueller and G. Ceder, J. Phys. Chem. B, 2005, 109, 17974 CrossRef CAS.
- T. Sagara, J. Klassen, J. Ortony and E. Ganz, J. Chem. Phys., 2005, 123, 014701(1–4).
- T. Sagara, J. Klassen and E. Ganz, J. Chem. Phys., 2004, 121, 12543 CrossRef CAS.
- G. Garberoglio, A. I. Skoulidas and J. K. Johnson, J. Phys. Chem. B, 2005, 109, 13094 CrossRef CAS.
- A. I. Skoulidas and D. S. Sholl, J. Phys. Chem. B, 2005, 109, 15760 CrossRef CAS.
- S. S. Kaye and J. R. Long, J. Am. Chem. Soc., 2005, 127, 6506 CrossRef.
- M. Fichtner, Adv. Eng. Mater., 2005, 7, 443 CrossRef CAS.
- J. L. C. Rowsell and O. M. Yaghi, Microporous Mesoporous Mater., 2004, 73, 3 CrossRef CAS.
- J. L. C. Rowsell and O. M. Yaghi, Angew. Chem., Int. Ed., 2005, 44, 4670 CrossRef CAS.
- L. Zhou, Renewable Sustainable Energy Rev., 2005, 9, 395 Search PubMed.
- S. Barrett, Fuel Cells Bull., 2005, 12.
- B. Bogdanovitch, M. Felderhoff, S. Kaskel, A. Pommerin, K. Schlichte and F. Schüth, Adv. Mater., 2003, 15, 1012 CrossRef CAS.
- F. Schüth, B. Bogdanovitch and M. Felderhoff, Chem. Commun., 2004, 2249 RSC.
- A. G. Wong-Foy, A. J. Matzger and O. M. Yaghi, J. Am. Chem. Soc., 2006, 128, 3494 CrossRef CAS.
- H. W. Langmi, A. Walton, M. M. Al-Mamouri, S. R. Johnson, D. Book, J. D. Speight, P. P. Edwards, I. Gameson, P. A. Anderson and I. R. Harris, J. Alloys Compd., 2003, 356–357, 710 CrossRef CAS.
- B. Panella, M. Hirscher and S. Roth, Carbon, 2005, 109, 13094.
- P. Benard and R. Chahine, Langmuir, 2001, 17, 1950 CrossRef CAS.
- R. Chaine and T. K. Bose, Int. J. Hydrogen Energy, 1994, 19, 161 CrossRef CAS.
- Ch. Miller, P. Rudolf and H. J. Teles, WO 2004/009523, 2004, BASF Aktiengesellschaft.
- J. Johnson, Chem. Eng. News, 2004, 82, 36.
- Z. Yong, V. Mata and A. E. Rodrigues, Sep. Purif. Technol., 2002, 26, 195 CrossRef CAS.
- W. Mori, F. Yoshida, H. Nakayama, S. Takamizaka and M. Kishita, Chem. Lett., 1997, 1219 CAS.
- S. Takamizaka, E. Nakata, H. Yokoyama, K. Mochizuki and W. Mori, Angew. Chem., Int. Ed., 2003, 42, 4331 CrossRef CAS.
- S. Takamizaka, T. Saito, T. Akatsuba and E. Nakata, Inorg. Chem., 2005, 44, 1421 CrossRef CAS.
- S. Takamizawa, E.-I. Nakata and T. Akatsuka, Angew. Chem., Int. Ed., 2006, 45, 2216 CrossRef CAS.
- H. Li, M. Eddaoudi, T. L. Groy and O. M. Yaghi, J. Am. Chem. Soc., 1998, 120, 8571 CrossRef CAS.
- A. R. Millward and O. M. Yaghi, J. Am. Chem. Soc., 2005, 127, 17998 CrossRef CAS.
- P. L. Llewellyn, S. Bourelly, C. Serre, Y. Filinchuk and G. Férey, Angew. Chem., Int. Ed., 2006, 45, 7751 CrossRef CAS.
- S. Bourrelly, P. L. Llewellyn, C. Serre, S. Surblé, A. Vimont, M. Daturi, G. de Weireld, L. Hamon, J.-H. Lee, J.-S. Chang, S.-H. Jhung and G. Férey, J. Am. Chem. Soc. Search PubMed , submitted.
- C. G. V. Burgess, D. H. Everett and S. Nuttall, Pure Appl. Chem., 1989, 61, 1845 CAS.
- C. Serre, P. L. Llewellyn, S. Bourrelly, G. Maurin, Y. Filinchuk, A. Vimont, M. Daturi, O. Leynaud, P. Barnes and G. Férey, Adv. Mater., 2007, 19, 2246 CrossRef.
- A. Vimont, H. Leclerc, F. Maugé, M. Daturi, J. C. Lavalley, S. Surblé, C. Serre and G. Férey, J. Phys. Chem. C, 2007, 111, 383 Search PubMed.
- S. Noro, S. Kitagawa, M. Kondo and T. Seki, Angew. Chem., Int. Ed., 2000, 39, 2082 CrossRef CAS.
- R. Kitaura, K. Fujimoto, S. Noro, M. Kondo and S. Kitagawa, Angew. Chem., Int. Ed., 2002, 41, 133 CrossRef CAS.
- R. Kitaura, S. Kitagawa, Y. Kubota, T. C. Kobayachi, K. Kindo, Y. Mita, A. Matsuo, A. Kobayachi, M. C. Chang, T. C. Ozawa, M. Suzuki, M. Sakata and M. Takata, Science, 2002, 298, 2358 CrossRef CAS.
- R. Kitaura, K. Seki, G. Akiyama and S. Kitagawa, Angew. Chem., Int. Ed., 2003, 42, 428 CrossRef CAS.
- T. Uemura and S. Kitagawa, J. Am. Chem. Soc., 2003, 125, 7814 CrossRef CAS.
- S. Horike, R. Matsuda, R. Kitaura, S. Kitagawa, T. Ijima, K. Endo, Y. Kubota and M. Takata, Chem. Commun., 2004, 2152 RSC.
- Y. Kubota, M. Takata, R. Matsuda and S. Kitagawa, Angew. Chem., Int. Ed., 2005, 44, 920 CrossRef CAS.
- R. Matsuda, R. Kitaura, S. Kitagawa, Y. Kubota, R. V. Belovsludov, T. C. Kobayachi, H. Sakamoto, T. Chiba, M. Takata, Y. Kawazoe and Y. Mita, Nature (London), 2005, 436, 238 CrossRef CAS.
- R. Kitaura, R. Matsuda, Y. Kubota, S. Kitagawa, M. Takata, T. C. Kobayachi and M. Suzuki, J. Phys. Chem. B, 2005, 109, 23378 CrossRef CAS.
- R. Matsuda, R. Kitaura, S. Kitagawa, Y. Kubota, R. V. Belosludov, T. C. KObayashi, H. Sakamoto, T. Chiba, M. Takata, Y. Kawazoe and Y. Mita, Nature (London), 2006, 441, 584 CrossRef CAS.
- J. L. C. Rowsell, E. C. Spencer, J. Eckert, J. A. K. Howard and X. O. M. Yaghi, Science, 2005, 309, 1350 CrossRef CAS.
- A. J. Fletcher, K. M. Thomas and M. J. Rosseinsky, J. Solid State Chem., 2005, 178, 2491 CrossRef CAS and refs. therein.
- D. Bradshaw, J. B. Claridge, E. J. Cussen, T. J. Prior and M. J. Rosseinsky, Acc. Chem. Res., 2005, 38, 273 CrossRef CAS.
- X. Zhao, B. Xiao, A. J. Fletcher, K. M. Thomas and M. J. Rosseinsky, Science, 2004, 306, 1012 CrossRef CAS.
- L. Pan, D. H. Olson, L. R. Ciemnolonski, R. Heddy and J. Li, Angew. Chem., Int. Ed., 2006, 45, 616 CrossRef CAS.
- H. Chun, D. N. Dybstev, H. Kim and K. Kim, Chem.–Eur. J., 2005, 11, 3521 CrossRef CAS.
- B. Ma, K. L. Mulfort and J. T. Hupp, Inorg. Chem., 2005, 44, 4912 CrossRef CAS.
- B. Chen, C. Liang, J. Yang, D. S. Contreras, Y. L. Clancy, E. B. Lobkovsky, O. M. Yaghi and S. Dai, Angew. Chem., Int. Ed., 2006, 45, 1390 CrossRef CAS.
- X. Q. Wang, L. M. Liu and A. J. Jacobson, Angew. Chem., Int. Ed., 2006, 45, 6499 CrossRef CAS.
- L. Alaerts, C. E. A. Kirschhock, M. Maes, M. A. van der Veen, V. Finsy, A. Depla, J. A. Martens, G. V. Baron, P. A. Jacobs, J. E. M. Denayer and D. E. De Vos, Angew. Chem., Int. Ed., 2007, 46, 4293 CrossRef CAS.
- P. Horcajada, C. Serre, M. Vallet-Regi, M. Sebban, F. Taulelle and G. Férey, Angew. Chem., Int. Ed., 2006, 45, 5974 CrossRef CAS.
- P. Horcajada, A. Ramila, G. Férey and M. Vallet-Regi, Solid State Sci., 2006, 8, 1243 CAS.
- S. Freiberg and X. X. Zhu, Int. J. Pharm., 2004, 282, 1 CrossRef CAS.
- K. S. Soppimath, T. M. Aminabhavi, A. R. Kulkarni and W. E. Rudzinski, J. Controlled Release, 2001, 70, 1 CrossRef CAS.
- P. Dhivanand and O. L. Sprockel, Int. J. Pharm., 1998, 167, 83 CrossRef.
- A. Rivera and T. Farias, Microporous Mesoporous Mater., 2005, 80, 337 CrossRef CAS.
- M. Vallet-Regi, A. Ramila, R. P. del Real and J. Perez-Pariente, Chem. Mater., 2001, 13, 308 CrossRef CAS.
- B. Munoz, A. Ramila, J. Perez-Pariente, I. Diaz and M. Vallet-Regi, Chem. Mater., 2003, 15, 500 CrossRef CAS.
- S. Hermes, M.-K. Schröter, R. Schmid, L. Khodeir, M. Muhler, A. Tissler, R. W. Fischer and R. A. Fischer, Angew. Chem., Int. Ed., 2005, 44, 2.
- R. Becker, H. Parala, F. Hipler, A. Birkner, C. Wöll, O. Hinrichsen, O. P. Tkachenko, K. V. Klementiev, W. Grünert, S. Schäfer, H. Wilmer, M. Muhler and R. A. Fischer, Angew. Chem., Int. Ed., 2004, 43, 2839 CrossRef CAS.
- M. P. Suh, H. R. Moon, E. Y. Lee and S. Y. Jang, J. Am. Chem. Soc., 2006, 128, 4710 CrossRef CAS.
- E. Spano, S. Hamad and C. R. A. Catlow, Chem. Commun., 2004, 864 RSC.
- A. Aumüller, P. Erk, G. Klebe, S. Hünig, J. U. von Schütz and H. P. Werner, Angew. Chem., Int. Ed. Engl., 1986, 25, 740 CrossRef.
- H. Kitagawa, Y. Nagao, M. Fujishima, R. Ikeda and S. Kanda, Inorg. Chem. Commun., 2003, 6, 346 CrossRef CAS.
- G. Férey, F. Millange, O. Morcrette, J. M. Grenèche, M. L. Doublet and J. M. Tarascon, Angew. Chem., Int. Ed., 2007, 46, 3259 CrossRef CAS.
- J. B. Goodenough, Magnetism and the chemical bond, Wiley-Interscience, New-York, 1963 Search PubMed.
- O. Kahn, Molecular Magnetism, VCH, New-York, 1993 Search PubMed.
- M. Drillon and J. S. Miller, Magnetism: Molecules to Materials, Wiley-VCH, Weinheim, 2001–2005, vol. I–V Search PubMed.
- H. Shrikanth, R. Hajndl, B. Moulton and M. Zaworotko, J. Appl. Phys., 2003, 93, 7089 CrossRef CAS.
- K. Barthelet, D. Riou and G. Férey, Chem. Commun., 2002, 1492 RSC.
- D. Maspoch, D. Ruiz-Molina and J. Veciana, J. Mater. Chem., 2004, 14, 2713 RSC.
- D. Maspoch, D. Ruiz-Molina, K. Wurtz, N. Domingo, M. Cavallini, F. Biscarini, J. Tejada, C. Rovira and J. Veciana, Nat. Mater., 2003, 2, 190 CrossRef CAS.
- D. Maspoch, N. Domingo, D. Ruiz-Molina, K. Wurtz, G. Vaughan, J. Tejada, C. Rovira and J. Veciana, Angew. Chem., Int. Ed., 2004, 43, 1828 CrossRef CAS.
- D. Maspoch, D. Ruiz-Molina, K. Wurtz, C. Rovira and J. Veciana, Dalton Trans., 2004, 43, 1073 RSC.
- D. Maspoch, N. Domingo, D. Ruiz-Molina, K. Wurtz, J.-M. Hernandez, G. Vaughan, C. Rovira, F. Lloret, J. Tejada and J. Veciana, Chem. Commun., 2005, 5035 RSC.
- G. J. Halder, C. J. Kepert, B. Moubarki, K. S. Murria and J. D. Cashion, Science, 2002, 298, 1762 CrossRef CAS.
- V. Niel, A. L. Thomson, M. C. Munoz, A. Galet, A. S. E. Goeta and J. A. Real, Angew. Chem., Int. Ed., 2003, 42, 3760 CrossRef CAS.
- S. Bonhommeau, G. Molnar, A. Galet, A. Zwick, J. A. Real, J. J. McGarvey and A. Bousseksou, Angew. Chem., Int. Ed., 2005, 44, 4069 CrossRef CAS.
- Z. Wang, B. Zhang, H. Fujiwara, H. Kobayashi and M. Kurmoo, Chem. Commun., 2004, 416 RSC.
- H. Z. Kou, B. C. Zhou and R. J. Wang, Inorg. Chem., 2003, 42, 7658 CrossRef CAS.
- S. A. Bourne, J. Lu, A. Mondal, B. Moulton and M. J. Zaworotko, Angew. Chem., Int. Ed., 2001, 40, 2111 CrossRef CAS.
- N. Guillou, S. Pastre, C. Livage and G. Férey, Chem. Commun., 2002, 2358 RSC.
- N. Guillou, C. Livage, M. Van Beek, M. Nogues and G. Férey, Angew. Chem., Int. Ed., 2003, 42, 644 CAS.
- N. Guillou, C. Livage, P. Rabu, M. Drillon and G. Férey, Angew. Chem., Int. Ed., 2003, 42, 5314 CrossRef.
- F. Serpaggi, T. Luxbacher, G. Férey and A. K. Cheetham, J. Solid State Chem., 1999, 145, 580 CrossRef CAS.
- S. I. Weissman, J. Chem. Phys., 1942, 10, 214 CrossRef CAS.
- J.-C. G. Bunzli and C. Piguet, Chem. Rev., 2002, 102, 1897 CrossRef.
- G. F. de Sá, O. L. Malta, C. de Mello Donegá, A. M. Simas, R. L. Longo, P. A. Santa-Cruz and E. F. da Silva, Jr., Coord. Chem. Rev., 2000, 196, 165 CrossRef CAS and references therein.
- D. Imbert, S. Comby, A. Chauvin and J. C. Bünzli, Chem. Commun., 2005, 1432 RSC and references therein.
- D. T. de Lill, N. S. Gunning and C. L. Cahill, Inorg. Chem., 2005, 44, 258 CrossRef CAS.
- F. Pellé, S. Surblé, C. Serre, F. Millange and G. Férey, J. Lumin., 2007, 122–123, 492 CrossRef CAS.
- T. Uemura, S. Horike and S. Kitagawa, Chem.–Asian J., 2006, 1, 36 CrossRef CAS.
- B. H. Hong, S. C. Bae, C.-W. Lee, S. Jeong and K. S. Kim, Science, 2001, 294, 348 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2008 |