Porphycenes: synthesis and derivatives†
David
Sánchez-García
ab and
Jonathan L.
Sessler
*a
aDept. of Chem. & Biochem., 1 University Station-A5300, The University of Texas, Austin, TX 78712-0165, USA. E-mail: Sessler@mail.utexas.edu; Fax: 1 512 471 7550; Tel: 1 512 471 5009
bInstitut Européen de Chimie et Biologie (IECB), UMR 5248 CBMN, CNRS-Université Bordeaux 1-ENITAB, IECB, 2 rue Robert Escarpit, 33607 Pessac, France. E-mail: d.sanchez@iecb.u-bordeaux.fr; Fax: 33(0)540002215; Tel: 33 5 40 00 22 19
First published on 9th July 2007
Abstract
Porphycene is an aromatic macrocycle and a constitutional isomer of porphyrin. It and its derivatives display unique physical and optical properties, including strong absorptions in the red region of the UV-vis spectrum. These features have made porphycene and porphycene analogues appealing molecules for use in biomedical applications and in the design of new materials. This critical review provides a concise overview of the most important syntheses of porphycenes, as well as those of various functionalized derivatives and heteroatom containing analogues. (95 references.)
 David Sánchez-García | David Sánchez García was born in Barcelona, Catalonia, Spain. He studied chemistry at the Institut Químic de Sarrià (Universitat Ramon Llull) in Barcelona and received his PhD in 2003 for research on the chemistry of 2,2′-biimidazole and the synthesis of tetraazaporphycenes. He carried out his postdoctoral research (2004–2006) with Professor Jonathan L. Sessler (University of Texas at Austin) developing new strategies for the synthesis of pyrrolic macrocycles. He is currently working with Dr Ivan Huc (Institut Européen de Chimie et Biologie, France) on foldamer chemistry. In addition to chemistry, he enjoys playing the violin. |
 Jonathan L. Sessler | Professor Jonathan L. Sessler was born in Urbana, Illinois, USA on May 20, 1956. He received a BS degree (with highest honors) in chemistry in 1977 from the University of California, Berkeley. He obtained a PhD in organic chemistry from Stanford University in 1982 (supervisor: Professor James P. Collman). He was an NSF-CNRS and NSF-NATO Postdoctoral Fellow with Professor Jean-Marie Lehn at L'Université Louis Pasteur de Strasbourg, France. He was then a JSPS Visiting Scientist in Professor Iwao Tabushi’s group in Kyoto, Japan. In September 1984 he accepted a position as an Assistant Professor of Chemistry at the University of Texas at Austin, where he is currently the Roland K. Pettit Professor. Dr Sessler has authored or co-authored over 400 research publications, written two books (with Dr Steven J. Weghorn, and Drs Won-Seob Cho and Philip A. Gale, respectively ) and edited a third (with Drs Susan Doctrow, Tom McMurry, and Stephen J. Lippard). He is the inventor of record on more than 70 issued US Patents. Dr Sessler is also a co-founder (with Dr Richard A. Miller, MD) of Pharmacyclics, Inc., a publicly traded company (pcyc; NASDQ) dedicated to develping biomedical applications of expanded porphyrins. In conjunction with Dr Martin R. Johnson, Dr Sessler has recently co-founded a second company, Anionics, Inc., that is targeting various commercial opportunities associated with anion recognition chemistry. Dr Sessler has served as the co-organizer of several international conferences and numerous symposia at ACS meetings, including, recently, the 3rd International Conference on Porphyrins and Phthalocyanines that was held in July of 2004. In addition to English, he speaks French, German, Spanish, and Hebrew and can get by in Japanese. He serves as Editor of Supramolecular Chemistry and is an Associate Editor for ChemComm. |
Introduction
Over the years, porphyrins , the so-called pigments of life, have attracted the interest of organic chemists because of their critical role in biological processes and their structural and aesthetic appeal. Impelled, in part, by this fascination, the synthetic chemistry of porphyrins has developed almost exponentially in recent decades. This has helped provide explanations for a number of biological phenomena while, at the same time, giving access to a plethora of new compounds with applications in fields that range from material chemistry to biomedicine. Three main features characterize porphyrins : They are macrocyclic , highly colored, and aromatic . A desire to understand and extend these characteristics has inspired considerable interest in the preparation of new porphyrin analogues. One of the most important of these is the so-called porphyrin isomers.The first constitutional isomer of porphyrin and the focus of this review is porphycene (Fig. 1). It was prepared in 1986 by Vogel and coworkers.1 Structurally, this new compound consists of two 2,2′-bipyrrole subunits linked by two double bonds. The result is a planar, aromatic macrocycle formally known as [18]porphyrin-(2.0.2.0).2 In fact, the name porphycene was suggested because this compound possesses structural features of both porphyrins and acenes . Porphycene is not only the first porphyrin isomer to be reported, it is also the most stable,3,4 at least among those systems with an N-four central core (as opposed, e.g., to containing N-confused pyrrolic subunits). Hence, its chemistry has been closely studied, especially as a ligand to form metal complexes.5 These complexes have been investigated in the context of many applications, including catalysis,6protein mimicry,7 and material chemistry.8 They have also attracted interest because of their unique optical properties. As a consequence of their lower symmetry relative to porphyrins , porphycenes generally display strong absorbance bands in the far-red portion of the visible spectrum (depending on the porphycene in question, the lowest energy λmax is found anywhere from 620–760 nm). These spectral characteristics have made functionalized porphycenes attractive for use in photodynamic therapy (PDT).9,10 Another biomedical application that has emerged recently involves the use of porphycenes in the photoinactivation of viruses and bacteria.11 However, in addition to their potential practical utility, porphycenes remain objects of fascination because of their relationship to porphyrins and because they provide experimental test beds for theoretical studies focused on electronic structure, aromaticity ,12 and proton exchange,13 to name a few.
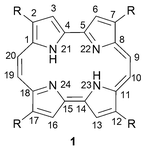 | ||
| Fig. 1 Core structure of porphycene. | ||
Preparation of porphycenes
In Vogel's seminal paper,1 porphycene 1 was obtained by using McMurry-type coupling conditions to effect the reductive dimerization14,15 of diformyl-2,2′-bipyrrole 2a (Scheme 1; R = H), followed by spontaneous oxidation with atmospheric O2. This coupling, which is thought to proceed through the 20 π-electron intermediate 3, can be regarded as the key step that enabled the synthesis of porphycenes. Although effective, the requisite McMurry reaction demands some precautions from an experimental point of view, especially pure reagents and anhydrous conditions. On the other hand, the starting material for the coupling reaction, the dialdehyde 2a can be obtained by subjecting 2,2′-bipyrrole16 to formylation under Vilsmeier–Haack17 conditions (Scheme 1).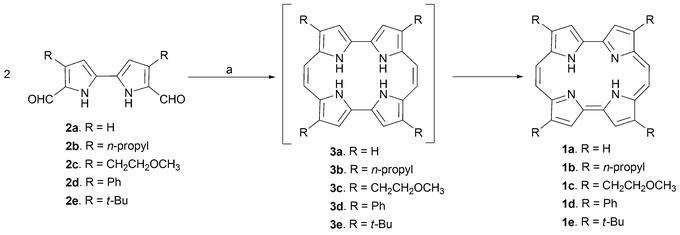 | ||
| Scheme 1 Vogel's synthesis of porphycene (1a, R = H; 1b, R = n-propyl; 1c, R = CH2CH2OCH3; 1d, R = Ph). Reaction conditions (yields in parentheses): a) TiCl4/Zn/pyridine, THF, reflux (R = H, (2%); R = n-propyl, (10%); R = CH2CH2OCH3, (25%); R = Ph (30%)). | ||
In order to enhance the solubility and crystallinity of porphycene, Vogel and coworkers also prepared derivatives bearing substituents in the 2,7,12 and 17 positions. In particular, tetrasubstituted porphycenes of general structure 1 bearing methyl, ethyl, tert-butyl, n-propyl (1b),18 and β-methoxyethyl groups (1c)19 were synthesized and characterized early on. Subsequently, derivatives bearing a greater number of substituents on the β-pyrrolic positions, the bridging meso-like alkylidene bridges, or both were prepared and studied. These latter systems are discussed later on in this review.
The general strategy introduced by Vogel remains the method of choice for preparing porphycenes. It takes advantage of the symmetry of the macrocycle and involves two coupling reactions, namely an Ullmann coupling and a subsequent McMurry coupling (Scheme 2). The first coupling provides the requisite substituted 2,2′-bipyrrole moiety,20 whereas the second serves to close the ring.
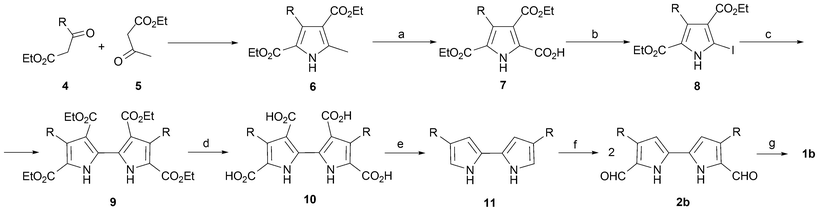 | ||
| Scheme 2 Synthesis of β-alkyl substituted porphycenes. The specific conditions and yields (in parentheses) are for R = n-propyl, yielding 1b. a) Br2, SO2Cl2, AcOH/HCOOH, 0 °C, 4 h (50–58%). b) KI/I2, EtOH/H2O, 75 °C, (75–77%). c) Cu, dimethylformamide (DMF), 20 °C, 17 h (67–71%). d) NaOH, EtOH/H2O, 15 h, reflux (96%). e) Sublimation at 230 °C, 0.2 torr (89%). f) POCl3/DMF then NaOAc/H2O (86%). g) TiCl4/Zn/pyridine, THF, reflux, 0.5 h (10%). | ||
Although, many methods have been described for the preparation of 2,2′-bipyrroles,21 the Ullmann coupling22 of iodopyrroles was, and remains, a particularly attractive choice. This is because, in contradistinction with some other methods, it provides the synthetic flexibility needed to incorporate a large range of substituents on the β-pyrrolic positions. In general, the starting iodopyrrole required for the bipyrrole-forming Ullmann coupling (8) is prepared via the decarboxylative iodination23 of a pyrrole monacid, such as 7. Such acids can, in turn, be prepared by oxidation of the corresponding α-methylpyrrole 6. Typically, these latter species are obtained by means of a classical Knorr24 reaction involving the appropriate β-ketoesters (4, 5). Once the coupling is complete, to give a tetraester, such as 9, further transformations, involving intermediates 10 and 11, provide the requisite diformyl species 2.
As noted above, the second dimerization serves to close the ring. It is based on a McMurry-type reductive coupling that is followed presumably by a spontaneous, in situ oxidation process. This gives the porphycene in variable yields. While the unsubstituted parent compound 1a is obtained in only ca. 2% yield, the more substituted porphycenes are routinely obtained in yields on the order of 10–25% (1; R = alkyl). This is ascribed to the greater solubility of the latter species and the corresponding intermediates during synthesis.
Porphycenes bearing aromatic groups have been investigated, with the 2,7,12,17-phenyl derivative (1d) being prepared by the groups of Vogel25 and Nonell.26 The main difference in the synthetic approaches used by these two researchers lies in the conversion of the tetraester 9 (R = Ph) into the 2,2′-bipyrrole 11 (R = Ph). Specifically, Nonell avoids the sublimation step present in Vogel's preparation; this is done by converting the tetraester into the corresponding 2,2′-bipyrrole in a single step by treatment with NaOH/ethylene glycol at reflux.27 In principle, the suppression of the sublimation step should facilitate the scale up of the synthesis, thereby making this series of compounds more accessible to those interested in studying their chemical and spectroscopic properties. With this same goal in mind, Nonell's group reported28 a non-decarboxylative procedure based on 2,2′-bipyrroles bearing two sets of orthogonal esters (e.g., 13; Scheme 3). Thus, the use of this kind of tetraester, prepared from iodopyrrole 12, allows the diester 14 to be prepared under readily accessible conditions (i.e., via intermediate 13). The result is a species that, it was envisioned, could be subsequently reduced selectively to afford the corresponding dialdehyde 2d. Unfortunately, this planned reduction proved difficult to achieve using conventional reductants. Nonetheless, the desired dialdehyde (2d) could be obtained in good yield by use of a MacFayden–Stevens reaction sequence (Scheme 3).29
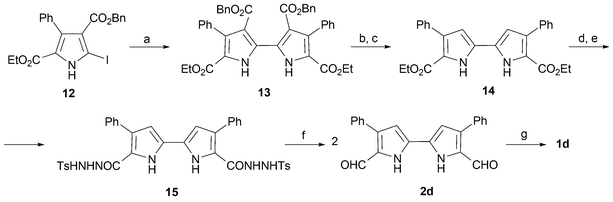 | ||
| Scheme 3 Non-decarboxylative synthesis of tetraphenylporphycene, 1d. Reaction conditions (yields in parentheses): a) Cu, DMF, 23 °C, 20 h (89%). b) Pd/C, HCOOH/HCOONH4, reflux, 4 h (100%). c) ethylene glycol, 170 °C, 5 h (93%). d) NH2NH2, EtOH, reflux, 48 h, (93%). e) TsCl, pyridine, 23 °C, 1 h (96%). f) diethylene glycol, Na2CO3, 170 °C (92%). g) TiCl4/Zn/CuCl, THF, reflux, 3 h (30%). | ||
The introduction of phenyl groups in positions 2,7,12 and 17 leads to a dramatic bathochromic shift (30 nm) compared with the unsubstituted porphycene. It has been suggested that the presence of aromatic rings could modify the position of the Q bands, thereby allowing an optimization of those optical properties considered desirable for applications such as PDT.9,30 With these objectives in mind, and cognizant of the emerging concepts of combinatorial chemistry, Borrell and Nonell31 reported a new strategy for preparing substituted bipyrroles. It is based on the use of palladium chemistry and relies on the thienobipyrrole 16.32 This precursor acts as a masked bipyrrole with positions 3 and 3′ blocked. It thus allows the remaining positions to be halogenated readily. After SEM protection (SEM = [β-(trimethylsilyl)ethoxy]methyl) the resulting dibrominated compound 17 can participate in Suzuki couplings. This permits a higher level of functionalization if so desired. Once such further chemistry is effected, the final substituted 2,2′-bipyrrole 18 is obtained following first removal of the sulfur atom and then the SEM protecting group. This is done by treating with Raney Ni followed by tetra-n-butylammonium fluoride (TBAF) (Scheme 4).
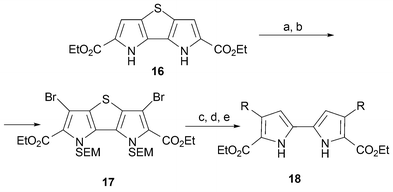 | ||
| Scheme 4 Reaction conditions (yields in parentheses): a) Br2, AcOH/AcOEt (89%). b) SEMCl, NaH, THF (94%). c) RB(OH)2, Pd(PPh3)4Na2CO3, 1,4-dioxane; R = Ph (100%), R = C5H4N (95%), R = p-MeOC6H4 (100%). d) Ni Raney, EtOH; R = Ph (100%), R = C5H4N (100%), R = p-MeOC6H4 (94%). e) TBAF (1 M in THF), 1,4-dioxane; R = Ph (93%), R = C5H4N (89%), R = p-MeOC6H4 (91%). | ||
Although the McMurry coupling is a well-established efficient reaction, and is one that has been used to effect other kinds of macrocyclizations, in the context of porphycene syntheses yields are only modest. On the basis of the experimental observations noted above, it was concluded that a lack of solubility is largely responsible for the low yields. Predicated on this assumption, an improved method for preparing unsubstituted porphycene1a was developed by Waluk and coworkers; this procedure, reported at the 4th International Conference on Porphyrins and Phthalocyanines (ICPP-4),33 involves using tert-butyl substituents as temporary solubilizing groups.
Thus, starting with pyrrole19, tosylation and functionalization gives 21, which is coupled to provide bipyrrole 22 that, after deprotection (23) and formylation, yields the key intermediate 2e (R = tert-butyl) required for reductive cyclization. After cyclization, the tert-butyl substituents may be removed in 65% yield by heating in H2SO4 (Scheme 5). Another novel aspect of this synthesis is that as actually implemented it involved a replacement of the Ullmann reaction used to prepare bipyrrole 22 by an oxidative coupling process.34,35
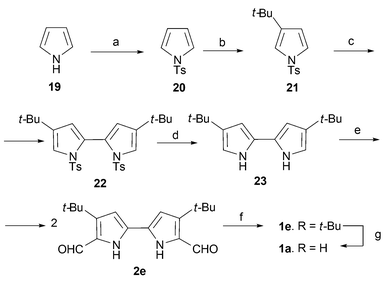 | ||
| Scheme 5 Improved synthesis of porphycene1a of Waluk and coworkers. Reaction conditions (yields in parentheses): a) TsCl, NaOH. b) t-BuCl, AlCl3, CH2Cl2. c) n-BuLi, TMP, CuCl2, THF. d) KOH, CH3OH. e) POCl3/DMF, CHCl3. f) TiCl4/Zn/CuCl, THF. g) H2SO4(aq), 15 min (65%). | ||
In 2006, Sessler and Sánchez-García36 reported at the ICPP-4 a simplification of the classic porphycene synthesis that is based on the use of a new protocol for preparing the requisite iodopyrroles (26) from simple aldehydes , such as 24. It takes advantage of the recognition by Suzuki et al.37 that α-free pyrrole diesters, such as 25, can be prepared via the reaction of an aldehyde with an alkyl isocyanoacetate in the presence of DBU. This preparation furnishes both alkylic and aromatic pyrroles of general structure 25 in good yields (50–71%). Treatment with iodine in a biphasic DCM–water mixture then furnishes the iodopyrroles 26 (Scheme 6). Ullmann coupling (to give 27) and removal of the ester functionality then provides a disubstituted bipyrrole (11) suitable for formylation and cyclization.
![Simplified synthesis of porphycenes developed by Sánchez-García and Sessler. Reaction conditions (R = C7H15 and p-MePh-) (yields in parentheses): a) ethyl isocyanoacetate (2 equiv.), 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) (2 equiv.), 50 °C (75%, 51%). b) I2/NaI, dichloroethane, 100 °C, (71%, 63%). c) (BOC)2O, DCM, 23 °C, filtration through a short pad of Celite™ and then Cu, DMF, 110 °C (43%, 35%). d) NaOH, deoxygenated ethylene glycol, 180 °C (60%, 70%). e) POCl3, DMF, 60 °C, then NaOAc, 100 °C (86%, 79%). f) TiCl4/Zn/CuCl, THF, reflux (24, 16%).](/image/article/2008/CS/b704945e/b704945e-s6.gif) | ||
| Scheme 6 Simplified synthesis of porphycenes developed by Sánchez-García and Sessler. Reaction conditions (R = C7H15 and p-MePh-) (yields in parentheses): a) ethyl isocyanoacetate (2 equiv.), 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) (2 equiv.), 50 °C (75%, 51%). b) I2/NaI, dichloroethane, 100 °C, (71%, 63%). c) (BOC)2O, DCM, 23 °C, filtration through a short pad of Celite™ and then Cu, DMF, 110 °C (43%, 35%). d) NaOH, deoxygenated ethylene glycol, 180 °C (60%, 70%). e) POCl3, DMF, 60 °C, then NaOAc, 100 °C (86%, 79%). f) TiCl4/Zn/CuCl, THF, reflux (24, 16%). | ||
The main features of this new protocol are 1) a reduction in the number of steps (from 8 to 6) compared with the traditional synthesis and 2) the considerable flexibility permitted in terms of the substituents, alkyl or aryl, that can be introduced in positions 2, 7, 12 and 17. In support of this latter assertion, two new porphycenes of general structure 1 were prepared, namely those with R = C7H15 and R = p-MePh.
Reactivity and functionalization of porphycenes
As illustrated in Scheme 7, the double bonds in porphycene undergo selective reduction when treated with H2 or Na/ROH; this yields the reduced analogues 28 and 29.38 Based on this reactivity, porphycenes can be regarded as aza polyenes,39 nitrogenated counterparts of the aromatic [18]annulene3012 (Fig. 2).![Structures of Sondheimer's [18]annulene and Vogel's 1,6-methano[10]annulene.](/image/article/2008/CS/b704945e/b704945e-f2.gif) | ||
| Fig. 2 Structures of Sondheimer's [18]annulene and Vogel's 1,6-methano[10]annulene. | ||
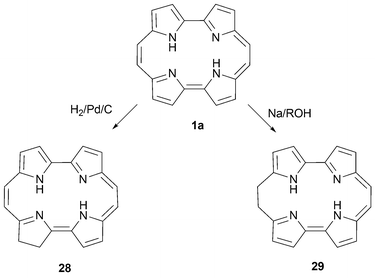 | ||
| Scheme 7 Hydrogenation and metal-based reduction of porphycene1a. | ||
One of the characteristics of [18]annulene30 is that it is more prone to polymerize than to undergo electrophilic substitution, a finding that is ascribed to its conformational freedom. In porphycenes, the rigidity imposed by the pyrrolic nitrogens, gives rise to benzenoid reactivity similar to that observed in the case of 1,6-methano[10]annulene31.40
Thus, halogenation of porphycene can be achieved by direct reaction with Br2 or I2.41–43 When the porphycene substrate is unsubstituted, attack of the halogen takes place exclusively at position 2 to give 33, whereas for 2,7,12,17 substituted porphycenes, halogenation takes place at the remaining β-pyrrolic positions. In general, two compounds can be isolated readily, namely the tri- and tetrabrominated porphycene species (e.g., 32). The use of bromine on a polymeric support has, however, been used to effect mono substitution in position 3 (34) (Scheme 8).41
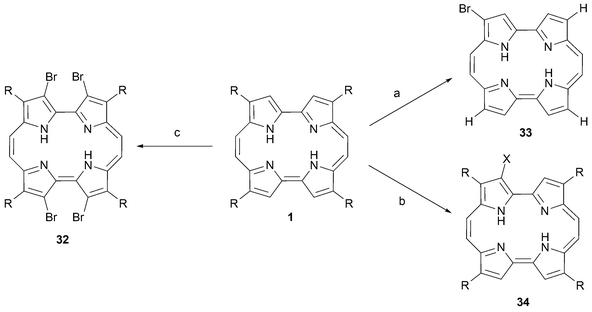 | ||
| Scheme 8 Halogenation of porphycenes. Reaction conditions (yields in parentheses): a) (R = H) N-bromosuccinimide (NBS), CHCl3, 10 °C, 10 min (23%). b) (R = n-propyl, X = Br) Amberlyst A-26, Br3-form, CH2Cl2, AcOH, 0 °C, 2 h (80%). (R = n-propyl, X = I) I2, 0 °C, 2 min (80%). c) (R = n-propyl) Br2,CCl4, NaAcO/H2O, 23 °C, 1 h (40%). | ||
Brominated pyrroles are not prone to react with nucleophiles. However, an unexpected rearrangement, resulting in the formation of the isocorrole 35 (Scheme 9), takes place when tetrabrominated porphycene 32b is heated under basic conditions.41,44
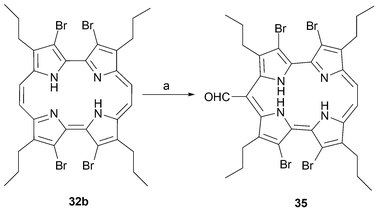 | ||
| Scheme 9 Contraction of porphycene 32b observed in basic media. a) K2CO3, DMF/H2O, 120 °C, 4 h, (34%). | ||
Treatment of 1b with fuming sulfuric acid affords a mixture of mono, di, and tri-β-sulfonated porphycenes (shown in Scheme 10 in the form of their sodium salts, 36–39).45 The mono and tri- substituted products can be isolated and purified from the crude reaction mixture using simple extraction methods. In contrast, the difunctionalized compounds are obtained as an inseparable mixture. The sodium salts of most of these sulfonated porphycenes are water-soluble, something that has made them attractive for use in biological applications.
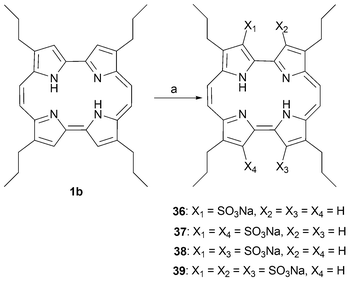 | ||
| Scheme 10 Sulfonated porphycenes. Reaction conditions (yields in parentheses): a) H2SO4 (SO3) 25%, CH2Cl2, 25 °C, 25 min, 36 (3%), 37 and 38 (29%), 39 (40%). | ||
Similarly, the Mak group have synthesized the 3-sulfonyl chloride derivative in 95% by reacting 1b with chlorosulfonic acid at room temperature.46 The resulting acid chloride reacts with amines to give sulfonamides 40 and 41 (Fig. 3). In the same report the authors showed that water-soluble cationic sulfonamides are effective mitochondrial localizers. An alternative means of placing a cationic amine (–CH2N(CH3)2·HCl) onto the periphery of the porphycene core (at position 3) has been disclosed; however, details of the methodology employed are lacking.47
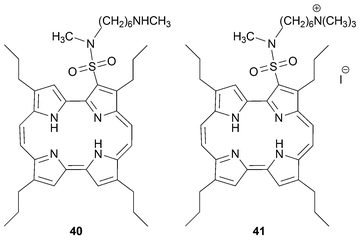 | ||
| Fig. 3 Porphycene sulfamides. | ||
Whereas halogens and SO3 react at the pyrrolic positions, the treatment of porphycenes with fuming HNO3 yields the 9-nitro substituted macrocycle 42 (Scheme 11).48 An alternative means of effecting this nitration involves reacting porphycene 1b with AgNO3/acetic acid in dichloromethane. The resulting 9-nitro group can then be reduced selectively to the corresponding amine (43) via the Zinin reduction,49 or by treatment with Raney Ni and hydrazine.
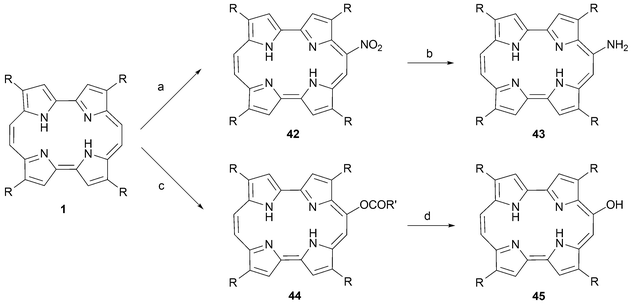 | ||
| Scheme 11 Nitration and dehydroacyloxylation of porphycenes: Reaction conditions (yields in parentheses): (R = n-propyl) a) AgNO3, HOAc, CH2Cl2, 60 °C, 15 min. (85%). b) Na2S2O4, NaOH, H2O, reflux, 1 h (50%). c) (R = n-propyl, R′ = Me) Pb(OAc)4, THF, reflux, 10 min (30%). (R′ = (CH2)3CO2t-Bu) PbO2, HO2C(CH2)3CO2t-Bu, CH2Cl2, 23 °C, 24 h (23%). d) (R = n-propyl, R′ = Me) NaOMe, MeOH, 23 °C, 5 min (89%). | ||
A different approach to functionalizing the 9 position involves forming the acyloxy derivatives (e.g., 44) by treatment with a Pb(IV) salt (PbO2 or Pb(OAc)4) in the presence of a carboxylic acid (e.g., acetic acid). The free 9-hydroxyporphycene 45 can then be easily prepared by subjecting the resulting acyloxy derivative to hydrolysis in basic media (Scheme 11).48
The 9-substituted porphycenes described above provide an important starting point for the preparation of a range of derivatives and conjugates.50 For instance, porphycenes bearing 9-hydroxyl groups can be used to prepare ethers , esters , and sulfonic acid esters, whereas 9-amino derivatives can be transformed into amido conjugates when treated with acid chlorides (cf. generic structures 46 and 47; Scheme 12). Many of the resulting systems have proved water soluble, and have attracted attention as possible photodynamic therapy photosensitizers. Not surprisingly, therefore, the basic physical parameters relevant to PDT have been published for many of these elaborated 9-substituted porphycene derivatives.47
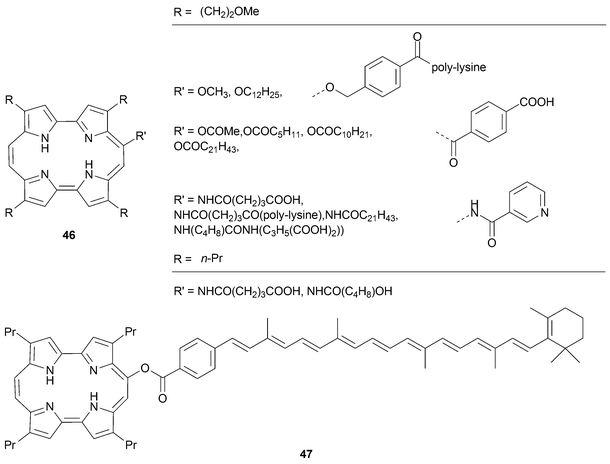 | ||
| Scheme 12 Selected derivatives generated from 9-amino and 9-hydroxy substituted porphycenes.47 | ||
Functionalization of positions 2, 7, 12 and 17
The β-methoxyethyl substituted porphycene 1c may be regarded as a pivotal compound in the preparation of a large number of peripherally functionalized derivatives. Removal of the methyl ether “protecting group” with a Lewis acid, such as BBr3, provides the corresponding alcohols . The availability of these latter species, in turn, allows several functional group interconversions, including, for instance, the preparation of other ether derivatives. It is noteworthy that demethylation of 1c can be carried out stepwise, allowing the preparation of mono, di and tri hydroxy derivatives.19Mixed McMurry couplings involving dialdehydes 2 bearing different substitutents (e.g., R = CH2CH2OMe and R = n-propyl) provide access to amphiphilic porphycenes after deprotection. Interestingly, exposure of the Ni complex of 1c to BBr3 in the presence of boric acid yields the mono-brominated derivative 49 instead of the corresponding alcohol , 48 (Scheme 13). This latter species can be obtained by treating 1c with 1 equiv. of BBr3. The utility of this monohydroxy product as an intermediate in its own right has been demonstrated via, for instance, its conversion to porphycene sugar derivatives, such as 50 (Scheme 14).51
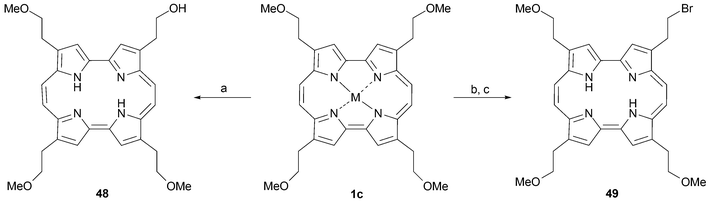 | ||
| Scheme 13 Transformations of porphycene (1c) effected by BBr3. Reaction conditions (yields in parentheses): a) (M = H2) BBr3, CH2Cl2, –30 °C, 16 h (18%). b) (M = Ni) BBr3, B(OH)3, CH2Cl2, –78 °C, 8 h (35%). c) (M = Ni) H2SO4, 10 min (90%). | ||
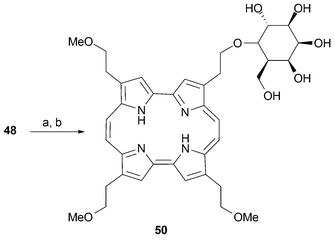 | ||
| Scheme 14 Synthesis of β-D-galactopyranoside of 2-hydroxyethyl-7,12,17-tris(methoxyethyl)-porphycene. Reaction conditions: a) AgCO3, Na2SO4, 2,3,4,6-tetra-O-acetyl-α-D-galactopyranosyl bromide, CH2Cl2, 23 °C, 3 d. b) NaOMe, THF, 23 °C, 2 h. | ||
β-Hydroxyethyl chains are versatile substituents that can be transformed into the corresponding chloride, acetate, or mesylate derivatives, as well as the bromides as described above. Both the bromide 49, as well as the mesylate 51 produced from 48 (Scheme 13), undergo substitution with NaCN to afford the corresponding cyano derivative 52. Treatment with HCl in methanol then yields the carboxylic ester , which after saponification gives the acid derivative 53 in good yield (Scheme 15).51
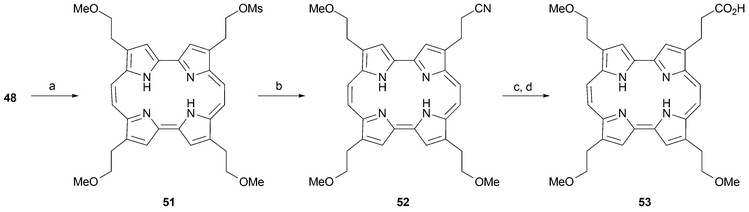 | ||
| Scheme 15 Functional groups interconversions. Reaction conditions (yields in parentheses): a) MsCl, pyridine, CH2Cl2, 2 h (91%). b) NaCN, DMSO, 3 h, (75%). c) MeOH/HCl(sat), 18 h, (81%). d) KOH, THF/H2O, 40 h then HCl (72%). | ||
On the other hand, full deprotection of 1c with BBr3 in the presence of boric acid affords the tetrabrominated derivative 54, which can be converted into 2,7,12,17-tetravinylporphycene55 by treatment with DBU in 20% yield (Scheme 16).51
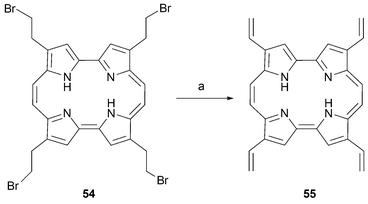 | ||
| Scheme 16 Preparation of 2,7,12,17-tetra-vinylporphycene55. Reaction conditions: a) DBU, THF, 23 °C, 2 d followed by HCl (20%). | ||
Meso and etio substituted porphycenes
The geometry of the porphycene core is dependent on the peripheral substituents. As a consequence, the metal cation coordination properties of this tetrapyrrolic ligand can be modified by changing the substituents on the periphery of the macrocycle .12 With a view to mapping out the effect of such modifications on the complexation properties of porphycene, derivatives such as 56 and 57 were prepared that bear different substituents in positions 9, 10, 19 and 20 (meso positions),52–54 as well as positions 3, 6, 13 and 16 (β-pyrrolic positions); the latter species bear a structural resemblance to the etioporphyrins and are referred to generically as etioporphycenes (Fig. 4).55,56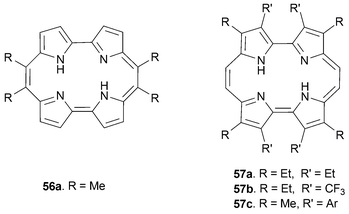 | ||
| Fig. 4 Meso and etio substituted porphycenes. | ||
Meso substituted porphycenes have generally been prepared from 5,5′-acyl substituted bipyrroles using the basic reductive coupling strategy introduced early on by Vogel. In this case, the requisite bipyrroles are usually obtained by treating an α,α′-unsubstituted bipyrrole with a Vilsmeier-type electrophile obtained by reacting the proper alkyl N,N′-amide with POCl3.57 Interestingly, oxidation of the intermediate annulene 58 (to produce the porphycene product 56) is relatively difficult to effect as compared to the analogous non-meso substituted porphycene precursor 3 (cf. Scheme 1). As a result, it can be isolated in the form of colorless crystals. It has been suggested that the alkyl chains on the alkene bridges slow down the rate of conformational motion and that this retardation, in turn, makes it difficult to access the near-planar geometry required for oxidation and aromatization (Scheme 17).
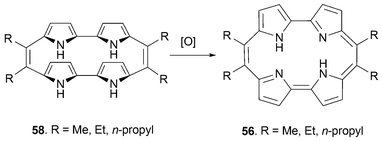 | ||
| Scheme 17 Reaction sequence meant to underscore the relative stability of meso substituted annulenes 58. | ||
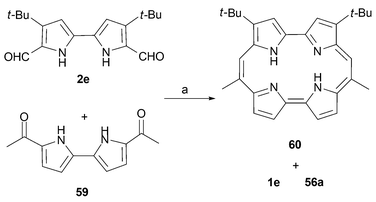
An example of an asymmetric porphycene 60 bearing methyl groups in two of the four meso positions has been reported recently.33 It was synthesized via the cross coupling of the diformyl and diacetyl bipyrroles 2e and 59, respectively. This preparative chemistry, for which a yield was not reported, is summarized in Scheme 18.
In the solid state, the cavity of 2,7,12,17-tetrasubstituted porphycenes are seen to be slightly rectangular. This geometry favors the formation of metal complexes with small cations. However, etioporphycenes (Fig. 5) have a larger cavity and are thus able to accommodate bigger cations. The more open cavity of etioporphycenes is rationalized in terms of the steric interactions of the alkyls chains in the β-positions that force the molecule to adopt a more square-like geometry.
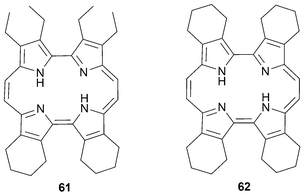 | ||
| Fig. 5 Etioporphycenes. | ||
To prepare etioporphycenes a route to β,β′-substituted pyrroles is required. The most versatile and efficient approach to these latter intermediates is via the Zard–Barton reaction.58–60 In this procedure, the pyrrole is prepared from the condensation of ethyl isocyanate with a nitro compound (64, prepared from 63, or 67, prepared from 66 and 63) in the presence of DBU; this provides an ethyl β,β′-disubstituted pyrrole 2-carboxylate (i.e., a pyrrole such as 65 or 68) bearing an ethyl ester group in position 2) (Scheme 19).61,62
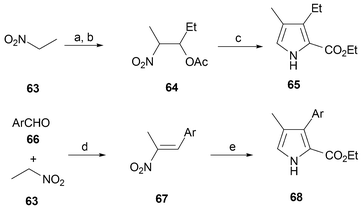 | ||
| Scheme 19 Zard–Barton synthesis of 3,4-substituted pyrroles. Reaction conditions (yields in parentheses): a) CH3CH2CHO, KF, i-PrOH, 24 h (80%). b) Ac2O, H2SO4, 1 h, (89%). c) CNCH2CO2Et, THF, i-PrOH, DBU, 4 h, 23 °C, (65%). d) (Ar = (p-H21C10OPh)) AcONH4 (70%). e) CNCH2COOEt, THF/i-PrOH, DBU (46%). | ||
Subsequent iodination affords the starting material required for the Ullmann coupling used to produce a tetra-β-substituted bipyrrole. N-protection of the iodopyrroles as the BOC (tert-butyloxycarbonyl) derivatives allows (Scheme 6) the yield of the dimerization to be increased up to 20% compared with the coupling of unprotected iodopyrroles.63
Although the pyrrolic positions in etioporphycenes are substituted by alkyl chains, some interesting chemistry involving these positions has been reported. In a few cases the result is the production of a new aromatic system. For instance, the two isomeric oxo porphycene derivatives 70 and 71 were prepared by treating octaethylporphycene57a with H2O2 in acidic media.64 The final oxygenated products are believed to arise via rearrangement of a dihydroxylated intermediate such as 69 (Scheme 20).
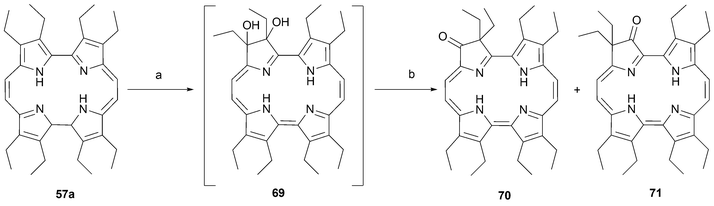 | ||
| Scheme 20 Preparation of octaethyl-2-oxoporphycene and octaethyl-3-oxoporphycene. Reaction conditions: a) H2O2. b) H2SO4. | ||
The nickel complex of octaethylporphycene57a undergoes mono-formylation under Vilsmeier–Haack conditions.65 The aldehyde produced in this way was used by Kister and Vogel to prepare the porphycene analogue of benzochlorin 74via intermediates 72 and 73 (Scheme 21).64 This macrocycle , which was subsequently prepared by Robinson,66 was termed octaethylbenzochloracene by these latter researchers.
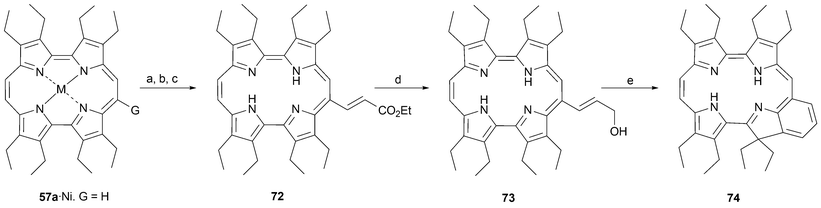 | ||
| Scheme 21 Synthesis of octaethylbenzochloracene. Reaction conditions (yields in parentheses): a) (M = Ni, G = H) DMF, POCl3, dichloroethane, 50 °C, 2 h, then NaOAc/H2O, 50 °C, 30 min (98%). b) (M = Ni, G = CHO) (carbethoxymethylene)triphenyl phosphorane, xylenes, reflux, 3.5 h (92%). c) H2SO4, CH2Cl2, 2.5 h, 23 °C, (97%). d) DIBALH, toluene, –78 °C, 1.5 h, (65%). e) phosphoric acid 85%, 23 °C, 3.5 h (45%). | ||
In 2002, Hayashi reported the reconstitution of myoglobin using a substituted etioporphycene (75).7 The ligand was designed to mimic the native protoporphyrin IX heme group (76) with two acid functions pendant from the macrocycle (Fig. 6). The reconstituted myoglobin prepared from the Fe(II) complex is blue and, like the native system, displays a high affinity for oxygen.
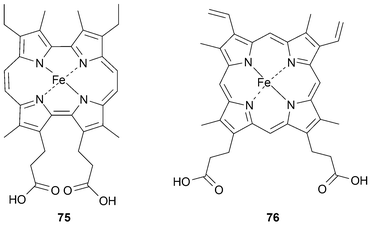 | ||
| Fig. 6 Artificial Fe(II) porphycene heme group (75) and native heme (76). | ||
Recently, the same group has synthesized an etioporphycene bearing trifluoromethyl groups.67 Although the synthetic plan follows the general sequence used by Vogel to prepare his original porphycenes, some modifications were made so as to adapt the synthesis to the fluoromethyl bearing pyrrolic precursor. Thus, starting with pyrrole 77, the first step involves oxidation of the α-methyl group to provide the corresponding formyl species 78. This latter group is then protected as an acetal and iodinated (to give 79 and 80, respectively) prior to carrying out the key bipyrrole-forming Ullmann coupling to provide 81. Deprotection and reductive coupling then provides the desired product 57b (Scheme 22).
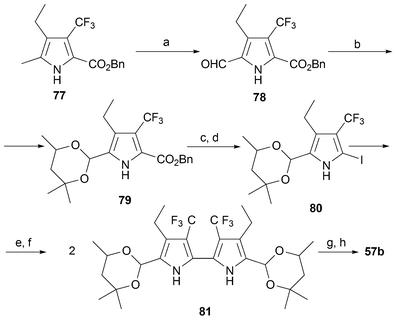 | ||
| Scheme 22 Synthesis of 57b. Reaction conditions: a) Br2, SO2Cl2, AcOH, trifluoroacetic acid, Ac2O, 0 °C, 3 h (84%). b) Hexylene glycol, p-TsOH, benzene (100%). c) Pd/C, H2, Et3N, THF (100%). d) KI, I2, NaHCO3, 50 °C, 2 h, H2O (80%). e) Boc2O, N,N-dimethylaminopyridine, CH2Cl2 (79%). f) Cu, DMF, 18 h, 23 °C, (64%). g) HCl(c), AcOH, 50 °C, 10 min (71%). h) TiCl4/Zn/CuCl, reflux, 3 h, THF then DDQ, CHCl3 (10%). | ||
Aromatic groups have been placed in the β-pyrrolic positions68 and even been used to connect adjacent pyrrolic rings69,70 (Scheme 23).
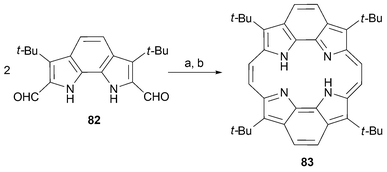
Benzoporphycene 83 was obtained by McMurry coupling of the corresponding aldehyde 82. As is true for meso-substituted porphycenes, such as 56, the non-aromatic intermediate leading to 83 is stable enough to be isolated. Oxidation of this N,N′-dihydroporphycene was effected by treatment with DDQ.
An interesting example, noteworthy because of its potential significance as a PDT photosensitizer, is benzoporphycene 85.19,71 This compound was prepared by a Diels–Alder reaction involving dimethyl acetylene carboxylate (DMAD) and the corresponding vinyl porphycene 84. The latter light sensitive intermediate can be prepared by subjecting the bromoethyl porphycene 49 to elimination in the presence of DBU (Scheme 24).
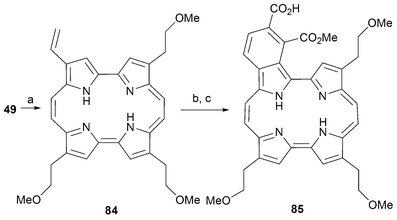 | ||
| Scheme 24 Synthesis of benzoporphycene 85. Reaction conditions (yields in parentheses): a) DBU, THF, 40 °C, 90 min (86%). b) dimethyl acetylene carboxylate (DMAD), 75 °C, 90 min (60%) c) LiOH, THF/H2O (65%). | ||
In 2007 Sessler and coworkers prepared a series of etioporphycenes bearing aromatic groups in positions 2, 7, 12 and 17 (e.g., 57c).62 These functionalized porphycenes were synthesized in order to study the possible formation of liquid crystals. Accordingly, the aromatic groups on the porphycene core were substituted with –OC10H21 alkoxy chains. Towards this end, β-substituted pyrroles were prepared using the Zard–Barton protocol and carried on to provide the highly organic soluble bipyrroles 86–88 (Scheme 25). It is noteworthy that the porphycenes in question (57c) were obtained in high yield. As in the case of other functionalized porphycene systems, this favorable result can be attributed to the presence of the highly solubilizing substituents.
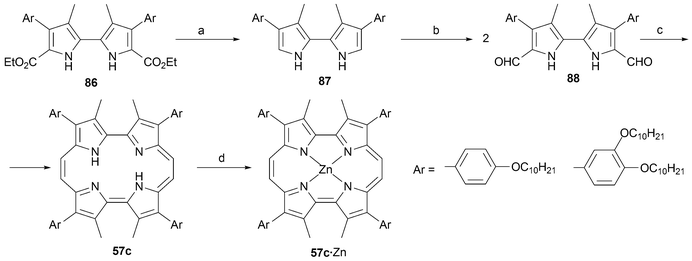 | ||
| Scheme 25 Synthesis of aryl substituted etio-type porphycenes (Ar = p-H21C10OPh, p,m-H21C10OPh). Reaction conditions (yields in parentheses): a) KOH, ethylene glycol, 200 °C (91%, 90%). b) POCl3, DMF/DCE, Δ, then AcONa, H2O, Δ, (82%, 76% two steps). c) TiCl4, Zn, CuCl, THF, Δ, (41%, 25%). d) Zn(OAc)2, CHCl3, MeOH, Δ. | ||
Porphycene analogues containing furan, thiophene, or imidazole subunits
In parallel with recent developments in porphyrin chemistry, structural variations of the tetrapyrrolic porphycene in such a manner that an 18 π-electron conjugation pathway is maintained gives rise to a potentially rich family of neutral as well as charged analogues. So far, efforts have focused on the synthesis of analogues with furan, thiophene, and imidazole subunits.The field was opened in 1988 by the Vogel group, which brought the furan-based analogue of porphycene, the tetraoxaporphycene dication 90, to light.72 This analogue was obtained in two steps by subjecting 5,5′-bi-2,2-furfuraldehyde73 to reductive coupling followed by oxidation. The coupling affords the cyclophane -type tetraoxa[20]annulene89 in 16% yield along with smaller amounts of higher order macrocyclic compounds. The tetraoxaannulene 89 is relatively stable in contrast to the corresponding pyrrole-derived 22, 24-dihydroporphycene (compound 3; Scheme 1). As expected, 89 on reaction with bromine in perchloric acid readily undergoes 2-electron oxidation to give the tetraoxaporphycene dication 90 as its perchlorate salt, a species that exhibits typical aromatic features (Scheme 26).
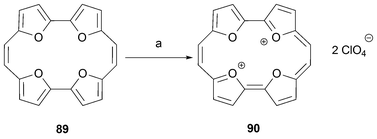 | ||
| Scheme 26 Synthesis of tetraoxaporphycene dication. Reaction conditions (yields in parentheses): a) Br2/HClO4 (90%). The 2-electron oxidation in question is effected using Br2 dissolved in perchloric acid. | ||
In 1991, Vogel and Cöln prepared a new porphycene analogue 93 containing two nitrogen and two oxygen atoms within the core.74 The starting furan–pyrrole dialdehyde was coupled using Vogel's modified McMurry conditions, a transformation that provided the stable annulene 92. Interestingly, the symmetric isomer 91 was not formed. However, as expected, oxidation of 92, using a mixture of concentrated nitric acid and 70% perchloric acid, then provided the 18 π-electron aromatic dioxaporphycene 93 in 94% yield (Scheme 27).
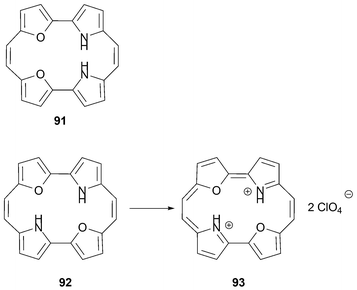 | ||
| Scheme 27 Oxidation of annulene 92 and synthesis of dioxaporphycene 93. A mixture of concentrated nitric acid in 70% perchloric acid was used as the oxidant leading to the isolation of 93 in its diprotonated form. | ||
The sulfur analogue of intermediate 3 and annulene 92, namely 94, was reported independently by Neidlein (1993) and Cava (1994). As expected based on this analogy, macrocycle 94 was found to be a distorted non-planar 20 π-electron annulene .75,76 This sulfur-containing cyclophane was synthesized by heating 2,2′-bithiophene-5,5′-dicarboxaldehyde77 at reflux in the presence of the titanium low-valent reagent; this gave 94 in 8–11% yield, along with a 3–14% yield of the corresponding hexathiophene trimer (Scheme 28). There is, however, no evidence that this procedure gave rise to the corresponding aromatic product, 95, a putative species that would be analogous to the dication 90 or porphycene 1. Presumably, this reflects an inherent difficulty in carrying out the requisite oxidation.
![Presumed resistance towards oxidation inferred for tetrathia[20]annulene94.](/image/article/2008/CS/b704945e/b704945e-s28.gif) | ||
| Scheme 28 Presumed resistance towards oxidation inferred for tetrathia[20]annulene94. | ||
Neidlein and co-workers78 obtained the alkynyl-substituted annulenes 97via Sonogashira coupling79 using the corresponding tetrabrominated macrocyle (obtained from the brominated bipyrrole 96) as the starting material (Scheme 29). However, there is no report of any successful oxidation to an aromatic species corresponding to the so-far hypothetical dication 95.
![Synthesis of a tetrakis alkynyl derivative, 97, of the tetrasulfido[20]annulene 94. Reaction conditions (yields in parentheses): a) TiCl4, Zn/Cu, pyridine, THF, reflux, 72 h followed by hydrolysis (26%). b) Me3Si–CCH, Pd(PhCN)2Cl2, Ph3P, CuI, i-Pr2NH, 70 °C, 2 h (76%).](/image/article/2008/CS/b704945e/b704945e-s29.gif) | ||
Scheme 29 Synthesis of a tetrakis alkynyl derivative, 97, of the tetrasulfido[20]annulene 94. Reaction conditions (yields in parentheses): a) TiCl4, Zn/Cu, pyridine, THF, reflux, 72 h followed by hydrolysis (26%). b) Me3Si–C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CH, Pd(PhCN)2Cl2, Ph3P, CuI, i-Pr2NH, 70 °C, 2 h (76%). CH, Pd(PhCN)2Cl2, Ph3P, CuI, i-Pr2NH, 70 °C, 2 h (76%). | ||
While the tetrathiaporphycene dication 95 defies synthesis, the neutral dithiaporphycene 99, with the sulfur atoms in a trans-like configuration, is found to be a planar aromatic macrocycle , matching the parent porphycene in many respects. It is obtained from the spontaneous oxidation of 98 obtained upon subjecting 2-(2-thienyl)-1H-pyrrole to reductive coupling (Scheme 30).80
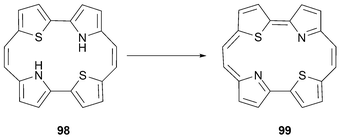 | ||
| Scheme 30 Thiophene and pyrrole containing annulene 98, and thiaporphycene 99. | ||
Mixed furan–thiophene porphycene analogues were prepared in 2000 in Dai's laboratory.81 In this case, McMurry coupling of 5,5′-diformyl-2,2′-furanthiophene100 in tetrahydrofuran at reflux afforded a separable mixture of the two possible isomers 101 and 102. Interestingly, isomer 102 (formed in 7.2% yield) is obtained in preference over 101 (generated in 2.6% yield). The authors suggest that the cyclization takes place via initial coupling of the furan “sides” of the biaryl precursor reflecting the relatively small size of oxygen compared to sulfur. However, there does not appear to be any independent support for this proposal.
In the same report detailing the synthesis of 101 and 102, Dai and co-workers disclosed the synthesis of the benzo substituted annulene 104. The synthesis of this latter product, using Pd-chemistry,82 is noteworthy because it provides an exception to the general “rule” wherein a biaryl moiety provides the starting material and a single McMurry coupling serves to close the ring in a “2 + 2” fashion (Scheme 31).
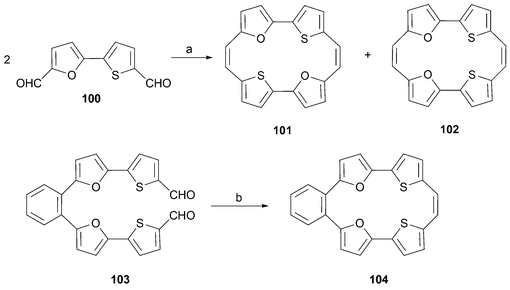 | ||
| Scheme 31 Reaction conditions (yields in parentheses): a) TiCl4/Zn/pyridine, reflux, THF, 2.5 h, 101 (2.6%), 102 (7.2%). b) TiCl4/Zn/pyridine, reflux, THF, 5 h (18%). | ||
Also in 2000, the first porphycene incorporating nitrogen in its periphery was reported.83 These kinds of compounds, embodied in structures 106, can be regarded as heteroporphycenes since their UV-vis and NMR spectral data is consistent with an aromatic system (Scheme 32). Again, this kind of porphycene derivative could be obtained by subjecting the appropriate reduced precursor, 105, to oxidation with 2,3-dichloro-5,6-dicyano-1,4-benzoquinone (DDQ).
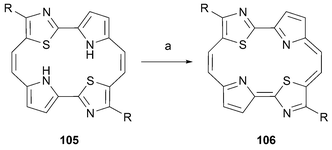 | ||
| Scheme 32 Examples of thia and aza porphycenes. Reactions conditions. (R = H, t-Bu, Ph) a) DDQ, THF, –20 °C to 23 °C, 30 min (17–87%). | ||
The synthesis of the key, reduced precursor 105 begins with a substituted 2-(1H-pyrrol-2-yl)thiazole. This intermediate, in turn, is prepared via the cyclocondensation of 1H-pyrrole-2-carbothioamide with various α-haloketones . In this latter step, a range of alkyl and aromatic substituents can be introduced. With this precursor in hand, the thiazole ring is formylated via selective generation of the anion with n-BuLi followed by quenching with N–formylmorpholine. Standard Vilsmeier–Haack conditions yield the corresponding dialdehyde 107. Treatment of these dialdehydes with a low-valent titanium (McMurry) reagent in THF at reflux, affords the corresponding annulenes 105 and its symmetrical isomer (nitrogens in positions 21 and 24), albeit in low yields (2–3%), along with the trimer 108 (Scheme 33).
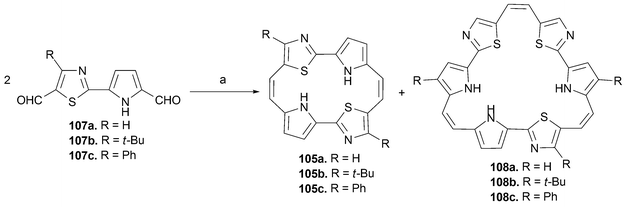 | ||
| Scheme 33 Reaction conditions (yields in parentheses): (R = H, R = t-Bu, R = Ph) a) TiCl4/Zn/pyridine, reflux, THF, 5 h (7–18%). | ||
During work-up of the McMurry reaction, the authors noticed some reduction of the porphycene core. However, they found that the removal of any residual Zn species by filtration prior to quenching the reductive coupling reaction obviated this undesirable reduction process, which was ascribed to hydrogenation at the meso positions.
In this same year Neidlein reported the preparation of a macrocycle (110) containing four thiazole units.84 The synthesis is very straightforward and consists of three steps: preparation of the 2-(thiazol-2-yl)thiazole from the double condensation of ethanebis(thioamide) and 1-bromo-3,3-dimethylbuta-2-one followed by formylation (to give 109), and standard McMurry-type reductive coupling. However, in contrast to what was found in the case of compound 105, annulene 110 could not be oxidized to produce the corresponding aromatic product (equivalent to the hypothetic species 95); this proved true in spite of testing a variety of conditions. On the other hand, the preparation was found to yield the corresponding trimer 111 in 10% yield (Scheme 34).
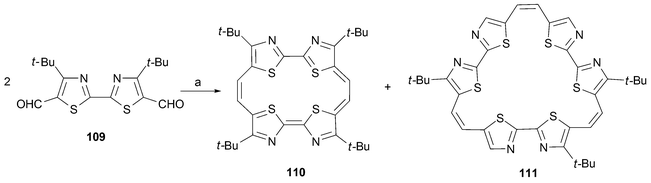 | ||
| Scheme 34 Reaction conditions (yields in parentheses): a) TiCl4/Zn/pyridine, reflux, THF, 65 h, 110 (6%), 111 (10%). | ||
In 2003 the groups of Sessler and Allen85 and Teixidó and Nonell86 reported independently the preparation of biimidazole-containing tetrazaporphycenes 119, compounds that have come to be referred to as “imidacenes”. The approach developed by Sessler leads to the alkyl derivatives, whereas that used by Nonell produces the aryl substituted counterpart (Scheme 35). In spite of these differences, in both cases a major effort was required to obtain the requisite 2,2′-bimidazole87 precursors.88,89 Sessler's approach to this class of building block involved the dimerization of iodoimidazoles 115 using a Pd-mediated Ullmann-type coupling. This gives a biimidazole 116 that is then formylated (to give 117) prior to being subject to reductive coupling to produce the annulene species 118. In contrast, the synthesis of Nonell is predicated on the use of the dibrominated biimidazole 113 (obtained from the tetrabromo species 112) as a scaffold for further elaboration. This novel strategy is impressive in that it allows for the introduction of aromatic substituents via the appropriate use of Pd coupling chemistry. This provides an aryl substituted intermediate (114) that is then subject to follow-up reductive coupling. Thus, like Sessler's approach, Nonell's strategy is based on the appropriate use of palladium coupling chemistry,90 albeit at a very different point in the synthesis.
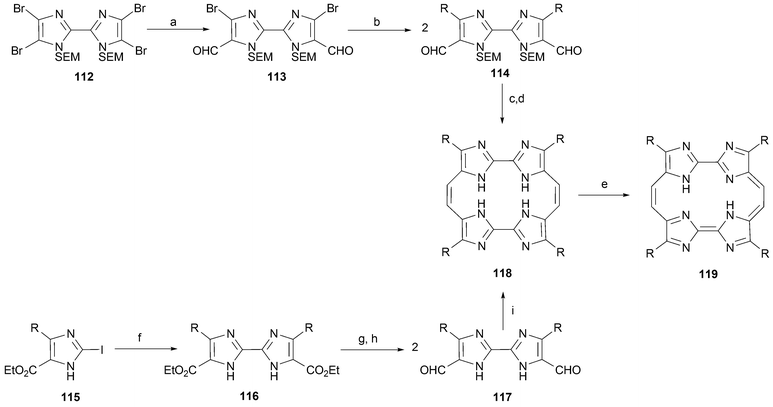 | ||
| Scheme 35 Reaction conditions (yields in parentheses): a) n-BuLi, –78 °C, THF then DMF (71%). b) (R = p-BuPh) RB(OH)2, Pd(PPh3)4, Na2CO3, toluene/EtOH (55%). c) HCl, EtOH/H2O (76%). d) TiCl4/Zn(Cu)/pyridine, THF (7%). e) DDQ, THF (60%). f) (R = n-propyl) Pd(PPh3)4, Et3N, toluene, 110 °C (28%). g) DIBALH, THF (98%). h) DDQ, THF (60%). i) TiCl4/Zn(Cu)/pyridine, THF, (2%). | ||
As is true for the dioxaannulenes discussed above, the reduced synthetic precursor produced after the McMurry ring-forming reductive coupling step (118) exhibits unusual stability. Needless to say, this stability stands in stark contrast to what is true in the case of the analogous reduced porphycene precursor 3 (Scheme 1). It is also noteworthy since a priori it would be expected that the corresponding oxidized imidacenes 119, benefiting from [18]annulene-type aromaticity , would be far more stable. To date, the photophysical properties of the aromatic tetraazaporphycenes (imidacenes), but not those bearing alkyl substituents, have been analyzed in detail.91 Here, it was noted that the λmax of the imidacene is shifted to the red by ca. 100 nm compared to the corresponding porphycene 1d. This behavior, attributable to the introduction of additional nitrogen atoms into the macrocyclic core, is in agreement with the trend observed for other porphycene analogues.83
Extended porphycenes
The insertion of an even number of sp or sp2 hybridized carbons into the linkers between the pyrrolic units of porphycenes provides an entry into a new class of conjugated macrocycle , the so-called extended porphycenes. The important consequence of this addition is that the main (4n + 2) π-electron conjugation pathway is lengthened such that n > 4. This leads to chromophores that absorb at longer wavelengths than regular porphycenes of general structure 1. This feature, in turn, has made extended porphycenes of interest as possible photosenzitizers for use in PDT applications.92As in the case of porphycene, it was the Vogel group that pioneered the synthetic development of extended porphycenes. Towards this end, two general strategies were developed that involve, respectively, the synthetic addition of alkylidene or alkylidyne moieties to either the meso bridge or between the 2,2′ positions of the bipyrrole subunits. To date, both strategies have been successfully implemented, albeit in neither case has the chemistry of the resulting systems been extensively pursued. The synthesis of the first type of expanded porphycene was achieved by subjecting the vinylogous dialdehyde 120 to reductive dimerization. The latter intermediate is readily obtained from 4,4′-dipropyl-2,2′-bipyrrole via reaction with 3-(dimethyl-1-amino)acrolein70 (Scheme 36).
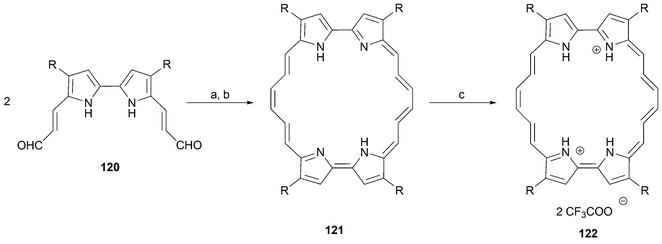
As an extension of this work, the monovinylogous diformyl bipyrrole 123 was subject to reductive coupling under the McMurry conditions followed by exposure to air (Scheme 37). This gave rise to the extended porphycene 124 as the dominant product in 7% yield. Also obtained as a minor by-product was the less symmetric isomer 126. This latter species could not be isolated. However, after electrocyclization and spontaneous dehydrogenation it produced 127 in 0.7% yield based on 123.54
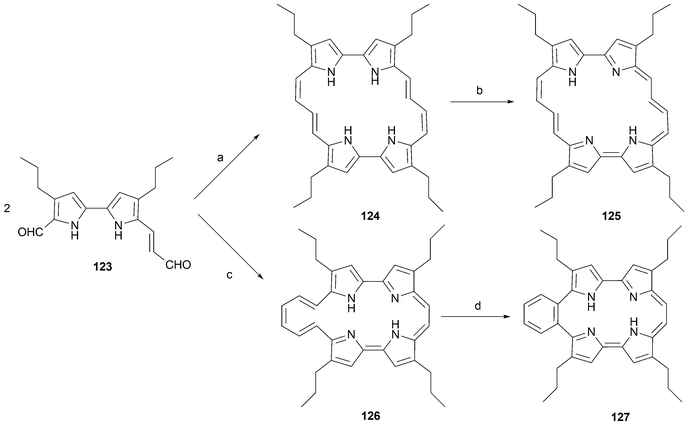 | ||
| Scheme 37 Formation of extended porphycene 125 and 9,10-benzotetrapropylporphycene, 127, from its putative ring opened precursor 126. Reaction conditions. a) TiCl4/Zn, THF. b) air. c) TiCl4/Zn. d) benzene reflux, 2 days.54 | ||
The first example of an expanded porphycene containing triple bonds inserted between the 2 and 2′ positions of the 2,2′-bipyrrole subunits, compound 133, was reported by Vogel in 1990.93 In this case, the resulting porphycene was found to be a 22 π-electron aromatic macrocycle (Fig. 7). Interestingly, this macrocycle displayed an acetylene–cumulene bonding motif, which is a feature in common with the Sondheimer–Nakagawa [4n + 2]dehydroannulenes.
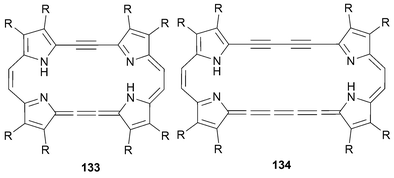 | ||
| Fig. 7 Extended porphycenes 133 and 134. | ||
In 1992 Vogel and coworkers also reported the synthesis of the 26 π-electron homologue, 134.94 In both cases (i.e, 133 and 134), the synthesis was effected via reductive dimerization of the corresponding dialdehyde, 131 or 132. The requisite acetylene linked bipyrroles were, in turn, prepared from iodopyrrole 128 and an acetylene source with the key bond attachment being made by means of a Sonogashira coupling (to give 129 and then 130 after deprotection). A refined procedure in the case where the β-pyrrolic substituent (R) is methyl was reported by Kim95 in 1999 (Scheme 38).
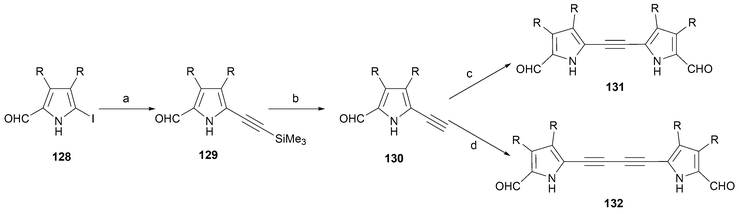 | ||
| Scheme 38 Reaction conditions: (R = CH3). a) Pd(PPh3)2Cl2, CuI, (trimethylsilyl)acetylene, diethylamine, 50 °C, (95%). b) Bu4NF, THF, 23 °C, 1 h, (99%). c) 128 (0.85. equiv.), Pd(PPh3)2Cl2, CuI, diethylamine, 50 °C, 3 h, (65%). d) (PPh3)4Pd, CuI, Et3N, chloroacetone, benzene, 23 °C, 16 h, (92%). | ||
A related class of extended porphycenes, containing “extra” sp2 hybridized linking atoms between the 2 and 2′ positions of the bipyrrole subunit, was obtained via hydrogenation of 133. This yields the 22 π-electron conjugated porphycene homologue 135. An alternative synthesis of this same macrocycle involves subjecting dialdehyde 136 to reductive oligomerization under McMurry conditions; the desired product 135 is obtained in this way, albeit in low yields (Scheme 39).
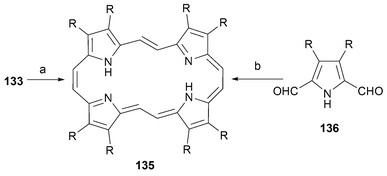
Conclusions
The most general method used to prepare porphycenes remains that introduced by Vogel roughly 20 years ago. It consists of two formal stages, namely the synthesis of a biheteroaryl moiety and dimerization of the corresponding dialdehyde by means of a McMurry coupling. The last step, although low-yielding in the case of less soluble systems is well established and is common to almost all preparations; hence the efforts to build new porphycenes have been focused on the optimization of the 2,2′-bipyrroles synthesis and the production of novel biheteroaryl systems. Nonetheless, the potential utility of new porphycenes and their derivatives provides an incentive to develop new, and more concise preparative methods, in part because such strategies offer a chance to improve the flexibility of current porphycene design strategies.To achieve these objectives two general approaches are envisioned. The first involves the direct synthesis of substituted porphycenes using appropriate biheteroaryls as the starting materials. This strategy, which is more classical, relies on the design of new preparations of biheteroaryls. Here, new synthetic transformations, including metal-catalyzed cross-coupling reactions, could prove advantageous.
The second strategy is based on the design of easy-to-prepare porphycenes followed by post-synthetic modification. This has the advantage that it could permit access to systems with specific requirements. However, it is unlikely to address the obvious synthetic challenge of finding new ways to make and modify porphycenes using approaches that differ radically from those introduced by Vogel. Nonetheless the current methods have provided a range of interesting structures and have allowed a variety of applications to be pursued. As such, the area of porphycene chemistry continues to attract an ever-larger number of practitioners, including increasingly researchers outside the synthetic organic chemical field. This attests to the fascination inherent in porphycene as a molecule, ligand, structural motif, and chromophore.
Acknowledgements
This work was supported by the National Science Foundation (grant CHE 0515670 to J.L.S.).References
- E. Vogel, M. Köcher, H. Schmickler and J. Lex, Angew. Chem., Int. Ed. Engl., 1986, 25, 257 CrossRef.
- In order to describe porphyrinoid structures, a systematic nomenclature system has been proposed; it consists of the number of π-electrons in brackets, the general name of the compound, and the numbers of C atoms between the pyrrole units for each of the bridges set off by commas and in parentheses. See: M. Gosmann and B. Franck, Angew. Chem., Int. Ed. Engl., 1986, 25, 1100 Search PubMed.
- J. L. Sessler and S. J. Weghorn, in Expanded, Contracted and Isomeric Porphyrins, Elsevier, Oxford, 1997 Search PubMed; J. L. Sessler, A. Gebauer and E. Vogel, in The Porphyrin Handbook, ed. K. M. Kadish, K. M. Smith and R. Guilard, Academic Press, New York, 2000, vol 2, 3–32 Search PubMed; A. Jasat and D. Dolphin, Chem. Rev., 1997, 97, 2267 Search PubMed.
- E. Vogel, Pure Appl. Chem., 1996, 68, 1355 CAS.
- C. J. Fowler, J. L. Sessler, V. M. Lynch, J. Waluk, A. Gebauer, J. Lex, A. Heger, F. Zuniga-y-Rivero and E. Vogel, Chem.–Eur. J., 2002, 8, 3485 CrossRef CAS and references therein. T. Baba, H. Shimakoshi, I. Aritome and Y. Hisaeda, Chem. Lett., 2004, 33, 906 Search PubMed; K. Rachlewicz, L. Latos-Grazynski, E. Vogel, Z. Ciunik and L. B. Jerzykiewicz, Inorg. Chem., 2002, 41, 1979 CrossRef CAS.
- T. Hayashi, K. Okazaki, N. Urakawa, H. Shimakoshi, J. L. Sessler, E. Vogel and Y. Hisaeda, Organometallics, 2001, 20, 3074 CrossRef CAS; W.-Ch. Lo, Ch.-M. Che, K.-F. Cheng and T. C. W. Mak, Chem. Commun., 1997, 1205 RSC.
- T. Hayashi, H. Dejima, T. Matsuo, H. Sato, D. Murata and Y. Hisaeda, J. Am. Chem. Soc., 2002, 124, 11226 CrossRef CAS; T. Hayashi, D. Murata, M. Makino, H. Sugimoto, T. Matsuo, H. Sato, Y. Shiro and Y. Hisaeda, Inorg. Chem., 2006, 45, 10530 CrossRef CAS; T. Matsuo, H. Dejima, S. Hirota, D. Murata, H. Sato, T. Ikegami, H. Hori, Y. Hisaeda and T. Hayashi, J. Am. Chem. Soc., 2004, 126, 16007 CrossRef CAS; H. Nakashima, J.-Y. Hasegawa and H. Nakatsuji, J. Comput. Chem., 2006, 27, 1363 CrossRef CAS; T. Matsuo, T. Ikegami, H. Sato, Y. Hisaeda and T. Hayashi, J. Inorg. Biochem., 2006, 100, 1265 CrossRef CAS; T. Matsuo, T. Tsuruta, K. Maehara, H. Sato, Y. Hisaeda and T. Hayashi, Inorg. Chem., 2005, 44, 9391 CrossRef CAS.
- A. Saoiabi, N. Maroufi, A. Laghzizil, J. M. Barbe and R. Guilard, J. Maro. Chim. Heterocycl., 2005, 4, 65 Search PubMed; V. Albrecht, A. Wiehe, B. Roeder and W. Neuberger, WO Pat., 2006007261, 2005; J. M. Barbe, P. Richard, M. A. Aukauloo, C. Lecomte, P. Petit and R. Guilard, J. Chem. Soc., Chem. Commun., 1994, 2757 Search PubMed.
- For general reviews on PDT, see I. J. MacDonald and T. J. Dougherty, J. Porphyrins Phthalocyanines, 2001, 5, 105 Search PubMed; E. D. Sternberg and D. Dolphin, Tetrahedron, 1998, 54, 4151 CrossRef CAS; T. J. Dougherty, C. J. Gomer, B. W. Henderson, G. Jori, D. Kessel, M. Korbelik, J. Moan and Q. Peng, J. Natl. Cancer Inst., 1998, 90, 889 CrossRef CAS.
- P. F. Aramendia, R. W. Redmond, S. Nonell, W. Schuster, S. E. Braslavsky, K. Schaffner and E. Vogel, Photochem. Photobiol., 1986, 44, 555 CrossRef CAS; M. Guardiano, R. Biolo, G. Jori and K. Schaffner, Cancer Lett., 1989, 44, 1 CrossRef CAS; C. Milanesi, R. Biolo, G. Jori and K. Schaffner, Laser Med. Sci., 1991, 6, 437 Search PubMed; A. Aicher, K. Miller, E. Reich and R. Hautmann, Urol. Res., 1994, 22, 25 CrossRef CAS; A. Villanueva, M. Cañete, S. Nonell, J. I. Borrell, J. Teixidó and A. Juarranz, Anti-Cancer Drug Des., 1996, 11, 89 CAS; M. Cañete, M. Lapeña, A. Juarranz, V. Vendrell, J. I. Borrell, J. Teixidó, S. Nonell and A. Villanueva, Anti-Cancer Drug Des., 1997, 12, 543 CAS; S. Karrer, C. Abels, R.-M. Szeimies, W. Baumler, M. Dellian, U. Hohenleutner, A. E. Goetz and M. Landthaler, Arch. Dermatol. Res., 1997, 289, 132 CrossRef CAS; S. Karrer, R.-M. Szeimies, A. Ebert, S. Fickweiler, C. Abels, W. Bäumler and M. Landthaler, Laser Med. Sci., 1997, 12, 307 Search PubMed; M. Cañete, C. Ortega, A. Gavaldà, J. Cristóbal, A. Juarranz, S. Nonell, J. Teixidó, J. I. Borrell, A. Villanueva, S. Rello and J. C. Stockert, Int. J. Oncol., 2004, 24, 1221 CAS; D. Kessel, M. Conley, M. C. H. Vicente and J. J. Reiners, Photochem. Photobiol., 2005, 81, 569 CrossRef CAS . Reviews, see: O. Arad, A. Gavaldá, O. Rey, N. Rubio, D. Sánchez-García, J. I. Borrell, J. Teixidó, S. Nonell, M. Cañete, A. Juarranz, A. Villanueva, J. C. Stockert and P. J. D. Jiménez, Afinidad, 2002, 500, 343 Search PubMed; J. C. Stockert, M. Cañete, A. Juarranz, A. Villanueva, R. W. Horobin, J. I. Borrell, J. Teixidó and S. Nonell, Curr. Med. Chem., 2007, 14, 997 Search PubMed.
- F. M. Lauro, P. Pretto, L. Covolo, G. Jori and G. Bertoloni, Photochem. Photobiol. Sci., 2002, 1, 468 RSC.
- E. Vogel, Pure Appl. Chem., 1990, 62, 557 CAS; E. Steiner and P. W. Fowler, Org. Biomol. Chem., 2003, 1, 1785 RSC; E. Vogel, J. Heterocycl. Chem., 1996, 33, 1461 CrossRef CAS.
- J. Waluk, Acc. Chem. Res., 2006, 39, 945 CrossRef CAS; A. Vdovin, J. Sepiol, N. Urbanska, M. Pietraszkiewicz, A. Mordzinski and J. Waluk, J. Am. Chem. Soc., 2006, 128, 2577 CrossRef CAS; J. Waluk and E. Vogel, J. Phys. Chem., 1994, 98, 4530 CrossRef CAS; K. Birklund-Anderson, E. Vogel and J. Waluk, Chem. Phys. Lett., 1997, 271, 341 CrossRef; A. Starukhin, E. Vogel and J. Waluk, J. Phys. Chem. A, 1998, 102, 9999 CrossRef CAS; J. Sepiol, Y. Stepaneko, A. Dovin, A. Mordzinski, E. Vogel and J. Waluk, Chem. Phys. Lett., 1998, 196, 549 CrossRef.
- T. Mukaiyama, T. Sato and J. Hanna, Chem. Lett., 1973, 1041 CAS; J. E. McMurry and M. P. Fleming, J. Am. Chem. Soc., 1974, 96, 4708 CrossRef CAS; J. E. McMurry, Acc. Chem. Res., 1983, 16, 405 CrossRef CAS; D. Lenoir, Synthesis, 1977, 553 CrossRef CAS.
- For reviews, see: J. E. McMurry, Chem. Rev., 1989, 89, 1513 Search PubMed; A. Fürstner and B. Bogdanovic, Angew. Chem., Int. Ed. Engl., 1996, 35, 2442 CrossRef CAS.
- H. Rapoport and N. Castagnoli, J. Am. Chem. Soc., 1962, 84, 2178 CrossRef CAS; G. R. Geier, III and S. C. Grindrod, J. Org. Chem., 2004, 69, 6404 CrossRef.
- V. J. Bauer, D. L. J. Clive, D. Dolphin, J. B. Paine, III, F. L. Harris, M. M. King, J. Loder, S.-W. Chien Wang and R. B. Woodward, J. Am. Chem. Soc., 1983, 105, 6429 CrossRef CAS.
- E. Vogel, M. Balci, K. Pramod, P. Koch, J. Lex and O. Ermer, Angew. Chem., Int. Ed. Engl., 1987, 26, 928 CrossRef.
- C. Richert, J. M. Wessels, M. Müller, M. Kisters, T. Benninghaus and A. E. Goetz, J. Med. Chem., 1994, 37, 2797 CrossRef CAS.
- M. J. Broadhurst, R. Grigg and A. W. Johnson, J. Chem. Soc., Perkin Trans. 1, 1972, 2111 RSC; A. H. Corwin, W. A. Bailey and P. Viohl, J. Am. Chem. Soc., 1942, 64, 1267 CrossRef CAS.
- R. Grigg, A. W. Johnson and J. W. F. Wasley, J. Chem. Soc., 1963, 359 RSC; R. Crigg and A. W. Johnson, J. Chem. Soc., 1964, 3315 RSC; J. L. Sessler, M. Cyr and A. K. Burrell, Tetrahedron, 1992, 48, 9661 CrossRef CAS and references cited therein; W. Hinz, R. A. Jones, S. U. Patel and M.-H. Karatza, Tetrahedron, 1986, 42, 3753 Search PubMed; H. Rapoport and J. Bordner, J. Org. Chem., 1964, 29, 2727 CrossRef CAS; J. Bordner and H. Rapoport, J. Org. Chem., 1965, 30, 3824 CAS; H. Bauer, Chem. Ber., 1968, 101, 1286 CrossRef; D. L. Boger and M. Patel, J. Org. Chem., 1988, 53, 1405 CrossRef CAS; H. Falk and H. FIödl, Monatsh. Chem., 1988, 119, 247 CrossRef; T. Benincori, E. Brenna, F. Sannicolò, G. Zotti, S. Zecchin, G. Schiavon, C. Gatti and G. Frigerio, Chem. Mater., 2000, 12, 1480 CAS.
- P. E. Fanta, Chem. Rev., 1964, 64, 613 CrossRef CAS.
- J. B. Paine, III and D. Dolphin, J. Org. Chem., 1988, 53, 2787 CrossRef.
- H. Fischer, M. Goldschmidt and W. Nüssler, Justus Liebigs Ann. Chem., 1931, 486, 1 CAS; M. R. Johnson, J. Org. Chem., 1997, 62, 1168 CrossRef CAS; M. R. Johnson, D. C. Miller, K. Bush, J. J. Becker and J. A. Ibers, J. Org. Chem., 1992, 57, 4414 CrossRef CAS.
- W. Hennig, PhD Thesis, University of Cologne, 1992.
- S. Nonell, N. Bou, J. I. Borrell, J. Teixidó, A. Villanueva, A. Juarranz and M. Cañete, Tetrahedron Lett., 1995, 36, 3405 CrossRef CAS.
- H. Ikeda and J. L. Sessler, J. Org. Chem., 1993, 58, 2340 CrossRef CAS.
- A. Gavaldá, J. I. Borrell, J. Teixidó, S. Nonell, O. Arad, R. Grau, M. Cañete, A. Juarranz, A. Villanueva and J. C. Stockert, J. Porphyrins Phthalocyanines, 2001, 5, 846 CrossRef CAS.
- H. Rapoport and K. G. Holden, J. Am. Chem. Soc., 1962, 84, 635 CrossRef.
- M. Cañete, A. Ortiz, A. Juarranz, A. Villanueva, S. Nonell, J. I. Borrell, J. Teixidó and J. C. Stockert, Anti-Cancer Drug Des., 2000, 15, 143 CAS.
- O. Arad, J. Morros, X. Batllori, J. Teixidó, S. Nonell and J. I. Borrell, Org. Lett., 2006, 8, 847 CrossRef CAS.
- M. Farnier, S. Soth and P. Fournari, Can. J. Chem., 1976, 54, 1074 CrossRef CAS; M. Farnier, S. Soth and P. Fournari, Can. J. Chem., 1976, 54, 1083 CrossRef CAS.
- N. Urbanska, M. Pietraszkiewicz and J. Waluk, Presented at 4th International Conference on Porphyrins and Phthalocyanines, Rome, Italy, July 2–7, 2006.
- G. W. Gribble, D. H. Blank and J. P. Jasinski, Chem. Commun., 1999, 2195 RSC.
- T. Itahara, J. Chem. Soc., Chem. Commun., 1980, 49 Search PubMed.
- J. L. Sessler and D. Sánchez-García, Presented at 4th International Conference on Porphyrins and Phthalocyanines, Rome, Italy, July 2–7, 2006. For published abstract see: J. Porphyrins Phthalocyanines, 2006, 10, 854 Search PubMed.
- M. Suzuki, M. Miyoshi and K. Matsumoto, J. Org. Chem., 1974, 39, 1980 CrossRef CAS.
- E. Vogel, M. Köcher, M. Balci, I. Teichler, L. Lex, H. Schmickler and O. Ermer, Angew. Chem., Int. Ed. Engl., 1987, 26, 931 CrossRef.
- For N-substituted porphycenes, see: J. Mühlhoff, PhD Thesis, University of Cologne, 1994; C. K. Chang, I. Morrison, W. Wu, S.-S. Chern and S.-M. Peng, J. Chem. Soc., Chem. Commun., 1995, 1173 Search PubMed; J.-I. Setsune and K. Hazama, Tetrahedron Lett., 1997, 38, 2513 RSC.
- P. von Ragué Schleyer, Chem. Rev., 2001, 101, 1115 CrossRef.
- S. Will, A. Rahbar, H. Schmickler, J. Lex and E. Vogel, Angew. Chem., Int. Ed. Engl., 1990, 29, 1390 CrossRef.
- I. Aritome, H. Shimakoshi and Y. Hisaeda, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2002, 58, o563 CrossRef.
- E. Vogel, P. A. Koch, A. Rahbar and A. D. Cross, US Pat., 5 244 671, 1993.
- H. J. Callot, A. Rohrer and T. Tschamber, New J. Chem., 1995, 19, 155 Search PubMed; E. Vogel, M. Bröring, P. Scholz, R. Deponte, J. Lex, H. Schmickler, K. Schaffner, S. E. Braslavsky, M. Müller, S. Pörting, S. J. Weghorn, C. J. Fowler and J. L. Sessler, Angew. Chem., Int. Ed. Engl., 1997, 36, 1651 CrossRef CAS.
- T. Baba, H. Shimakoshi and Y. Hisaeda, Tetrahedron Lett., 2004, 45, 5973 CrossRef CAS.
- N.-K. Mak, T.-W. Kok, R. N.-S. Wong, S.-W. Lam, Y.-K. Lau, W.-N. Leung, N.-H. Cheung, D. P. Huang, L.-L. Yeung and C. K. Chang, J. Biomed. Sci., 2003, 10, 418 CrossRef CAS.
- S. E. Braslavsky, M. Müller, D. O. Mártire, S. Pörting, S. G. Bertolotti, S. Chakravorti, G. Koç-Weier, B. Knipp and K. Schaffner, J. Photochem. Photobiol., B, 1997, 40, 191 Search PubMed; M. Müller, PhD Thesis, University of Cologne, 1994.
- E. Vogel, M. Mueller, O. Halpern and A. D. Cross, US Pat., 5 637 608, 1997.
- H. K. Porter, Org. React., 1973, 20, 455 CAS.
- A. Segalla, F. Fedeli, E. Reddi, G. Jori and A. Cross, Int. J. Cancer, 1997, 72, 329 CrossRef CAS; L. Polo, A. Segalla, G. Bertoloni, G. Jori, K. Schaffner and E. Reddi, J. Photochem. Photobiol., B, 2000, 152 CrossRef CAS; R.-M. Szeimies, S. Karrer, C. Abels, P. Steinbach, S. Fickweiler, H. Messmann, W. Bäumler and M. Landthaler, J. Photochem. Photobiol., B, 1996, 34, 67 CrossRef CAS; K. Scherer, C. Abels, W. Baeumler, G. Ackermann and R.-M. Szeimies, Arch. Dermatol. Res., 2004, 295, 535 CrossRef CAS.
- E. Vogel, C. Richert, T. Benninghaus, M. Muller and A. D. Cross, US Pat., 5 179 120, 1993.
- E. Vogel, I. Grigat, M. Köcher and J. Lex, Angew. Chem., Int. Ed. Engl., 1989, 28, 1655 CrossRef.
- E. Vogel, M. Köcher, J. Lex and O. Enmer, Isr. J. Chem., 1989, 29, 257 CAS.
- H.-J. Dietrich, PhD Thesis, University of Cologne, 1994.
- E. Vogel, P. Koch, X.-L. Hou, J. Lex, M. Lausmann, M. Kisters, A. M. Aukauloo, P. Richard and R. Guilard, Angew. Chem., Int. Ed. Engl., 1993, 32, 1600 CrossRef.
- R. Guilard, M. A. Aukaloo, C. Tardieux and E. Vogel, Synthesis, 1995, 12, 1480 CrossRef.
- J. L. Sessler, A. Aguilar, D. Sánchez-García, D. Seidel, T. Köhler, F. Arp and V. M. Lynch, Org. Lett., 2005, 7, 1887 CrossRef CAS.
- D. H. R. Barton and S. Z. Zard, J. Chem. Soc., Chem. Commun., 1985, 1098 RSC; D. H. R. Barton, J. Kervagoret and S. Z. Zard, Tetrahedron, 1990, 46, 7587 CrossRef CAS.
- J. L. Sessler, M. J. Cyr and A. K. Burrell, Synlett, 1991, 127 CrossRef CAS.
- Application of Zard–Barton reaction in porphyrins : C. P. Gros, L. Jaquinod, R. G. Khoury, M. M. Olmstead and K. M. Smith, J. Porphyrins Phthalocyanines, 1997, 1, 201 Search PubMed.
- J. L. Sessler, M. R. Johnson, S. E. Creager, J. C. Fettinger and J. A. Ibers, J. Am. Chem. Soc., 1990, 112, 9310 CrossRef CAS.
- M. Stępień, B. Donnio and J. L. Sessler, Chem.–Eur. J., 2007 DOI:10.1002/chem.200700125.
- J. L. Sessler and M. C. Hoehner, Synlett, 1994, 3, 211 CrossRef.
- M. Kisters, PhD Thesis, University of Cologne, 1995.
- F. D'Souza, P. L. Boulas, M. Kisters, L. Sambrotta, A. M. Aukauloo, R. Guilard and K. M. Kadish, Inorg. Chem., 1996, 35, 5743 CrossRef CAS.
- M. N. Masuno, B. C. Robinson and A. S. Phadke, J. Porphyrins Phthalocyanines, 2001, 5, 177 CrossRef CAS; K. M. Barkigia, B. C. Robinson and M. W. Renner, J. Porphyrins Phthalocyanines, 2005, 9, 864 CrossRef CAS.
- T. Hayashi, Y. Nakashima, K. Ito, T. Ikegami, I. Aritome, A. Suzuki and Y. Hisaeda, Org. Lett., 2003, 16, 2845 CrossRef; T. Hayashi, Y. Nakashima, K. Ito, T. Ikegami, I. Aritome, K. Aoyagi, T. Ando and Y. Hisaeda, Inorg. Chem., 2003, 42, 7345 CrossRef CAS.
- D. Donnerstag, PhD Thesis, University of Cologne, 1993; J. Dobkowski, Y. Lobko, S. Gawinwski and J. Waluk, Chem. Phys. Lett., 2005, 416(1–3), 128 Search PubMed; J. Dobkowski, V. Galievsky, A. Starukhin, E. Vogel and J. Waluk, J. Phys. Chem. A, 1998, 102(26), 4966 CrossRef CAS.
- P. K. Panda, Y.-J. Kang and C.-H. Lee, Angew. Chem., Int. Ed., 2005, 44, 4053 CrossRef CAS.
- E. Vogel, Pure Appl. Chem., 1993, 65, 143 CAS.
- R. K. Pandey, J. Porphyrins Phthalocyanines, 2000, 4, 368 CrossRef CAS.
- E. Vogel, M. Sicken, P. Röhrig, H. Schmickler, J. Lex and O. Ermer, Angew. Chem., Int. Ed. Engl., 1988, 27, 411 CrossRef; R. Bachmann, F. Gerson, G. Gescheidt and E. Vogel, J. Am. Chem. Soc., 1993, 115, 10286 CrossRef CAS.
- R. Grigg, J. A. Knight and M. V. Sargent, J. Chem. Soc. C, 1966, 976 RSC.
- D. Cöln, PhD Thesis, University of Cologne, 1991.
- F. Ellinger, A. Gieren, T. Hübner, J. Lex, F. Lucchesini, A. Merz, R. Neidlein and J. Salbeck, Monatsh. Chem., 1993, 124, 931.
- Z. Hu, J. L. Atwood and M. P. Cava, J. Org. Chem., 1994, 59, 8071 CrossRef CAS.
- J. Nakayama and T. Fujimori, Sulfur Lett., 1990, 11, 29 CAS.
- U. Dahlmann, C. Krieger and R. Neidlein, Eur. J. Org. Chem., 1998, 63, 525 CrossRef.
- T. X. Neenan and G. M. Whitesides, J. Org. Chem., 1988, 53, 2489 CrossRef CAS.
- G. DeMunno, F. Lucchesini and R. Neidlein, Tetrahedron, 1993, 49, 6863 CrossRef CAS.
- W.-M. Dai and W. L. Mak, Tetrahedron Lett., 2000, 41, 10277 CrossRef CAS.
- Reviews see: A. Suzuki, Acc. Chem. Res., 1982, 15, 178 Search PubMed; A. Suzuki, Pure Appl. Chem., 1985, 57, 1749 CrossRef CAS; A. Suzuki, Pure Appl. Chem., 1986, 58, 629 CrossRef CAS; A. Suzuki, Pure Appl. Chem., 1991, 63, 419 CrossRef CAS; A. Suzuki, Pure Appl. Chem., 1994, 66, 213 CAS.
- T. Nüssbaumer, C. Krieger and R. Neidlein, Eur. J. Org. Chem., 2000, 13, 2449.
- T. Nüssbaumer and R. Neidlein, Helv. Chim. Acta, 2000, 83, 1161 CrossRef CAS.
- A. L. Sargent, I. C. Hawkins, W. E. Allen, H. Liu, J. L. Sessler and C. J. Fowler, Chem.–Eur. J., 2003, 9, 3065 CrossRef CAS.
- S. Nonell, J. I. Borrell, S. Borrós, C. Colominas, O. Rey, N. Rubio, D. Sánchez-García and J. Teixidó, Eur. J. Org. Chem., 2003, 9, 1635 CrossRef.
- H. Debus, Justus Liebigs Ann. Chem., 1858, 107, 199.
- W. E. Allen, C. J. Fowler, V. M. Lynch and J. L. Sessler, Chem.–Eur. J., 2001, 7, 721 CrossRef CAS.
- D. Sánchez-García, S. Borrós, S. Nonell, J. I. Borrell, C. Colominas and J. Teixidó, J. Heterocycl. Chem., 2002, 39, 733 CrossRef CAS.
- W. M. Sharman and J. E. Van Lier, J. Porphyrins Phthalocyanines, 2000, 4, 441 CrossRef CAS.
- N. Rubio, D. Sánchez-García, A. Jiménez-Banzo, O. Rey, J. I. Borrell, J. Teixidó and S. Nonell, J. Phys. Chem. A, 2006, 110, 3480 CrossRef CAS.
- E. Vogel, A. D. Cross, N. Jux, E. Rodriguez-Val, S. Boehm and W. Henning, US Pat., 5 132 101, 1992.
- N. Jux, P. Koch, H. Schmickler, J. Lex and E. Vogel, Angew. Chem., Int. Ed. Engl., 1990, 29, 1385 CrossRef; E. Vogel, N. Jux, E. Rodriguez-Val, J. Lex and H. Schmickler, Angew. Chem., Int. Ed. Engl., 1990, 29, 1387 CrossRef.
- D. O. Martire, N. Jux, P. F. Aramendia, R. M. Negri, J. Lex, S. E. Braslavsky, K. Schaffner and E. Vogel, J. Am. Chem. Soc., 1992, 114, 9969 CrossRef CAS.
- D. H. Cho, J. H. Lee and B. H. Kim, J. Org. Chem., 1999, 64, 8048 CrossRef CAS.
Footnote |
| † This work is dedicated to Prof. Emanuel Vogel in commemoration of the 20th anniversary of the first porphycene synthesis. |
| This journal is © The Royal Society of Chemistry 2008 |