In situ hydrothermal synthesis of tetrazole coordination polymers with interesting physical properties
Hong
Zhao
,
Zhi-Rong
Qu
,
Heng-Yun
Ye
and
Ren-Gen
Xiong
*
Ordered Matter Science Research Center, College of Chemistry and Chemical Engineering, Southeast University, Nanjing 211189, P. R. China. E-mail: xiongrg@seu.edu.cn
First published on 1st October 2007
Abstract
Tetrazole compounds have been studied for more than one hundred years and applied in various areas. Several yeas ago Sharpless and his co-workers reported an environmentally friendly process for the preparation of 5-substituted 1H-tetrazoles in water with zinc salt as catalysts. To reveal the exact role of the zinc salt in this reaction, a series of hydrothermal reactions aimed at trapping and characterizing the solid intermediates were investigated. This study allowed us to obtain a myriad interesting metal–organic coordination polymers that not only partially showed the role of the metal species in the synthesis of tetrazole compounds but also provided a class of complexes displaying interesting chemical and physical properties such as second harmonic generation (SHG), fluorescence, ferroelectric and dielectric behaviors. In this tutorial review, we will mainly focus on tetrazole coordination compounds synthesized by in situhydrothermal methods. First, we will discuss the synthesis and crystal structures of these compounds. Their various properties will be mentioned and we will show the applications of tetrazole coordination compounds in organic synthesis. Finally, we will outline some expectations in this area of chemistry. The direct coordination chemistry of tetrazoles to metal ions and in situ synthesis of tetrazole through cycloaddition between organotin azide and organic cyano group will be not discussed in this review.
 Ren-Gen Xiong | Ren-Gen Xiong was born in 1961 in Jiangxi, P. R. China, and obtained his PhD in 1994 from the University of Logistical Engineering. He then pursued research, firstly at the Coordination Chemistry Institute at Nanjing University (1994–1996), then at the Department of Chemistry at Puerto Rico University (1996–1997) and finally at the Chemistry Department, Brandeis University (1997–1998). In 1999 and 2006, he was a visiting scholar at Boston College and Kyoto University as well as Hokkaido University. Since then he has been a Professor at the University of Nanjing. He has now been appointed as the head of the Ordered Matter Science Research Center, SEU. His research interests mainly cover molecular-electronics based on homochiral compounds. |
1 Introduction
Just as with many scientific discoveries, the preparation of the first tetrazole complex by the Swedish chemist Bladin in 1885 was accidental.1 Since this discovery, interest in this area of chemistry has remained strong over the last one hundred years and to date, studies on the synthesis and applications of tetrazoles remain very popular.2 This is quite unsurprising in view of the functional group's role in coordination chemistry as a ligand with various coordination modes,3 in medicinal chemistry as a metabolically stable surrogate for a carboxylic acid group,4 and in various materials science applications, including specialty explosives.5While Bladin and co-workers established the first synthetic route to tetrazoles, the most widely used method of preparation is [2+3] cycloaddition of an azide to a nitrile. The addition of hydrazoic acid to the cyanide group resulting in the formation of 5-substituted tetrazole derivatives was first reported by Hantzsch and Vagt6 and led to the birth of [2+3] cycloaddition as the protocol of choice for the synthesis of tetrazoles. However, these traditional cycloaddition reactions have many drawbacks; namely the need to employ expensive and toxic metal–organic azide complexes such as tin or silicon organic azides, the application of highly moisture-sensitive reaction conditions, or the use of hydrazoic acid, which is extremely toxic, volatile, and explosive. In addition, all of the known methods involve the use of organic solvents.
In 2001, Sharpless and co-workers described a safe, convenient, and environmentally friendly synthetic route to 5-substituted 1H-tetrazoles, which can be accomplished with water as a solvent and zinc salts as catalyst (Scheme 1).7 In later works, Sharpless proposed a mechanism for the tetrazole formation reaction that involved azide addition to the nitrile functionality and the coordination role of the Lewis acid catalyst (Scheme 2).8 However, the postulate was based solely on theoretical calculations with no experimental evidence.
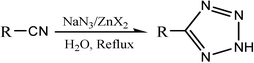 | ||
| Scheme 1 | ||
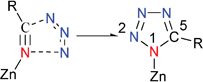 | ||
| Scheme 2 | ||
Works in our group have found that single crystals of coordination polymers can often be generated under hydrothermal conditions through in situ synthesis. In 2002, as part of an ongoing program on hydrothermal reactions, we repeated the Demko–Sharpless reaction under hydrothermal conditions and successfully isolated the intermediates of these conditions. This has lead to the establishing of a number of methods for the synthesis of tetrazoles and coordination compounds containing this ligand that has provided not only strong mechanism insight into the Demko–Sharpless reaction but also new complexes with technologically useful properties and applications.
In situ ligand synthesis, as a new approach in crystal engineering of metal coordination complexes, was first proposed by Champness and Schroder in 1997.9 In comparison with the direct tetrazole-synthesis method using tetrazole precursor derivatives (usually a cyano group) as ligands, the in situ hydrothermal was shown to initiate [2+3] cycloaddition of azides with nitriles. The advantage of the in situ method (meaning “in the original position” in Latin), making all the reactions take place just in one step from the reactants, was of course the ability to assemble the desired product in one step, as oppose to the direct method which required the use of more than one step. In addition, the in situ method was shown to be: (1) highly efficient in that there was no need for ligand synthesis; (2) able to ensure the sufficient growth of large single crystals; and (3) environmentally friendly.10Hydro(solvo)thermal reactions, which are well established strategies for the synthesis of zeolites and typically carried out in sealed conditions and in the temperature range 120–200 °C under autogenous pressure (generally 10–30 atm), are known to facilitate the self-assembly of the product from soluble precursors. The combination of hydrothermal methods with in situ synthesis has demonstrated increasing success in providing alternative pathways to crystalline complexes that otherwise are difficult to obtain by normal direct synthetic methods.
In this review, we will mainly focus on tetrazole coordination compounds synthesized by in situhydrothermal methods. First, we will discuss the synthesis and crystal structures of these compounds. Their various properties will be mentioned and we will show the applications of tetrazole coordination compounds in organic synthesis. Finally, we will outline some expectations in this area of chemistry. The direct coordination chemistry of tetrazoles to metal ions and in situ synthesis of tetrazoles through cycloaddition of organotin azides with organocyanides will not be covered in this review.
2 Tetrazole coordination complexes (TCC)
According to the mechanism proposed by Sharpless and co-workers, the intermediates in the [2+3] cycloaddition of an azide anion to a nitrile group should involve a 1-metal-coordination compound. However the reaction mechanism seems much more complicated based on subsequent works by us and others. Most of the coordination compounds, which were synthesized by the in situhydrothermal method, were found not only to possess the 1-coordination mode but also several other modes. Thus, before we describe the structures of these coordination compounds, we will first discuss their coordination modes.2.1 Coordination modes
Putting aside the mechanism of the [2+3] cycloaddition and in situhydrothermal method, the tetrazole ligand has been shown to be able to participate in at least nine distinct types of coordination modes with metal ions in the construction of metal–organic frameworks (Scheme 3). Tetrazoles have attracted increasing attention in recent years in coordination chemistry due to the excellent coordination ability of the four nitrogen atoms of the functional group to act as either a multidentate or a bridging building block in supramolecular assemblies. Thus, it is not difficult to find numerous examples in the literature of tetrazole coordination compounds with these nine coordination modes.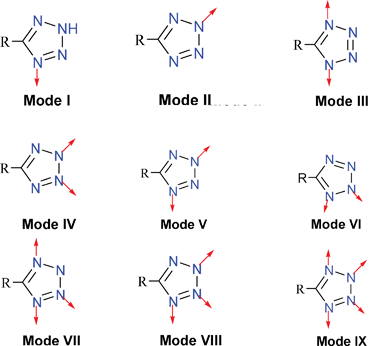 | ||
| Scheme 3 | ||
2.2 Pyridyl-tetrazole (PTZ) coordination compounds
There are a myriad studies in the literature of tetrazole coordination compounds containing the pyridyl functionality. They can be prepared from the corresponding cyanopyridine, NaN3, and MX2. Due to the donor–acceptor system between the pyridine and cyano groups, the azide anion can easily attack the N atom of the cyano group. This reactivity means that the reaction can be carried out at room temperature.11 In this review, we will mainly focus on complexes synthesized under hydrothermal conditions.[Zn(OH)(3-PTZ)] (1) (3-PTZ = 5-(3-pyridyl)tetrazolate) was prepared by reaction of 3-cyanopyridine with NaN3 and ZnCl2 under hydrothermal condition at 160 °C.12 This complex is a two-dimensional (2D) polymer (Fig. 1). It should be emphasized that the 3-PTZ ligand acts as a bidentate bridge through the 2,3 N atoms of the tetrazole (Mode IV). Its coordination mode does not agree with the mechanism proposed by Sharpless and co-workers. Using the same reactants as for 1 but at a lower temperature (105 °C) afforded [(3-PTZ)2Zn] (2).13 The solid structure of complex 2 revealed the Zn atom to coordinate only to four of the N atoms from the tetrazoles of the 3-PTZ ligands (Mode III). This results in the formation of a 3D diamondoid framework. Similarly, [CdN3(3-PTZ)] (3) was prepared in the same manner as that for 1 but with CdCl2 to replace ZnCl2.12 However, 3 was shown to contain azide ligands and a 3D polymer structure in which these ligands adopted two distinct bridging modes based on X-ray analysis. The complex structure is perhaps best understood by considering zig-zagging chains of cadmium atoms extending in the c direction (Fig. 2). The two unique types of azide ions alternate as bridging ligands along the chain. The complex also contains one tetrazole–metal coordination in Mode VI (Scheme 4).
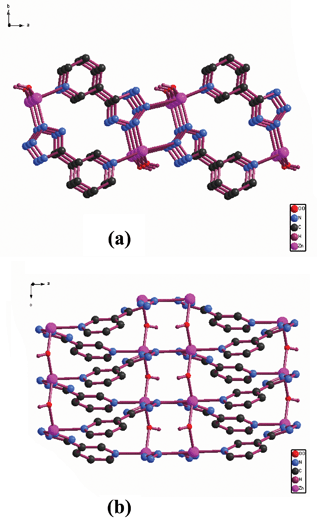 | ||
| Fig. 1 2D structure of 1. (a) A view almost normal to the axis of the two fold helices that extend in the c direction. (b) A perspective view of the sheet structure from a direction parallel to the c axis. | ||
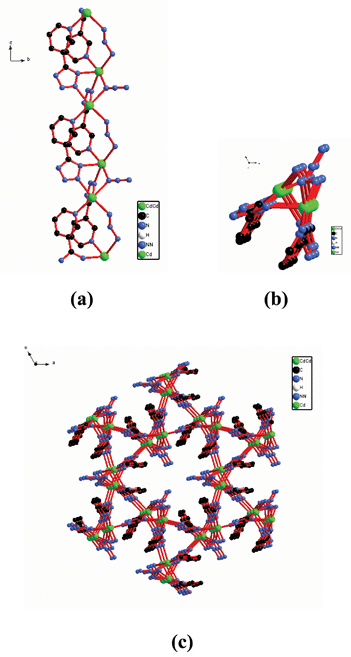 | ||
| Fig. 2 3D structure of 3. (a) Part of the zig-zag chain of Cd atoms bridged by azide ions and 3-PTZ ligands. (b) The zig-zag chains viewed down the c axis. (c) 3D structure of 2 viewed down the c axis. The interchain connections are almost normal to the c axis. Only one orientation for each disordered azide ion is shown. | ||
 | ||
| Scheme 4 | ||
[ZnCl(4-PTZ)](4) was prepared in the same manner as that for 1 at 160 °C but with 4-cyanopyridine to replace 3-cyanopyridine.12 Interestingly, with this small change, a different coordination environment was obtained. Compound 4 has a 3D network structure (Fig. 3a). This complex coordination polymer is perhaps best understood in terms of [ZnCl(4-PTZ)] strips that extend in the c direction, that possess two distinct kinds of tetrazole coordination modes (Modes III and V). Inspection of Fig. 3b reveals large channels with an approximate hexagonal cross section. These intraframework voids are occupied by a second identical network. The two networks are represented in Fig. 3c. Simply changing the reaction temperature from 160 to 105 °C was found to give [(4-PTZ)Zn(OH)(H2O)] (5).13 The solid state structure of compound 5 showed that the Zn atom not only coordinates to two atoms from the pyridyl ring and tetrazole of the 4-PTZ ligand (Mode II), but also binds to two hydroxy groups that are presumably formed in situ from water. Thus, compound 5 shows a two-dimensional layered structure with an intercalated water molecule through hydrogen bonding between the two layers. This result is unexpected, probably suggesting that water is also a reactant. [Cd3(OH)2Cl2(4-PTZ)2] (6) was obtained by the treatment of CdCl2 with 4-cyanopyridine in the presence of NaN3 and water under hydrothermal conditions at 110 °C.14 The structure incorporates not only the anticipated PTZ ligand which acts as a tridentate bridging ligand but also hydroxide and chloride ions. For each Cd, the transchloride and transhydroxide ions form an approximate square planar arrangement while the remaining trans positions are occupied by N atoms from the two 4-PTZ ligands (Mode IV) (Scheme 5).
![3D interpenetrating structure of 4. (a) [ZnCl(4-PTZ)] strips extending in the c direction. (b) A view down the c axis of 4 showing the connections between strips. The central strip is highlighted with red spheres. (c) A space-filling model showing the interpenetration of the independent networks within the crystal.](/image/article/2008/CS/b616738c/b616738c-f3.gif) | ||
| Fig. 3 3D interpenetrating structure of 4. (a) [ZnCl(4-PTZ)] strips extending in the c direction. (b) A view down the c axis of 4 showing the connections between strips. The central strip is highlighted with red spheres. (c) A space-filling model showing the interpenetration of the independent networks within the crystal. | ||
 | ||
| Scheme 5 | ||
[(2-PTZ)2Zn(H2O)2] (7) was prepared by hydrothermal reaction of 2-cyanopyridine, NaN3, and ZnCl2 at 105 °C (Scheme 6).13 The local coordination geometry around the Zn center can be best described as a slightly distorted octahedron with four equatorial nitrogen atoms from two 2-PTZ ligands (Mode I) and two apical water molecules, resulting in the formation of a monomeric Zn complex.
 | ||
| Scheme 6 | ||
[(2-NH2-5-PTZ)2Zn] (8) was prepared under hydrothermal conditions by reacting 2-amino-5-cyanopyridine with ZnCl2 in the presence of NaN3 (Scheme 7).15 The three-dimensional diamondoid polymeric structure of 8 was determined by X-ray crystallography (Fig. 4). The Zn(II) center is bonded to the four N atoms of the four 2-NH2-5-PTZ ligands (Mode III). It is worth noting that the amino and pyridyl groups present in 8 are uncoordinated to the Zn atom.
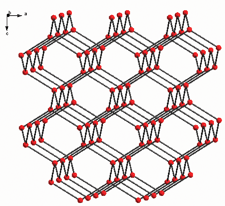 | ||
| Fig. 4 3D diamondoid-like network (the ligands between Zn atom are simplified as bonds). | ||
 | ||
| Scheme 7 | ||
[(3-PTZ)Ag] (9) was prepared under very different conditions.16 In view of the fact that Ag(I) can be easily reduced in the in situ [2+3] tetrazole reaction system, the expected product cannot be obtained by directly reacting the silver salt with other reactants. A solution to this synthetic problem is the use of a Ag+ coordination polymer with cyanopyridine to replace AgNO3 as the Lewis acid for the in situ [2+3] cycloaddition reaction of the cyano and azide groups in the presence of water (Scheme 8). This gave the anticipated Ag tetrazole coordination polymer 9. The local coordination geometry around each Ag center is a slightly distorted tetrahedron defined by four N atoms from three different tetrazole(Mode VIII) and one pyridyl groups. Thus, each 3-pyridyltetrazole acts as a tetradentate ligand coordination to four different silver centers leading to the formation of the 2D layered network.
 | ||
| Scheme 8 | ||
2.2 Other simple tetrazole coordination compounds
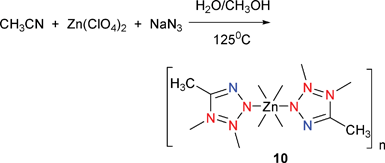
[Zn-(CH3CN4)2](H2O)4 (10) was prepared by reaction of Zn(ClO4)2 and MeCN under hydrothermal conditions at a temperature of 125 °C (Scheme 9).17 The 5-methyltetrazole ligand in 10 acts as a tridentate bridging ligand that is Mode VIII coordinated to the Zn2+ ions. The local coordination environment around the Zn center forms a perfect octahedron. The network of 10 has the topology of a diamond net with a diamond subunit as the connecting node that can be called a super-diamondlike framework (Fig. 5). To our knowledge, 10 is the first example of such a diamondlike network with a diamond subunit as the connecting node.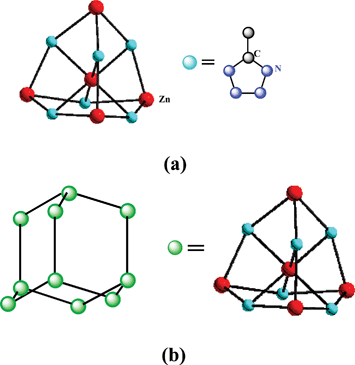 | ||
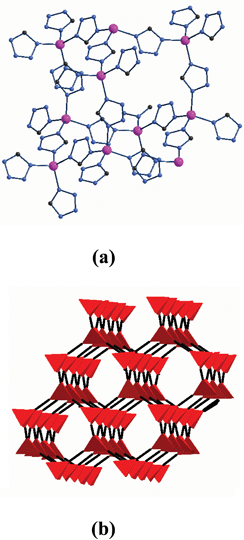
| Fig. 5 (a) Zn lying in a tetrahedron center composed of other Zn atoms as four corners. (b) Diamond-like net representation of 10 with a supertetrahedron as the connecting node. | ||
 | ||
| Scheme 9 | ||
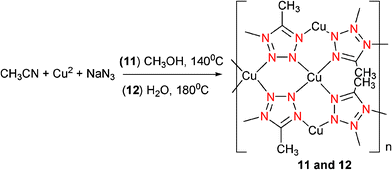
Under solvothermal conditions at 140 °C, reaction of equimolar NaN3 and Cu(NO3)2 in acetonitrile–methanol afforded pale-green crystals of [Cu(Mtta)](0.17H2O) (11) (Mtta = 5-methyltetrazolate), as shown in Scheme 10.18 In contrast, the analogous reaction in acetonitrile–water gave its pseudopolymorph [Cu(Mtta)] (12) (Scheme 10).18 The structures of both complexes consist of a 3D neutral network based on tetraconnected copper centers that are linked through µ4-tetrazolate anions (Mode IX). Interestingly, the different dihedral angles and distorted direction of tetrazolate rings in the two complexes result in two different nanoporous structures. Complex 11 contains a large hexagonal 1D channel running along the c axis. In contrast, complex 12 contains a square 1D channel running along the b axis. An interesting phenomenon is that complex 12 without guest water molecules was produced from the hydrothermal reaction, whereas complex 11 contains guest water molecules presumably from the solvothermal reaction. This might be due to the structural differences between the two complexes. The hexagonal channel in complex 11 is large enough to accommodate a water molecule, whereas the square 1D channel in 12 is too small to accept such guest molecules.
 | ||
| Scheme 10 | ||
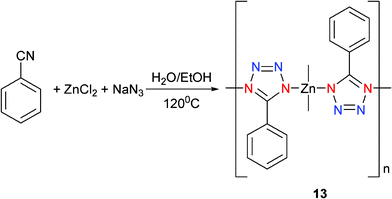
[(5-Phenyltetrazole)2Zn] (13) was furnished from in situhydrothermal reaction of cyanobenzene with ZnCl2 in the presence of NaN3 and shown to possess a 3D polymeric structure on the basis of X-ray analysis (Scheme 11).15 Each of the Zn centers was revealed to have the same four coordinates and a slightly distorted tetrahedron. The 5-phenyltetrazolato ligand was found to act as a bidentate bridging linker that connected two Zn centers together (Mode III) and the formation of a three-dimensional diamondoid network (Fig. 6).
 | ||
| Fig. 6 (a) A diamondoid-like net representation of 13 in which each 10-Zn center is composed of one diamond node, and the phenyl groups on the tetrazole ring are omitted for clarity. (b) A simplified diamond net representation of 13 in which the highlighted areas represent Zn tetrahedra and the long lines stand for 5-phenyltetrazolato ligands. | ||
 | ||
| Scheme 11 | ||
[Zn(4-MPTZ)2] (4-MPTZ = 4-methylphenyltetrazole) (14) was prepared by reaction of Zn(NO3)2 and NaN3 with 4-methylbenzonitrile at 125 °C (Scheme 12).17 The 4-MPTZ ligands in 14 act as bidentate bridging ligands that are connected to the Zn2+ ions in the Mode III and Mode IV coordination modes. This leads to the formation of a 2D network containing a hexagonal net with a dimer unit as the connecting node.
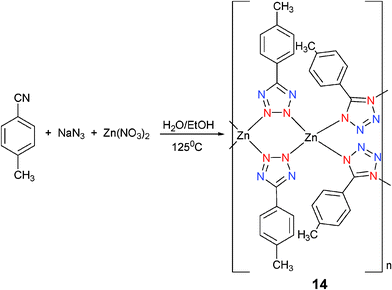 | ||
| Scheme 12 | ||
2.3 Tetrazole coordination compounds obtained from dicyano reactants
Thus far, all the complexes mentioned in the above sections are monotetrazole coordination compounds derived from monocyano precursors. The main purpose of these studies was to examine the scope and generality of the Demko–Sharpless reaction. From this point on, we will discuss recent advances in the chemistry and applications of coordination complexes containing ligand systems with more than one tetrazole moiety, an area of research that has become increasingly popular due to the presence of the four nitrogen atoms inherent in this class of compounds to serve as either a multidentate or a bridging building block in supramolecular assemblies.[Zn(2,2′-bpy)(BTZ)(H2O)](H2O)2 (bpy = bipyridine, BTZ = bistetrazole) (15) was prepared by reaction of ZnCl2 with NaN3 and malononitrile in the presence of 2,2′-bpy in water at 160 °C (Scheme 13).19 The local coordination geometry around each zinc atom can be best describe as a slightly distorted octahedron formed by five nitrogen atoms of one 2,2′-bpy, two BTZ ligands (Mode I and III) and an oxygen atom from a water molecule. It is worth noting that strong H-bonds have been found between the nitrogen atoms of the bistetrazole ligand and uncoordinated water molecules and this results in the formation of a three-dimensional network.
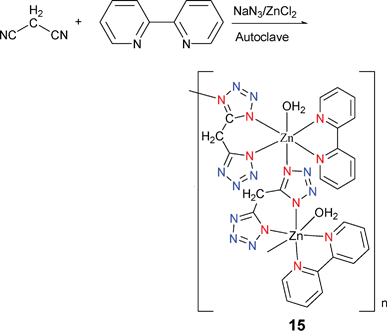 | ||
| Scheme 13 | ||
[Zn(4,5-DCIM)(Phen)(H2O)]n (16) was formed by the hydrothermal reaction of ZnCl2 with (4,5-dicyano)imidazole (DCIM), NaN3, and 1,10-phenanthroline (Phen) in water (Scheme 14).20 In complex 16, the local coordination around the zinc atom can be best described as a distorted octahedron formed by six nitrogen atoms in which two are from one phen group and four are from two 4,5-DTIM ligands (Mode I and III). Each DTIM ligand bridges two zinc atoms to result in the formation of a 1D chain polymer. It is also interesting to note that strong hydrogen bonds have been found between the tetrazole ring of 4,5-DTIM and the uncoordinated water molecules. This results in the formation of a 3D network.
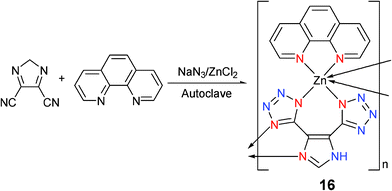 | ||
| Scheme 14 | ||
[(trans-1,2-DCEL)Na2(H2O)5] (17) was obtained by reaction of trans-1,2-dicyanoethylene (DCEL) with ZnCl2 in the presence of NaN3 and NaOH under hydrothermal conditions (Scheme 15).21 Unexpectedly, the metal involved in coordination is not Zn but Na. Compound 17 represents the first sodium tetrazole compound with a 1D chain structural coordination polymer. Strong hydrogen bonds are formed between water and the nitrogen atom of the tetrazole rings, and π–π stacking effects of the adjacent tetrazole rings result in the formation of a 3D network. It contains Na–N coordination in modes I and VI.
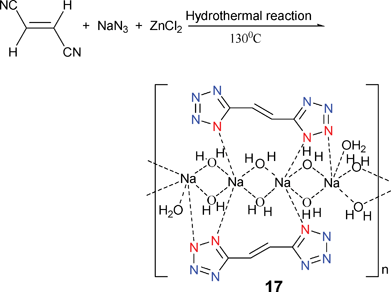 | ||
| Scheme 15 | ||
Not all the dicyano precursors can convert to bis-tetrazole coordination compounds. It is possible for a dicyano compound to undergo reaction and for only one of the cyano groups to be converted to a tetrazole group. Here we have two such examples; the first is monoaqua-4-tetrazolylbenzenecarboxylate cadmium(II) [(Te-Ph-CA)Cd(H2O)] (18) which was prepared by hydrothermal treatment of CdCl2 with 1,4-dicyanobenzene, NaN3 and water at 160 °C (Scheme 16).22 Analysis of 18 found it to contain only a mono-tetrazole group while the fate of the second cyano group was hydrolysis leading to the formation of a carboxylate group. This compound has a 3D polymeric structure. The Cd center is bonded to three O atoms from the carboxylate group and a water molecule, as well as to three N atoms from three different tetrazole moieties (Mode VII). Its three dimensionally condensed polymeric structures leads to a significant enhancement of fluorescent intensity.
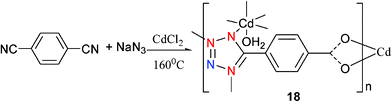 | ||
| Scheme 16 | ||
The second example is [Cu(cptz)]n (19) (Hcptz = 5-(4-cyanophenyl)tetrazole) which was prepared by hydrothermal reaction of CuCl2 with NaN3 and 1,4-dicyanobenzene in water–ethanol (v/v 1 ∶ 1) (Scheme 17).23 Again, only one of the two cyano groups of 1,4-dicyanobenzene undergoes in situ [2 + 3] cycloaddition to give the cptz ligand of 19, coupled with reduction of Cu(II) to Cu(I). Each Cu atom is bound to one cyano and three tetrazolyl groups from four different cptz ligands (Mode VII) to adopt a slightly distorted tetrahedral geometry. The structure of 19 possesses a 3D network and emits very strong yellow luminescence at room temperature.
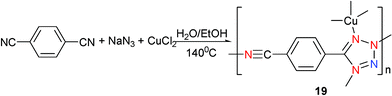 | ||
| Scheme 17 | ||
2.4 Tetrazole coordination complexes prepared from chiral precursors
The synthesis of noncentrosymmetric metal–organic coordination polymers (MOCPs) and organic solids (OS) using crystal engineering strategies has come under increasing scrutiny in recent years.24 Indeed, we and others have demonstrated that bulky noncentrosymmetric MOCP and OS are essential for a number of technologically important properties. With chiral and noncentrosymmetric ligands, coordination complexes tend to crystallize in an acentric space group. This has led to the introduction of chiral substrates as precursors for the synthesis of novel tetrazole coordination complexes. By combining chiral centers with the excellent coordination ability of tetrazoles, it can be envisaged that such a strategy would hold promise for the construction of new complexes with a wide range of properties and potential applications.Reaction of (S)-3-CNPHA [(S)-3-cyanophenylalanine] and NaN3 with either ZnCl2 or CdCl2 under hydrothermal conditions gave the corresponding coordination polymers mono[(S)-5-(3-tetrazoyl)phenylalaninato]-zinc(II) [Zn((S)-TPA)] (20) and mono[(S)-5-(3-tetrazoyl)phenylalaninato]cadmium(II) monoaqua [Cd((S)-TPA](H2O)] (21) (Scheme 18).25 Crystal structure determination of 20 reveals a complex chiral 3D coordination polymer that contains one unique ligand and one zinc center. It is worth noting that 20 is the first structurally characterized homochiral-tetrazole-based metal coordination polymer. Interestingly, compound 21 is essentially isostructural with 20. A minor difference between compounds 20 and 21 is the inclusion of water within the intranetwork voids of 21.
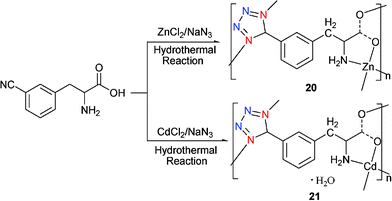 | ||
| Scheme 18 | ||
Reaction of CdCl2 with NaN3, racemic {trans-2,3-dihydro-2-(4′-pyridyl)-3-(3′-cyanophenyl)benz[e]indole} (L) in water under hydrothermal conditions at 160 °C for 4 days gave [Cd(L–N3)2(H2O)2]n (22) (Scheme 19).26 The Cd(II) metal center is bonded to the four N atoms of the L ligands (two from the pyridyl rings and two from the tetrazole rings in Mode II), and to two water molecules through the oxygen atoms. It is interesting to note that the adjacent collinear 1D chains align in the same direction with respect to each other which prevents cancellation of the dipolar moment. As expected, compound 22 exhibits excellent physical properties (this will be covered in the next section). In contrast, reaction of L with ZnCl2 under the same conditions at 140 °C afforded the dehydrogenated L coordination complex [(CN4–C6H4–C12H7NC5H4N)2Zn](1.5H2O) (23).15 Compound 23 is a 2-dimensional coordination polymer that contains a local coordination environment around the Zn center that can be best described as a slightly distorted tetrahedron in which each Zn atom is coordinated by four N donors from four different ligands (Mode II).
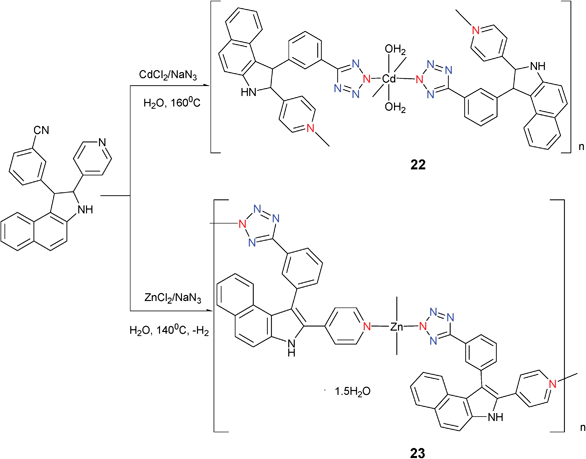 | ||
| Scheme 19 | ||
Hydrothermal reaction of N-(4-cyanobenzyl)-(S)-proline with CdCl2 and excess NaN3 at 110 °C for 2 days furnishes complex 24 (Scheme 20).27 X-Ray single crystal determination of 24 reveals that the Cd center sits in a slightly distorted octahedron that is composed of three N atoms from two tetrazolyl groups and a pyrrolidinyl group, and a terminal Cl atom as well as two O atoms from two carboxylate groups (Fig. 7). Each N-(4-(1H-tetrazol-5-yl)benzyl)proline (H-TBP) ligand acts as a pentadentate bridging linker that connects five Cd atoms together to give a 3D framework. Notably, an O atom from the carboxylate group of the H-TBP ligand binds to a Cd center that is also linked to a N atom from the pyrrolidinyl ring to give a stable five-membered ring. Fig. 8 shows a simplified net representation of 24 along the b-axis that consists of three types of nets: (a) a fifteen-membered ring formed from six N atoms, two C atoms, two O atom, and four Cd atoms; (b) a five-membered ring formed from two N atoms, an O atom, and two Cd atoms; and (c) two different three-membered rings, the first arising from a tetrazolyl group acting as a µ2-linker and the second arising from chelation of the five membered proline ring. Complex 24 crystallizes in the noncentric space group (Cc), which belongs to the polar point group (Cs).
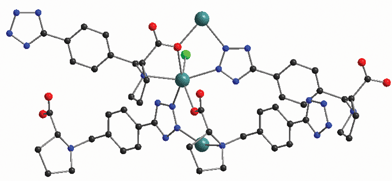 | ||
| Fig. 7 Asymmetric unit representation of 24 with a slightly distorted octahedral Cd coordination geometry. | ||
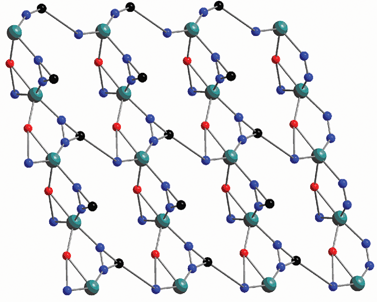 | ||
| Fig. 8 A simplified 2D network along the b-axis in which the straight lines, and triangular C–N–N and Cd–O–N units represent the benzyl, tetrazolyl, and pyrrolinyl groups, respectively. | ||
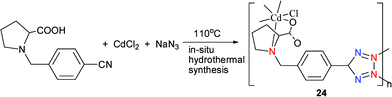 | ||
| Scheme 20 | ||
There are numerous examples in the literature of tetrazole coordination compounds prepared by [2+3] cycloaddition using other methods such as solvothermal conditions, mixing reactants with stirring at room temperature or refluxing. In our work, we used an indirect method to obtain complex 9. Under solvothermal conditions, Li and coworkers reported that the use of [2+3] cycloaddition for the preparation of Ag-tetrazole coordination complexes [Ag(Mtta)]n (Mtta = 5-methyltetrazolate) (25) (Mode VII) and [Ag(Etta)]n (Etta = 5-ethyltetrazolate) (26) could be accomplished (Mode IX) (Scheme 21).28 It should be noted, however, that the hydrothermal method is the one that most resembles the Demko–Sharpless reaction.
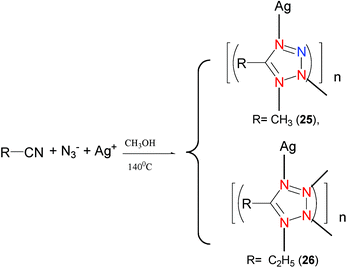 | ||
| Scheme 21 | ||
3 On the mechanism of the Demko–Sharpless reaction
On the mechanism of the Demko–Sharpless reaction, Sharpless and co-workers suggested that the intermediates of this reaction obeyed N–metal coordination Mode I. According to calculations reported by Sharpless,8b initial coordination to the nitrile functionality has a lower energy barrier. Indeed, the following examples demonstrate that the 1-coordination is most favorable. Under solvothermal and hydrothermal conditions, direct reaction of 4-HPTZ with a metal salt was recently reported to give the complexes [Cd(4-PTZ)2(H2O)2]14 (27) and [Zn(HCN4)2]17 (28) (Scheme 22). X-Ray crystal structure analysis of 27 and 28 showed ligand coordination to the metals only at the 1-position. However, it seems much more complicated when the hydrothermal method was used to mimic the Demko–Sharpless reaction environment and to trap the intermediates.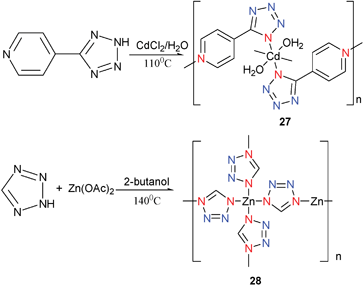 | ||
| Scheme 22 | ||
Even if we assumed that all the N–metal coordination complexes containing 1-coordination (Mode I, III, V, VI, VII, VIII and IX) followed the proposed mechanism, there are still five compounds that do not follow this mechanism among all the mentioned 24 complexes prepared by the hydrothermal method. It is possible that the syntheses of these complexes follow the proposed mechanism but under the hydrothermal conditions, the intermediates are destroyed and result in other N atom coordination modes. However, the five exceptions imply that there are possibly other competing mechanisms in the Demko–Sharpless reaction.
Here we have another interesting example, {[(4-POTZ)2(H2O)3(UO2)](H2O)}29 (29) which was obtained by treatment of UO2(NO3)2 with 4-cyanopyridine oxide and NaN3 using H2O and methanol as solvent under hydrothermal conditions at 90 °C. Surprisingly, while the POTZ (pyridine oxide tetrazole) ligand was synthesized in the reaction, the coordination mode of the U ions with the ligand differed significantly from other examples obtained by the Demko–Sharpless tetrazole synthesis reaction. In this compound, only the O atoms were found to coordinate with the U ions, as shown in Scheme 23. In addition, Demko–Sharpless reaction of UO2(NO3)2 with 4-cyanopyridine oxide and NaN3 occurs only once, unlike complex 17, where the metal involved in coordination is Na and not Zn, which was surmised to be possibly due to the initial Zn-containing product reacting further with Na to give the Na-tetrazole coordination complex.
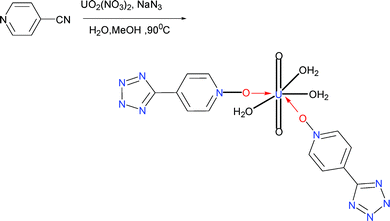 | ||
| Scheme 23 | ||
By comparing these four examples with the five complexes mentioned at the beginning of this section that do not follow the proposed Sharpless mechanism, we can conclude that: (1) the intermediates may be not stable due to the reaction environment and natures of the reactants; (2) there may be other possible competing mechanisms in the Demko–Sharpless reaction; (3) due to the excellent coordination ability of tetrazoles, there is still a degree of uncertainty regarding whether complexes obtained can be fully considered as intermediates of [2+3] cycloaddition reaction. Therefore, our work can only be considered as a reference for the study of the Demko–Sharpless reaction. It remains a challenge to develop studies that will allow for a better understanding of the mechanism of this reaction.
Finally, regarding the above-mentioned products obtained through in situ [2+3] cycloaddition reactions between a Lewis acid and an organic nitrile in the presence of NaN3, we have summarized the TCC composition changes with anions and metal ions in the presence of a Lewis acid and an organic nitrile in Table 1. From Table 1 we can draw some important conclusions for the design and construction of TCCs as follows: (1) if L is bent (not linear), the TCCs most likely contain X–, OH– and N3– while if the cation radius (such as Cd2+) is large, the apical position around the central metal ion may be occupied by water; (2) if L contains an ortho-coordinating atom (easily forming a 5-membered ring with an N atom of the tetrazole), the TCCs most likely are zero-dimensional molecules; (3) if the cation in the Lewis acid contains Cu2+, the product TCC mostly contains the reduced Cu+; (4) if a coordination complex with an organic cyano group is used as the cyano starting material, the TCC may contain another metal anion resulting in the formation of a heterometal tetrazole. However, detailed rules should be revised after more structures of TCCs are determined.
| Lewis acids | TCC Composition | Nitrile starting material | Cases | Reference |
|---|---|---|---|---|
| a In these cases, TTCs were obtained from dicyano reactants, and needed assisting ligands. b In these cases, the copper(II) was easily reduced to copper(I) under hydrothermal conditions. c The Ag-complex with an organic nitrile is used as the nitrile starting material. d The N atoms of the tetrazole failed to coordinate to the U atom. | ||||
| MX2 (M = Zn2+, Cd2+ X = Cl–, Br–, ClO4–, NO3–Y = OH–, N3– L = tetrazole ligands) | MXL | Organic nitrile | 4, 24 | 12, 27 |
| MXL (H2O) | Organic nitrile | 30, 32, 33 | 46 | |
| [ML2](H2O)0–1.5 | Organic nitrile | 2, 8, 10, 13, 14, 23 | 13, 15, 17 | |
| ML2(H2O)2 | Organic nitrile | 7, 22, 27 | 13, 26, 14 | |
| MYL | Organic nitrile | 1, 3 | 12 | |
| MYL(H2O) | Organic nitrile | 5 | 13 | |
| ML | Organic nitrile | 20, 21, 28 | 25, 17 | |
| ML(H2O) | Organic nitrile | 15,a16,a18 | 19, 20, 22 | |
| Others | Organic nitrile | [Cd3(OH)2Cl2L2] (6) | 14 | |
| Organic nitrile | Na2(H2O)5L] (17) | 21 | ||
| CuII X2 (X = Cl–, NO3–) | CuI L | Organic nitrile | 11,b12,b19b | 18, 23 |
| Ag+ | AgL | Organic nitrile | 25, 26 | 28 |
| Coordination complexc | 9 | 16 | ||
| UO2(NO3)2 | (UO2)L2(H2O)3 | Organic nitrile | 29 d | 29 |
4 Properties and applications
An attractive feature of highly stable tetrazole coordination polymers is their suitability for various functional applications. Functionalities can be readily introduced onto the metallosupramolecular structure by employing functional tetrazoles in the assembly processes. On the other hand, tetrazole coordination polymers containing metal ions are generally more sensitive and responsive to electro- and photochemical stimuli compared to metal-free tetrazole molecules.Therefore, the employment of highly stable tetrazole coordination polymers may open up new opportunities to develop novel molecular switches and devices. In Gaponik's review on metal–tetrazole complexes there are detailed descriptions of applications of metal–tetrazole complexes.3 In the following section we mainly focus on the properties and applications of the complexes mentioned in this review.
4.1 Fluorescent sensing
Fluorescence is a luminescence that occurs when a molecule or quantum dot relaxes to its ground state after being electronically excited. There are many natural and synthetic compounds (liquid or solid-state) that exhibit fluorescence, and they have a number of applications: lighting, biochemistry, medicine, gemology, mineralogy, geology and forensics, etc. Fluorescence efficiency can be correlated with many structural features of chemicals including π–π* and n–π* transitions, structural rigidity, noncovalent interactions (e.g.hydrogen bonds, π–π interactions, and hydrophilic and hydrophobic interactions), interior intermolecular energy transfer, and photoinduced electron transfer.30 In this context, tetrazoles may be considered to be excellent fluorescent compounds due to the aromaticity of these compounds. Mixed inorganic–organic hybrid coordination polymers have also been investigated for fluorescence properties. Thus, tetrazole coordination polymers have potential use as fluorescent solid-state materials.Complex 6 shows a strong emission peak at 390 nm that can be assigned to an intraligand fluorescent emission since a weak similar emission at 353 nm is observed for the ligand (Fig. 9). A relatively weak peak at 470 nm is also observed for 6. Because of the longer emission lifetime of 6 at 470 nm, this peak may be tentatively assigned to phosphorescence. On the other hand, the Cd–OH–Cl sheet of 6 may be responsible for the phosphorescent emission since complex 27 displays only a strong emission peak at 380 nm. To our knowledge, a metal coordination polymer demonstrating both fluorescent and phosphorescent emission is unknown. Thus complex 6 is an attractive candidate for use as a blue fluorescent and phosphorescent material.
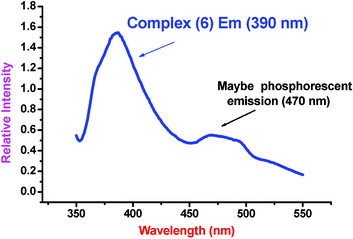 | ||
| Fig. 9 Solid state fluorescent spectrum of 6 at room temperature in which the emission at 390 nm is fluorescent while the emission at 470 nm may be phosphorescent. | ||
The solid-state fluorescence spectrum of complex 9 at room temperature shows that maximal emission peaks occur at 495 and 521 nm (Fig. 10), respectively. Complex 9 has a 2D condensed polymeric structure and this unique structure perhaps lends itself to the significant enhancement of the fluorescent intensity, which is approximately two or three times larger than that of the corresponding free ligand. The enhancement could be a result of coordination of the ligand to Ag, which increases the conformational rigidity of the ligand, thereby reducing the nonradiative decay of the intraligand excited state of 9. This situation also happens in other tetrazole coordination complexes.
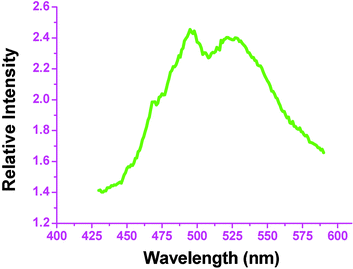 | ||
| Fig. 10 Solid state fluorescent emission spectrum of 9 at room temperature | ||
Complexes 8, 13 and 23 display strong fluorescent emissions at 415 nm (a blue fluorescent emission, Fig. 11a), 390 nm (blue fluorescent emission, Fig. 11b), and 495 and 532 nm (pale yellow-green fluorescent emission, Fig. 11c) in the solid states. The solid-state fluorescent spectra of 10 and 14 at room temperature reveal maximal emission peaks at 396 and 397 nm, respectively. The results suggest that these complexes could potentially be good blue-light emitted materials. The photoluminescent mechanism is tentatively attributed to ligand-to-ligand transitions that are in reasonable agreement with literature examples on this class of zinc coordination polymers.
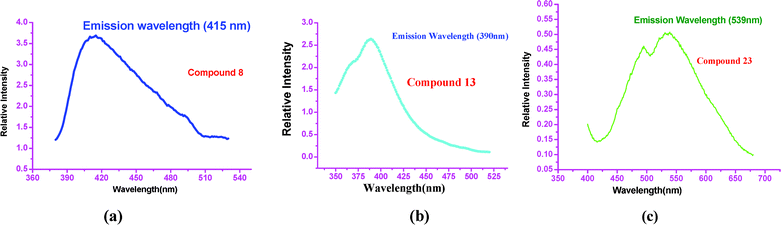 | ||
| Fig. 11 Fluorescent emission spectra, in the solid state at room temperature, of: (a) 8, (b) 13, (c) 23. | ||
Complexes 15 and 22 possess strong blue fluorescence at 410 and 470 nm. The solid state fluorescence of 16 at room temperature shows that the maximum emission peaks occur at 458 and 540 nm. This suggests that compound 16 is a good yellow emitting material. Compound 18 has a three-dimensionally condensed polymeric structure leading to significant enhancement of fluorescent intensity, 5–6 times larger than that of the free ligand. This is probably due to coordination of the ligand to Cd(II) increasing the conformational rigidity of the ligand.22 Complex 19 also emits a very strong yellow luminescence at room temperature in the solid state. A broad emission peak with a maximum at 563 nm was observed. The long emission lifetimes could be assigned to metal-to-ligand charge transfer triple excited states.
4.2 Second harmonic generation (SHG)
Recently, with the development of the new high technology and the permeation of solid physical materials, research focusing on functional coordination polymers with optical, electric, thermal and magnetic etc. properties has attracted many chemists' attention. Many solid-state physical properties, such as pyroelectricity, piezoelectricity, ferroelectricity, nonlinear optical second harmonic generation (SHG), and triboluminescence are only found in chiral or noncentrosymmetric bulk material. Of particular importance are nonlinear optical (NLO) function (especially SHG) because of its practical importance in areas such as telecommunications, optical storage and information processing.31SHG describes the capacity of a material to double the frequency of incident light. Viable SHG materials must possess the following attributes: transparency in the relevant wavelengths, ability to withstand laser irradiation, and chemical stability. Most importantly, the material in question must be crystallographically chiral or noncentrosymmetric. Of the 21 acentric crystal classes,32 only compounds found in class 432 do not posses SHG behavior.
SHG, or frequency doubling, can be defined as the conversion of a specific wavelength of light into half its original, i.e.λ1 → 1/2λ1, or with respect to frequency ω, ω1 → 2ω2.32 The first report of SHG was described by Franken and co-workers in 1961, who reported SHG on a quartz crystal using a ruby laser.33 TCCs have their own inherent advantages as NLO materials.
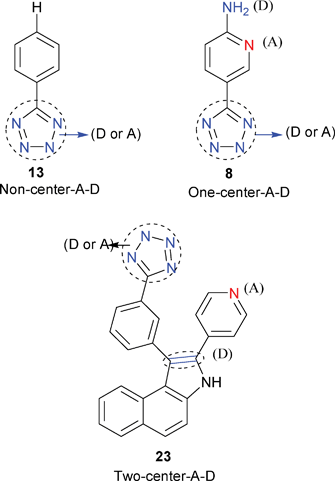
Potassium dideuteriophosphate (KDP) is a typical example of a NLO-active diamondoid network that crystallizes in a chiral space group (P212121) (Scheme 24). In this review, complexes 2, 8, 10, 13 and 14 possess diamondoid networks similar to that of KDP. Complex 2 crystallizes in an acentric space group and displays a moderate powder SHG response of ca. 4 times that of KDP. This feature is similar to that of the diamond-like network in KDP. Both 8 and 13 crystallize in a noncentrosymmetric space group. Complexes 8 and 13 display strong powder SHG efficiencies of 5 and 1 times that of urea (50 and 10 times that of KDP), respectively, presumably as a result of their KDP-like diamondoid structures. Moreover, the presence of strong donor–acceptor substituents in the free ligand in 8 allows for a high nonlinearity or substantial SHG response. Asymmetric ligands have been used to introduce electronic asymmetry (push–pull effect or one-center A–D system), an essential criterion for a SHG response. The combination of a KDP-like structure34 and an excellent push–pull effect (or one-center A–D system) of the free ligand in 8 further optimizes the performance of pure organic and inorganic NLO materials. Such hybrid inorganic–organic NLO materials have also recently been the subject of much investigation as NLO materials. This is one possible reason for the SHG response of 8 being significantly larger than that of 13 (Scheme 25).
 | ||
| Scheme 24 | ||
 | ||
| Scheme 25 | ||
Compound 23 crystallizes in a noncentrosymmetric space group (Fdd2) in the crystal class mm2, where its optical activity can occur as a specific physical effect. Complex 23 has very strong SHG responses in the powdered state; ca. 50 and 500 times of those of urea and KDP, respectively. As mentioned above, dehydrogenation in 23 results in the formation of a large conjugated system in the ligand of 23. On the other hand, the resulting ligand in 23 is highly asymmetric and can best be induced by the two-functional electronic asymmetry that is essential for second-order optical nonlinearity. Thus, the building block is an excellent two-A–D chromophore that is essential for strong SHG materials. Furthermore, the presence of two push–pull effect centers (or a two-center A–D system) (Scheme 25) is much effective in the enhancement of the dipolar moment than the presence of a one-center A–D system. To our knowledge, a one-center A-D system also leads to the noncancellation of the molecular dipolar moment for a compound with a noncentrosymmetric space group (such as in the two cases of 8 and 13 in which they all crystallized in an acentric space group and their dipolar moments could not be canceled). Evidently, the two-center A–D system results in the non-cancellation of the dipolar moment if the compound crystallizes in an acentric space group. At the same time, the dipolar moment should be larger than that of the one-center A–D system. Because SHG responses depend on charge separation or molecular dipolar moment (hyperpolarizability), the combination of a multicenter A–D system and metal–ligand coordination could be responsible for a synergistic effect that leads to a strong enhancement of the SHG response.
The same situation is found in complexes 20, 21 and 22. Complexes 20 and 21 are representative examples of homochiral inorganic–organic hybrid coordination polymer networks with SHG efficiency. Complex 22 displays strong SHG efficiency, 80 times than that of urea, which is due to the molecule bearing two chiral centers within the two donor–acceptor systems. Pyridyltetrazole (PTZ) can form an excellent D–A system. Indeed, a series of PTZ coordination compounds, such as complexes 1–4, have been reported by us to display strong SHG efficiency. Since complex 24 crystallizes in the noncentric space group (Cc), its optical property was examined. Our measurements on a powdered sample of 24 suggest that it is SHG active with an approximate response of 10 times that of KDP; this implies that 24 is a dipolar active compound.
4.3 Ferroelectric and dielectric properties
A ferroelectric material is one that can be switched rapidly between different states by means of an external electric field. Such materials may be useful in a variety of new technologies e.g. electric-optical devices, information storage, switchable NLO (nonlinear optical) devices and light modulators.35,36 In crystallography, the ferroelectric behavior requires the adoption of a space group that is associated with one of 10 polar crystal classes (C1, C2, Cs, C2ν, C3, C3ν, C4, C4ν, C6, C6ν). Furthermore, strong H-bonds are desirable. One well-known ferroelectric compound that is also second harmonic generation (SHG) active is the Fdd2 phase of KDP (KH2PO4); another is TGS (triglycine sulfate).Ferroelectricity may be formally defined as a pyroelectric material that has reversible, or ‘switchable’, polarization.30 Of course this would imply that the material must be polar, possess a permanent dipole moment, and be capable of having this moment reversed in the presence of an applied voltage. One important characteristic of ferroelectricity is electric hysteresis loop, which may be measured through a Sawyer–Tower circuit. Fig. 12 is a typical electric hysteresis loop (polarization vs. electric field) of 180° domain ferroelectricity.37
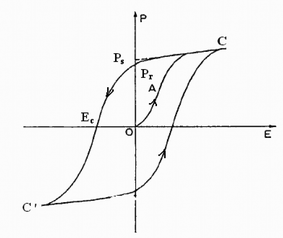 | ||
| Fig. 12 Ferroelectric hysteresis loop. Ps, spontaneous polarization. Pr, remnant polarization. Ec, coercive field. | ||
In recent years, a significant amount of effort has been placed on developing ferroelectric coordination polymers38 and organic compounds.39 However, examples of TCCs with ferroelectric properties have remained rare.
In this review, complex 22 crystallizes in the polar point groupC2v, by introducing a ligand containing two chiral centers. It exhibits moderate ferroelectric behavior, with a weak electric hysteresis loop, a ferroelectric feature with a remnant polarization (Pr) of 0.12–0.28 µC cm–2 and coercive field (Ec) of 10 KV cm–1 (Fig. 13). The saturation spontaneous polarization (Ps) of 22 is estimated to be 4.5 µC cm–2, which is comparable to that found in KH2PO4 (5.0 µC cm–2). This example provides a new facet of tetrazole coordination complexes with ferroelectric properties.
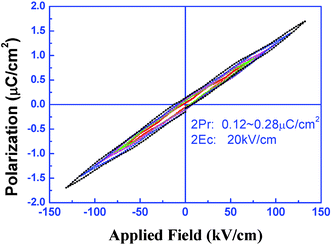 | ||
| Fig. 13 Electric hysteresis loop observed by Virtual Ground Mode in a powdered sample of 22 in the form of a pellet using an RT6000 ferroelectric tester at room temperature with the sample immersed in insulating oil. The measurement was performed using an ac pulse. | ||
The ferroelectric behavior of complex 24 was also examined in view of the fact that its point groupCs is one of the 10 polar point groups required for such properties. Fig. 14 clearly shows there is an electric hysteresis loop in 24 (a typical ferroelectric feature) with a remnant polarization (Pr) of ca. 0.38 µC cm–2 and coercive field (Ec) of ca. 2.10 KV cm–1. Saturation of the spontaneous polarization (Ps) 24 occurs at ca. 0.50 µC cm–2, which is significantly higher than that for a typical ferroelectric compound (e.g., NaKC4H4O6·4H2O, Rochelle salt; usually Ps = 0.25 µC cm–2).
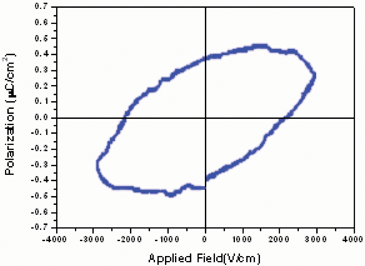 | ||
| Fig. 14 Electric hysteresis loop of a pellet obtained from a powdered sample of 24 observed by Virtual Ground Mode using an RT6000 ferroelectric tester at room temperature. | ||
It is worth noting that the high permittivity (dielectric constant [εo]) properties of ferroelectric materials have also made them important in the development of dielectric resonators and filters for microwave communication systems. A dielectric material is a substance that is a poor conductor of electricity, but an efficient supporter of electrostatic fields; such materials play an integral role in modern electronic devices. They are employed in capacitors, transducers, actuators, random access memories, and microwave filters.40
Dielectric permittivity (ε) = ε1(ω) + iε2(ω), where ε1(ω) and ε2(ω) are the real (dielectric constant) and imaginary (dielectric loss) parts. Dielectric constant (ε1) is a number relating the ability of a material to carry alternating current to the ability of vacuum to carry alternating current. In particular, when materials have different ε1(ω) and ε2(ω), they possess versatile properties and can be applied in many areas. For example, materials with high dielectric constant and low dielectric loss have been used in capacitance devices, compounds displaying high dielectric loss can be used in heating resonators, and the most interesting thing is that the materials with ultralow dielectric constant have found use in the fabrication of high performance integrated circuits.41
For complex 24, as shown in Fig. 15, a relaxation process was also observed, suggesting that dielectric loss changes with temperature at different frequencies with the peak maxima obeying the Arrhenius equation τ = τoexp(H/kT), where T = absolute temperature; k = Boltzmann's constant; H = activation energy; and τo = inverse of the frequency factor. For a Debye peak, the condition for the peak is ωτ = 1. Thus, the equation can also be re-written as: ln f = –ln(2πτ0) – H/kTp, where Tp = temperature of the peak, ω = 2πf, and f = vibration frequency.
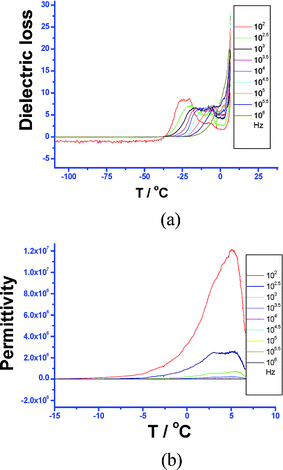 | ||
| Fig. 15 (a) Temperature dependence of the dissipation factor (or loss component) of the dielectric response at different frequencies. (b) Temperature dependence of the real part of the dielectric response of 24 at different frequencies (inset). | ||
Thus, we can estimate the activity energy H and relaxation time τo from Fig. 15a. The average H and τo are ca. 1.96 eV and 1.60 × 10–5.5 (s), respectively. This suggests that the relaxation process (peak) is probably associated with dipolar Cd–Cl bond vibration or the displacement of the proton on the tetrazolyl group. Notably, the permittivity at low frequencies reaches a maximum value (1.2 × 107, 5 °C, 100 kHz), which rapidly drops by 2 orders of magnitude at relatively high frequency (103.5 Hz); this is similar to that for perovskite-related oxide CaCu3Ti4O12 (105) which was reported to display a 1000-fold reduction.41 This discrepancy suggests the presence of a dipole relaxation at low frequencies and is in good agreement with the dielectric loss measurements; the frequency dependence of the dielectric constant ε1 at ca. 6.6 °C indicates that ε1 rapidly decreases with an increase in frequency, which further supports the presence of a dipole relaxation process while ε1 remains unchanged (εo = 38.6, estimated from the slope) at ca. –53.8 °C (Fig. 16). Interestingly, ε2 does not change with frequency.
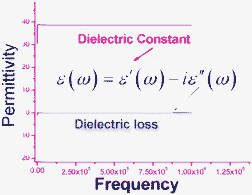 | ||
| Fig. 16 Frequency dependence of permittivity of 24 at –53.8 °C. | ||
In summary novel ferroelectric MOFs were prepared by employing cycloaddition chiral ligand synthesis under hydrothermal conditions. This class of materials provides new impetus to examine the potential applications of MOFs as ferroelectric materials.
Most of the TCCs mentioned above are stable up to up a temperature of 300 °C. The reported observed losses in weight of these complexes at low temperatures result from the thermal displacement of water molecules which does not affect the thermal stability of the dehydrated structure based on TGA analysis. Moreover, the majority of TCCs are insoluble in common organic and aqueous solvents. These two features make tetrazole coordination complexes potentially exciting materials possessing technologically desirable SHG, fluorescence and ferroelectric properties mentioned above.
4.4 Application in organic synthesis
A key application of TCC is in organic synthesis, where they are used as the starting and intermediate compounds for the preparation of valuable and practical substances. The synthesis of N-substituted tetrazoles N-alkyl-, N-aryl- and N-heteroaryltetrazoles42 from various metal tetrazolates, and as a method for introducing tetrazolyl groups into complex biologically active compounds,43 are prominent examples of can be accomplished with this synthetic strategy.At the very beginning of our research on tetrazoles, our main purpose was to gain some insight into the mechanism of the Demko–Sharpless reaction by trapping the intermediates of this functional group transformation. One consequence of these studies was the optimization of the reaction conditions for tetrazole complex synthesis. This subsequently lead to the application of the hydrothermal method in the synthesis of a precursor to Valsartan,44 which contains a tetrazole group and is a useful treatment for high blood pressure. In this synthetic approach, the intermediate (30) was furnished under hydrothermal conditions and shown to readily convert to the known Valsartan precursor (31) by fine-tuning the pH conditions, as shown in Scheme 26. This work has also provided new insights into the mechanistic role of zinc in the Demko–Sharpless reaction and a synthetic route to a new class of tetrazole compounds. As shown in Scheme 27, complexes 32 and 33 were also prepared by this study.45 However, this new method requires further optimization studies.
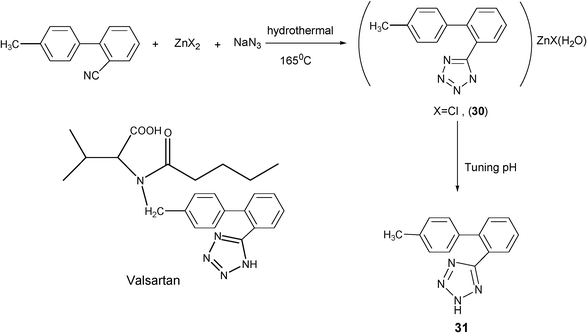 | ||
| Scheme 26 | ||
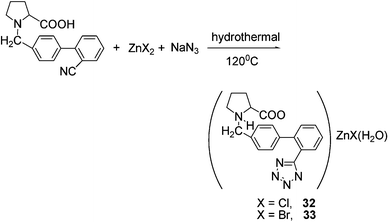 | ||
| Scheme 27 | ||
4.5 Application as porous materials
Tetrazoles possess four nitrogen atoms that make up the five-membered ring of the functional group. Thus, this ring system can serve as an excellent chelating ligand. In addition, tetrazole coordination complexes can easily form polymers due to the same four coordination sites and it is the porous structures which some of these polymer complexes can possess that has in recent years attracted the attention of chemists. More recently, a new class of ordered, three-dimensional extended solids composed of metal ions and organic linkers, known as metal–organic frameworks, has emerged as a promising storage alternative for high-pressure and liquefied hydrogen tanks, metal hydrides, and carbon-based adsorbents. Long and co-workers reported the synthesis of a porous metal–organic framework for hydrogen storage using 1,3,5-tritetrazolylbenzene as a building block.46 Among complexes mentioned in this review, complexes 3, 4, 10, and 20–22 possess open channels in their crystal structures. The porous nature of these latter complexes not only make them potentially useful materials as reversible zeolite analogues but also demonstrates the promise tetrazoles hold for the development of new porous materials.5 Summary
Due to tetrazole's various applications chemists have studied this class of compounds for more than 100 years. Numerous metal–tetrazole coordination complexes, which contain almost all known metals, have now been investigated. The complexes mentioned in this review form only a small corner of the whole tetrazole world. However, the method used to synthesis tetrazole complexes and discussed in this review, in situhydrothermal reaction, is a new route that was developed only several years ago. This method provides the advantages in the synthesis of TCCs mentioned in section 1. The hydrothermal reaction has been shown to afford a convenient synthetic route to a series of TCCs. Characterization of these intermediates has provided insight into the nature of the tetrazole compounds formed in the Demko–Sharpless reaction and has provided important clues to the mechanistic role of zinc in this reaction. This, in turn, may allow synthetic chemists to further optimize this reaction.Finally, metal–tetrazole coordination complexes have been shown to possess various useful physical and chemical properties, the nature of which have been demonstrated to depend on the nature of the metals and substituents. As a consequence, both coordination and organometallic chemists are continuing to explore the rich and diverse possibilities offered by such complexes in a wide range of applications. Although the area has progressed impressively over the past few decades, it remains in its infancy and there remain wonderful opportunities for further development. Future work can be expected to yield novel metal–tetrazole complexes such as heterometal or deuterated tetrazole complexes that may have even more fascinating properties.
Acknowledgements
We thank 973 Project (2006CB80614) and the National Natural Science Foundation of China. The former PhD students Xi-Sen Wang, Qiong Ye, Yun-Zhi Tang, Guo-Xi Wang, Yu-Mei Song, and master’s degree students Xiang Xue, Li-Zhong Wang and Ting Zhou are gratefully acknowledged for their distinct contributions. We also thank Dr Philip Wai Hong Chan for his hard work in revision of the English.References
- J. A. Bladin, Ber. Dtsch. Chem. Ges., 1885, 18, 1544 CrossRef
.
-
(a) F. R. Benson, Chem. Rev., 1947, 41, 1 CrossRef CAS
- P. N. Gaponik, S. V. Voitekhovich and O. A. Ivashkevich, Russ. Chem. Rev., 2006, 75, 507 Search PubMed
- H. Singh, A. S. Chawla, V. K. Kapoor, D. Paul and R. K. Malhotra, Prog. Med. Chem., 1980, 17, 151 Search PubMed
-
(a) V. A. Ostrovskii, M. S. Pevzner, T. P. Kofmna, M. B. Shcherbinin and I. V. Tselinskii, Targets Heterocycl. Syst., 1999, 3, 467 Search PubMed
- A. Hantzsch and A. Vagt, Justus Liebigs Ann. Chem., 1901, 314, 339 CrossRef CAS
- Z. P. Demko and K. B. Sharpless, J. Org. Chem., 2001, 66, 7945 CrossRef CAS
-
(a) F. Himo, Z. P. Demko, L. Noodleman and K. B. Sharpless, J. Am. Chem. Soc., 2002, 124, 12210 CrossRef CAS
- J. Blake, N. R. Champness, S. S. M. Chung, W.-S. Li and M. Schroder, Chem. Commun., 1997, 1675 RSC
- X.-M. Zhang, Coord. Chem. Rev., 2005, 249, 1201 CrossRef CAS
-
(a) P. Lin, W. Clegg, R. W. Harrington and R. A. Henderson, Dalton Trans., 2005, 2388 RSC
- R.-G. Xiong, X. Xue, H. Zhao, X.-Z. You, B. F. Abrahams and Z.-L. Xue, Angew. Chem., Int. Ed., 2002, 41, 3800 CrossRef CAS
- L.-Z. Wang, Z.-R. Qu, H. Zhao, X.-S. Wang, R.-G. Xiong and Z.-L. Xue, Inorg. Chem., 2003, 42, 3969 CrossRef CAS
- X. Xue, X.-S. Wang, L.-Z. Wang, R.-G. Xiong, B. F. Abrahams, X.-Z. You, Z.-L. Xue and C.-M. Che, Inorg. Chem., 2002, 41, 6544 CrossRef CAS
- Q. Ye, Y.-H. Li, Y.-M. Song, X.-F. Huang, R.-G. Xiong and Z.-L. Xue, Inorg. Chem., 2005, 44, 3619
- X.-S. Wang, Y.-Z. Tang and R.-G. Xiong, Chin. J. Inorg. Chem., 2005, 21, 1025 CAS
- X.-S. Wang, Y.-Z. Tang, X.-F. Huang, Z.-R. Qu, C.-M. Che, P. W. H. Chan and R.-G. Xiong, Inorg. Chem., 2005, 44, 5278 CrossRef CAS
- T. Wu, B.-H. Yi and D. Li, Inorg. Chem., 2005, 44, 4130 CrossRef CAS
- L.-Z. Wang, X.-S. Wang, Y.-H. Li, Z.-P. Bai, R.-G. Xiong, M. Xiong and G.-W. Li, Chin. J. Inorg. Chem., 2002, 18, 1191 CAS
-
(a) H. Zhao, Q. Ye, Q. Wu, Y.-M. Song, Y.-J. Liu and R.-G. Xiong, Z. Anorg. Allg. Chem., 2004, 630, 1367 CrossRef CAS
- X.-F. Huang, Y.-M. Song, Q. Wu, Q. Ye, X.-B. Chen, R.-G. Xiong and X.-Z. You, Inorg. Chem. Commun., 2005, 8, 58 CrossRef CAS
- X.-S. Wang, X.-F. Huang and R.-G. Xiong, Chin. J. Inorg. Chem., 2005, 21, 1020 CAS
- X.-H. Huang, T.-L. Sheng, S.-C. Xiang, R.-B. Fu, S.-M. Hu, Y.-M. Li and X.-T. Wu, Inorg. Chem. Commun., 2006, 9, 1304 CrossRef CAS
-
(a) T. J. Marks and M. A. Ratner, Angew. Chem., Int. Ed. Engl., 1995, 34, 155 CrossRef CAS
- Z.-R. Qu, H. Zhao, X.-S. Wang, Y.-H. Li, Y.-M. Song, Y.-J. Liu, Q. Ye, R.-G. Xiong, B. F. Abrahams, Z.-L. Xue and X.-Z. You, Inorg. Chem., 2003, 42, 7710 CrossRef CAS
- Q. Ye, Y.-Z. Tang, X.-S. Wang and R.-G. Xiong, Dalton Trans., 2005, 1570 RSC
- Q. Ye, Y.-M. Song, G.-X. Wang, K. Chen, D.-W. Fu, P. W. H. Chan, J.-S. Zhu, S.-P. D. Huang and R.-G. Xiong, J. Am. Chem. Soc., 2006, 128, 6554 CrossRef CAS
- T. Wu, R. Zhou and D. Li, Inorg. Chem. Commun., 2006, 9, 341 CrossRef CAS
- X.-S. Wang, Y.-M. Song and R.-G. Xiong, Chin. J. Inorg. Chem., 2005, 21, 1030 CAS
-
(a) L. Pu, Chem. Rev., 2004, 104, 1687 CrossRef CAS
-
(a)
Nonlinear Optical properties of Organic Molecules and Crystals, vol. 1, ed. J. Zyss and D. S. Chemla, Academic Press, New York, 1989, p. 23 Search PubMed
- K. M. Ok, E. O. Chi and P. S. Halasyamani, Chem. Soc. Rev., 2006, 35, 710 RSC
- P. A. Franken, A. E. Hill, C. W. Peters and G. Wienrich, Phys. Rev. Lett., 1961, 7, 118 CrossRef
- S. Endo, T. Chino, S. Tsuboi and K. Koto, Nature, 1989, 340, 452 CrossRef CAS
- R. P. Lemieux, Acc. Chem. Res., 2001, 34, 845 CrossRef CAS
- K. E. Maly, M. D. Wand and R. P. Lemieux, J. Am. Chem. Soc., 2002, 124, 7898 CrossRef CAS
- T. Lee and I. A. Aksay, Cryst. Growth Des., 2001, 1, 409
-
(a) Z.-R. Qu, H. Zhao, Y.-P. Wang, X.-S. Wang, Q. Ye, Y.-H. Li, R.-G. Xiong, B. F. Abrahams, Z.-G. Liu, Z.-L. Xue and X.-Z. You, Chem.–Eur. J., 2004, 10, 54
-
(a) H. Zhao, Y.-H. Li, X.-S. Wang, Z.-R. Qu, L.-Z. Wang, R.-G. Xiong, B. F. Abrahams and Z.-L. Xue, Chem.–Eur. J., 2004, 10, 2386–2390 CrossRef CAS
- T. A. Vanderah, Science, 2002, 298, 1182 CrossRef CAS
- C. C. Homes, T. Vogt, S. M. Shapiro, S. Wakimoto and A. P. Ramirez, Science, 2001, 293, 673 CrossRef
-
(a) A. Palazzi, S. Stagni, S. Selva and M. Monari, J. Organomet. Chem., 2003, 135, 669
-
(a) D. V. Davydov, I. P. Beletskaya, B. B. Semenov and Y. I. Smushkevich, Tetrahedron Lett., 2002, 43, 6217 CrossRef CAS
- L. J. GooBen, G.-J. Deng and L. M. Levy, Science, 2006, 313, 662 CrossRef
-
(a) D. W. Fu and H. Zhao, Chin. J. Inorg. Chem., 2007, 23, 122 CAS
- M. Dinca, A. Dailly, Y. Liu, C. M. Brown, D. A. Neumann and J. R. Long, J. Am. Chem. Soc., 2006, 128, 16876 CrossRef CAS
| This journal is © The Royal Society of Chemistry 2008 |