DOI:
10.1039/B614208G
(Tutorial Review)
Chem. Soc. Rev., 2008,
37, 68-83
Anion-π interactions
Received 27th June 2007
First published on 12th September 2007
Abstract
This tutorial review provides an overview of the theoretical and experimental investigations that resulted in the recognition of anion-π interactions, i.e., non-covalent forces between electron deficient aromatic systems and anions. Several pioneering theoretical studies revealed that these interactions are energetically favorable (∼20–50 kJ mol–1). Anion-π interactions are gaining significant recognition, and their pivotal role in many key chemical and biological processes is being increasingly appreciated. The design of highly selective anion receptors and channels represent important advances in this nascent field of supramolecular chemistry.
 Brandi L. Schottel | Brandi L. Schottel received a BS in Chemistry and Biology from the University of Missouri-Columbia in 2002, where she conducted research on the gas sorption properties of calixarene derivatives under the direction of Professor Jerry L. Atwood. She received a PhD from Texas A&M University in 2007. She performed her graduate research, which involved systems with anion interactions, in the laboratories of Professor Kim R. Dunbar. She currently is a Postdoctoral Fellow in the laboratories of Professor Kenneth N. Raymond at UC Berkeley. |
 Helen T. Chifotides | Helen T. Chifotides was born in Providence, RI, and received her BS and PhD degrees in Chemistry from the University of Athens, Greece. After a NATO Postdoctoral Fellowship at Michigan State University (1994–1995), she was appointed as a Lecturer in Chemistry at Oregon State University (1995–1997). She held a senior staff scientist position in the Biochemistry Laboratory of the Pulmonary Hospital ‘Sotiria’ in Athens, Greece, until 2001. Currently, she is a Senior Researcher at Texas A&M University. Her research interests span anti-cancer metal complexes with DNA and other biologically active molecules, as well as non-covalent interactions in supramolecular chemistry. |
 Kim R. Dunbar | Kim R. Dunbar was born in Mount Pleasant, PA, and received a BS from Westminster College in 1980 and a PhD from Purdue University in 1984. She joined the faculty of Michigan State University in 1987 and moved to Texas A&M University in 1999, where she is a Distinguished Professor and holds the Davidson Chair of Science. Her research spans topics in synthetic and structural inorganic chemistry, with a focus on the design of conducting and magnetic molecular materials, and the antitumor properties of metal complexes. She has been named a Fellow of the Alfred P. Sloan Foundation and American Association for the Advancement of Science, and a Camille and Henry Dreyfus Teacher–Scholar. She received Distinguished Alumna Awards from Westminster College in 2000 and Purdue University in 2004, and a Distinguished Faculty Award from Michigan State University in 1998. In 2006 she received the Inaugural ‘Texas A&M University Association of Former Students Distinguished Award for Graduate Mentoring’. She is an Associate Editor for the ACS journal Inorganic Chemistry and current Chair of the ACS Division of Inorganic Chemistry, for which she served as a former Secretary. |
Introduction
Supramolecular chemistry, the chemistry of non-covalent interactions, is a highly active interdisciplinary field with important implications in biology, chemistry, physics and engineering.1,2 A recently developed branch of supramolecular chemistry has unearthed a novel type of non-covalent forces between electron deficient aromatic systems and anions, namely the anion-π interaction. The vital role of anions in many key chemical and biological processes, and the involvement of π-rings in molecular anion recognition and transport (artificial highly selective anion receptors and channels), indicate that anion-π contacts could be prominent players in medicinal and environmental applications.3 The nascence, the theoretical and experimental investigations, as well as the importance of anion-π interactions, are highlighted in this tutorial review.In sharp contrast to the mature area of cation binding to aromatic systems, anion-π interactions had hitherto been overlooked, primarily due to their counterintuitive nature (anions are expected to exhibit repulsive interactions with aromatic π-systems due to their electron donating character) and the synthetically challenging factors inherent to the nature of anions (larger size and much higher free energies of solvation compared to cations, wide range of coordination geometries and electronic “saturation”).4 In 1968, Park and Simmons reported their pioneering account of katapinates, i.e., macrobicyclic ammonium host molecules interacting with chloride anions through electrostatic interactions and hydrogen bonds.5 The ensuing reports of anion receptors focused primarily on the previous interactions with a variety of cyclic, acyclic, inorganic and organic hosts.6,7 In the early 1990s, Schneider et al. reported weak but distinct attractive interactions (∼2 kJ mol–1) between negative charges and polarizable aryl parts of host–guest systems.8 Theoretical studies reported in the late 1990s revealed the favorable nature of interactions between an electron rich moiety of small molecules, i.e., hydrogen fluoride and hexafluorobenzene, preceding, in 2002, three other almost simultaneous pioneering theoretical studies by Alkorta et al.,9 Deyà et al.,10 and Mascal et al.11 The latter three studies confirmed the presence of favorable non-covalent interactions between electron deficient aromatic rings and anions, with binding energies comparable to hydrogen bonds (20–50 kJ mol–1); the term ‘anion-π interaction’ was coined by Deyà et al. to describe this type of contact.10
Definition
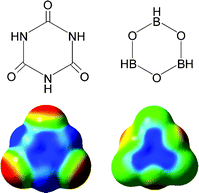
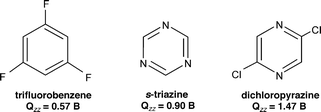
Typically, anion-π interactions are termed as favorable non-covalent contacts between an electron deficient (π-acidic) aromatic system and an anion.12 Elegant studies have revealed that the anion-π interaction is, in general, dominated by electrostatic and anion-induced polarization contributions.10,12,13 The electrostatic component of the interaction is correlated to the permanent quadrupole moment, Qzz, of the electron deficient aromatic ring (the quadrupole moment is a measure of the charge distribution of a molecule relative to a particular molecular axis), e.g., hexafluorobenzene has a large and positive quadrupole moment (Qzz(C6F6) = + 9.50 B; 1 B (Buckingham) = 3.336 × 10–40 C m2) due to the strong electronegativity of the fluorine atoms (Fig. 1a), whereas benzene has a large and negative quadrupole moment (Qzz(C6H6) = –8.48 B) (Fig. 1b).13 The aforementioned topological analysis of the electron density in anion-π interactions13 showed that a strong correlation exists between the magnitude of the aromatic ring Qzz and the electrostatic contribution to the anion-π interaction, with higher positive Qzz values leading to more favorable interactions (e.g., for trifluoro-s-triazine with [Cl]– ions: Qzz = + 8.23 B, Et = –15.0 kcal mol–1 and for 1,3,5-trifluorobenzene with [Cl]– ions: Qzz = + 0.57 B, Et = – 4.8 kcal mol–1, Et = total anion-π interaction energy).13 Further studies indicated that, for molecules with a very positive Qzz, e.g., 1,3,5-trinitrobenzene (Qzz = +20 B), the anion-π interaction is basically dominated by the electrostatic term, whereas for molecules with small Qzz values, e.g., s-triazine (Qzz = + 0.90 B), the anion-induced polarization contribution prevails.13 The anion-induced polarization (Fig. 2) correlates with the molecular polarizability, α||, of the aromatic compound, and this component has a significant contribution to the anion-π interaction for molecules with high α|| values, e.g., α|| (s-tetrazine) = 58.7 a.u.14 Interestingly, molecules with small magnitudes of Qzz were found to exhibit a dual behavior by binding to both anions and cations.12,14 Recent theoretical studies and crystallographic data indicate that favorable anion-π interactions, dominated by the electrostatic term, are established between positively charged seven-membered rings, e.g., the aromatic tropylium cation and various anions.15 There is evidence, however, that even non-electron deficient aromatic rings can establish anion-π interactions if the ring is simultaneously interacting with a cation on the opposite face of the ring.16 Furthermore, recent theoretical studies indicate that aromatic rings with negative quadrupole moments exhibit, albeit weak, anion-π interactions in the gas phase, e.g., benzene (Qzz = – 8.48 B) has a [F]– binding enthalpy of – 5.51 kcal mol–1,17 and 1,4,5,8,9,12-hexaazatriphenylene (HAT; Qzz = – 8.53 B, α|| = 54.03 a.u.) has a complexation energy of – 5.51 kcal mol–1 for binding [Br]– ions (Epol = –10.25 and Eele = 0.22 kcal mol–1, Epol, Eele: polarization and electrostatic energy contributions, respectively).18 The latter findings emphasize the importance of the polarization component to the anion-π interaction energy, in particular for non-electron deficient rings,12,17 a conclusion that has important implications for biological systems that contain aromatic rings.3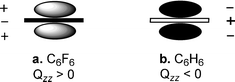 |
| | Fig. 1 Representation of the quadrupole moment for (a) hexafluorobenzene and (b) benzene. (Reproduced with permission10 from P. M. Deyà et al., Angew. Chem., Int. Ed., 2002, 41, 3389. Copyright 2002 Wiley-VCH Verlag GmbH & Co. KGaA.) | |
 |
| | Fig. 2 Aromatic π-electron density polarizability. Figure adapted from ref. 17. | |
Theoretical investigations
Mascal et al.,11 as well as other groups, employ electrostatic potential (ESP) maps to visualize the charge distribution of aromatic rings under consideration. An ESP map of a molecule at selected points on the 0.02 isodensity surface (a shell or 3D surface around the molecule, typically defining its shape and size, where the electron density is 0.02 a.u.) depicts the surface by color, wherein different colors are used to identify different potentials. The ESP at a point (x, y, z) is given by the ESP energy between an imaginary positively-charged (+1) ion located at (x, y, z) and the molecule. If the ion is attracted to the molecule, the potential is negative; if the ion is repelled by the molecule, the potential is positive. Since the imaginary ion has a +1 charge, it will be attracted to electron rich regions of molecules (negative potential) and repelled by electron poor regions (positive potential). Thus, electron rich regions have negative potentials and electron poor regions have positive potentials. Typically, a color scale is used, with the most negative potential colored red and the most positive potential colored blue, e.g., for 1,3,5-triazine and trifluoro-1,3,5-triazine (Fig. 3, top), the central areas of the rings (blue) correspond to positive potentials and thus are electron depleted regions (Fig. 3, bottom); the central area of trifluoro-1,3,5-triazine (B) has a higher positive potential than 1,3,5-triazine (A) due to the electron withdrawing fluorine atoms. It is notable, however, that in both rings, there is a substantial area of positive charge concentrated on the C3 axes, which renders the molecules good candidates for establishing anion-π interactions.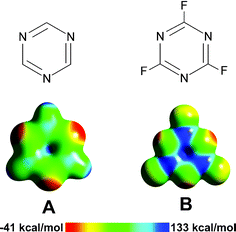 |
| | Fig. 3 (A) 1,3,5-Triazine and (B) trifluoro-1,3,5-triazine. Top: Schematic drawings; bottom: calculated ESP surfaces generated at an isodensity value of 0.02 a.u. Electrostatic potential surface energies range from –41 (red) to +133 (blue) kcal mol–1. Figure regenerated with computational parameters from ref. 11. | |
Mascal et al.11 performed an ab initio study of the interactions between the π-acidic rings 1,3,5-triazine and trifluoro-1,3,5-triazine (Fig. 3, top) and [Cl]–, [F]– and azide ([N3]–) ions by applying second-order Møller–Plesset perturbation theory (MP2).19 The authors ascertained favorable binding interactions between the aromatic rings and the anions, with stronger interactions and shorter non-covalent bond distances r (r is defined as the distance between the anion [X]– and the centroid of the ring; Fig. 4) observed for trifluoro-1,3,5-triazine as compared to 1,3,5-triazine for the same anion [X]–, e.g., triazine + chloride (EMP2 = –4.8 kcal mol–1, r = 3.2 Å) and trifluoro-1,3,5-triazine + chloride (EMP2 = –14.8 kcal mol–1, r = 3.0 Å).11 In the theoretical studies by Mascal et al.,11,20 Deyà et al.,10,12 as well as others,3 the distance r of the non-covalent interaction is defined as previously stated, and the angle θ is defined as the angle of the [X]–⋯aryl centroid axis and the line connecting the ring centroid with a ring carbon atom (Fig. 4). Although this definition has recently become a topic of controversy for anions that are positioned off-center,21 there is merit to it because electron deficient aromatic rings exhibit a substantial area of positive charge at the center of the ring in ESP maps (e.g., 1,3,5-triazine/trifluoro-1,3,5-triazine and cyanuric acid/boroxine in Fig. 3 and Fig. 5, respectively).
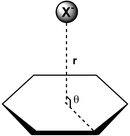 |
| | Fig. 4 Distance r and angle θ for an anion that is positioned above the centroid of an aromatic ring. | |
 |
| | Fig. 5 Left: Cyanuric acid and right: boroxine. Top: Schematic drawings; bottom: calculated ESP surfaces. Electrostatic potential surface energies range from –20 (red) to +20 (blue) kcal mol–1. (Reproduced with permission20 from M. Mascal, Angew. Chem., Int. Ed., 2006, 45, 2890. Copyright 2006 Wiley-VCH Verlag GmbH & Co. KGaA.) | |
Alkorta et al.9 performed DFT (Density Functional Theory) and MP219 calculations on the interactions between several electron deficient aromatic rings, e.g., hexafluorobenzene, octafluoronaphthalene and pentafluoropyridine and the anions [H]–, [F]–, [Cl]–, [Br]–, [CN]– and [CNO]–.9 The studies indicated in each case that the anion interacts favorably with the π-cloud of the aromatic ring. The DFT calculations, however, provided a reasonable qualitative description of the interactions, but longer distances and smaller interaction energies compared to the computationally more expensive MP2 method.9 The topological properties of the electron density for the complexes were analyzed by applying the AIM (Atoms in Molecules)22 methodology, which has been successfully used to understand non-covalent interactions.10
In AIM,22 the topology of the electron density, ρ, yields a reliable mapping of the molecule and is effectively described by a set of critical points (CPs); the CPs of the electron density distribution are associated with atomic nuclei, bonds, rings and cages.22 Critical points are labelled by two values (ω and σ), where ω is the rank of the critical point (typically, for energetically stable configurations, ω = 3) and σ is the sum of the algebraic signs λ of the second derivatives of the density ρ in three dimensions (λ is +1 or –1 if the density has a minimum or maximum, respectively).12,22
(3, –3) critical point: The electron density decreases in all three perpendicular directions of space. This is a local maximum of electron density and corresponds to the position of an atom.
(3, –1) critical point: The electron density decreases in two perpendicular directions of space and rises in the third. This is a saddle point of electron density with a maximum of electron density in two directions of space and a minimum in the third. This point is called a bond critical point.
(3, +1) critical point: The electron density decreases in one direction of space and rises in the two other perpendicular directions. This is a saddle point with a maximum in one and a minimum in two directions of space and is called a ring critical point.
(3, +3) critical point: This is a local minimum with electron density rising in all three directions of space and is called a cage critical point. It is common for closed-shell interactions, such as anion-π interactions, to be associated with a positive Laplacian (∇2) at the (3, –1) and (3, +3) CPs, indicating depletion of the electron density.12 Quantitative values of ρ and the Laplacian of ρ(∇2ρ) are good indicators of the character and strength of anion-π interactions.12
Deyà's initial study probed the interactions of hexafluorobenzene with halides, hydride, [CN]–, [NO3]– and [CO3]2– at the Hartree–Fock (HF)23 and MP219 levels of theory (the authors also performed a search of the Cambridge Crystal Database (CSD) for the presence of anion-π interactions in reported structures; the search will be discussed in the section with the experimental studies).10 The AIM analysis for all the anions (except for nitrate and carbonate ions) revealed six (3, –1) CPs connecting the anion with the carbon atoms of the C6F6 ring, and six (3, +1) CPs between the anion and the middle of a C–C bond (Fig. 6, top).10 All the anion-π interactions were further described by a cage (3,+3) critical point over the hexafluorobenzene ring, indicating depletion of electron density, which is common for closed-shell interactions (vide supra; Fig. 6, top). In the cases of the [NO3]– (Fig. 6, bottom) and [CO3]2– anions, only three bond and three ring CPs were found due to the geometry of the optimized complexes.10 The most stable interaction was established with the carbonate anion due to its –2 charge.10,12 Similar studies have been performed by the same group for a number of electron deficient aromatic rings, e.g., trifluoro-1,3,5-triazine, hexazine, s-triazine and others.12–14 A common feature of all the compounds upon complexation of the anion is the formation of a cage critical point, located along the line connecting the ion with the center of the ring.10,12–14 Another interesting trend derived from these studies is the direct correlation between the interaction energy and the values of the electron charge density at the cage CPs, indicating that the value of the electron density at the cage CP after complexation can be used as a measure of the strength of the anion-π interaction.12–14
![Representation of the AIM22-derived CPs located when [F]– (top) and [NO3]– (bottom) interact with C6F6. The interactions for both anions are shown with top (left) and side (right) views. The red, yellow and light blue circles represent bond (3, –1), ring (3, +1) and cage (3, +3) CPs, respectively. (Reproduced with permission10 from P. M. Deyà et al., Angew. Chem., Int. Ed., 2002, 41, 3389. Copyright 2002 Wiley-VCH Verlag GmbH & Co. KGaA.)](/image/article/2008/CS/b614208g/b614208g-f6.gif) |
| | Fig. 6 Representation of the AIM22-derived CPs located when [F]– (top) and [NO3]– (bottom) interact with C6F6. The interactions for both anions are shown with top (left) and side (right) views. The red, yellow and light blue circles represent bond (3, –1), ring (3, +1) and cage (3, +3) CPs, respectively. (Reproduced with permission10 from P. M. Deyà et al., Angew. Chem., Int. Ed., 2002, 41, 3389. Copyright 2002 Wiley-VCH Verlag GmbH & Co. KGaA.) | |
To determine whether polarization is important, and to analyze the binding properties of the aforementioned aromatic rings12–14 and the overall physical nature of their interactions with anions, the polarization contribution to the total interaction energy was computed using the recently developed Molecular Interaction Potential with polarization (MIPp).12 MIPp is an improved generalization of the molecular electrostatic potential (MEP), in which three terms contribute to the interaction energy, i.e., (a) an electrostatic term identical to that in MEPs, (b) a classical dispersion–repulsion term and (c) a polarization term derived from perturbational theory.12 MEPp calculations performed for hexafluorobenzene in the presence of [F]– ions indicated that the polarization and electrostatic components are comparable when the anion–ring distance is in the range 2.0–3.0 Å.10 Similar results have been obtained for 1,3,5-trinitrobenzene, s-tetrazine and other electron deficient aromatic systems.12–14 In particular, the MIPp calculations performed on a series of electron deficient aromatic systems with varying molecular polarizability (α||) values13 indicated that, apart from the direct correlation of the π-system Qzz and the electrostatic contribution to the total interaction energy, there is also a direct correlation between the molecular polarizability perpendicular to the molecular plane (α||) and the total interaction energy; this relationship emphasizes the importance of the polarization component in anion-π interactions,12,13 especially in the case of aromatic systems with small magnitudes of Qzz (e.g., for 1,3,5-trifluorobenzene with Qzz = + 0.9 B, the polarization component is dominant in the total anion-π interaction energy with [F]–).24
The importance of the polarization component of the aromatic ring was further exhibited in a study performed by Deyà et al.16 at the HF23 and MP219 levels of theory on aromatic ring systems that are non-electron deficient. In this study, the interaction between benzene and the halide ions was considered when the aromatic system was simultaneously interacting with [Na]+ on the opposite face of the ring (hexafluorobenzene was also studied for the purpose of comparison).16 The study showed that the established anion-π interactions are indeed favorable; the MIPp analysis further indicated the critical contribution of the polarization component to the interaction energy in the 2.0–2.5 Å range. The critical point analysis of the compounds conducted by AIM22 revealed, apart from the bond and ring CPs, the presence of two cage CPs located above and below the aromatic ring along the C6 axis connecting both the anion and the cation with the center of the ring (Fig. 7);16 these CPs indicate that the benzene ring is simultaneously participating in both anion-π and cation-π interactions on opposite sides of the ring. It is noteworthy, however, that the [X]–⋯ring centroid distance is shorter for C6F6 than for C6H6.16
![Diagram of the CPs determined by AIM22 for C6H6 interacting simultaneously with an [F]– anion and a [Na]+ cation. Red circles = (3, –1) bond CPs, green circles = (3, +1) ring CPs and blue circles = (3, +3) cage CPs. (Reproduced with permission16 from P. M. Deyà et al., New J. Chem., 2003, 27, 211. Copyright 2003 the Royal Society of Chemistry (RSC) on behalf of the Centre National de la Recherche Scientifique (CNRS).)](/image/article/2008/CS/b614208g/b614208g-f7.gif) |
| | Fig. 7 Diagram of the CPs determined by AIM22 for C6H6 interacting simultaneously with an [F]– anion and a [Na]+ cation. Red circles = (3, –1) bond CPs, green circles = (3, +1) ring CPs and blue circles = (3, +3) cage CPs. (Reproduced with permission16 from P. M. Deyà et al., New J. Chem., 2003, 27, 211. Copyright 2003 the Royal Society of Chemistry (RSC) on behalf of the Centre National de la Recherche Scientifique (CNRS).) | |
A system that nicely demonstrates the importance of the quadrupole moment and polarizability components in anion-π interactions is that of cyanuric acid and its sulfur-substituted derivatives (Fig. 8) in the presence of halides.25 The MP219 results indicate that the binding energies of the rings with the anions are not affected by the number of sulfur atoms present in the system, but depend only on the anion. The increase in the number of sulfur atoms induces an increase in the molecular polarizability (α||) of the rings; at the same time, however, it causes a decrease in the quadrupole moment Qzz.25 This compensating effect of the two main factors that contribute to the anion-π binding energy, i.e., the electrostatic and the anion-induced polarization contributions, are nicely visualized in Fig. 9.
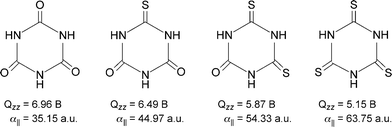 |
| | Fig. 8 The structures of cyanuric acid and its sulfur-substituted derivatives. (Reproduced with permission25 from P. M. Deyà et al., Chem.–Eur. J., 2005, 11, 6560. Copyright 2005 Wiley-VCH Verlag GmbH & Co. KGaA.) | |
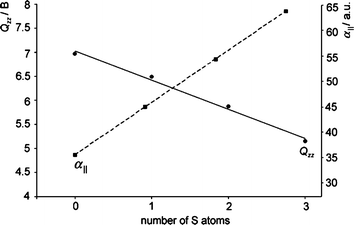 |
| | Fig. 9 Plot of the regression between the molecular polarizability (α||) and the quadrupole moment (Qzz) with the number of sulfur atoms present in the cyanuric derivatives shown in Fig. 8. (Reproduced with permission25 from P. M. Deyà et al., Chem.–Eur. J., 2005, 11, 6560. Copyright 2005 Wiley-VCH Verlag GmbH & Co. KGaA.) | |
Apart from the critical contributions of the electrostatic and anion-induced polarization components to the interaction energies of anion-π interactions, recent studies by Deyà et al.14 and Kim et al.26 showed that anion-π interactions (in contrast to cation-π interactions) have considerable contributions from dispersion energies. In particular, a theoretical study by Kim et al. showed that several molecular orbital interactions of a dispersive type are established between the occupied orbitals of C6F6 and the anion, especially in the case of organic anions (Fig. 10).26 The two previous reports also include comparative studies between anion-π and cation-π interactions.14,26 Kim et al.26 performed ab initio calculations on different types of π-systems, e.g., olefinic, aromatic and heteroaromatic, and concluded that, for the systems studied, the total interaction energies of anion-π and cation-π complexes are comparable. Furthermore, Deyà et al. detailed a theoretical investigation of the interaction of [Na]+ and [F]– ions with trifluorobenzene, s-triazine and dichloropyrazine rings (Fig. 11); the choice of [Na]+ and [F]– as interacting ions was made because they are isoelectronic, and consequently the complexes formed are also isoelectronic. The authors concluded that the relative strength of cation-π to anion-π interactions strongly depends on the quadrupole moment and polarizability of the aromatic system.14 For compounds with comparable quadrupole moments, the energies of cation-π are more negative than anion-π interactions, principally because cation complexes have shorter equilibrium distances that allow them to polarize the π-cloud of the aromatic ring in a more effective way.27 In contrast to cation-π interactions, notable features of anion-π interactions are the increase in aromaticity of the ring upon complexation of the anion,14,27 and an obvious interaction between the anion px and py orbitals and the molecular orbitals of the aromatic ring, e.g., [F]– ions with 1,3,5-trifluorobenzene, especially the pz orbitals of the carbon atoms;28 however, this clear bonding interaction is compensated by antibonding orbitals, rendering anion-π interactions non-bonding in character.28
![Dispersive type molecular orbital interactions between the highest occupied orbitals of hexafluorobenzene and (a) [Br]–, (b) [CN]– and (c) [NO3]–. (Reproduced with permission26 from K. S. Kim et al., J. Phys. Chem. A, 2004, 108, 1250. Copyright 2004 the American Chemical Society.)](/image/article/2008/CS/b614208g/b614208g-f10.gif) |
| | Fig. 10 Dispersive type molecular orbital interactions between the highest occupied orbitals of hexafluorobenzene and (a) [Br]–, (b) [CN]– and (c) [NO3]–. (Reproduced with permission26 from K. S. Kim et al., J. Phys. Chem. A, 2004, 108, 1250. Copyright 2004 the American Chemical Society.) | |
 |
| | Fig. 11 π-Aromatic systems examined by Deyà et al. to investigate the differences between anion-π and cation-π interactions. (Reproduced with permission14 from P. M. Deyà et al., J. Phys. Chem. A, 2004, 108, 9423. Copyright 2004 the American Chemical Society.) | |
Additivity of anion-π interactions
Deyà et al. investigated the additivity of anion-π interactions using ab initio calculations with the goal of designing useful neutral anion receptors.29 The authors studied the 1 : 1 (1–4), 1 : 2 (anion-π2; 5–8) and 1 : 3 (anion-π3; 9–12) anion : ring complexes of s-triazine and trifluoro-1,3,5-triazine with [Br]– and [Cl]– ions (Fig. 12). The emerging results confirm the additivity of anion-π interactions because the distance r from the anion to the ring centroid is practically insensitive to the stoichiometry of the complex, and the binding energies of the anion-π2 and anion-π3 complexes are approximately two and three times the energy of the 1 : 1 complex, respectively.29 Furthermore, AIM22 analysis of the results indicated that the values of the charge density at the (3, +3) cage CPs of the π-systems did not change with the addition of more aromatic rings, and thus the strength of each anion-π interaction, which is directly related to the charge density at the critical point (vide supra), is not affected by the presence of multiple rings.29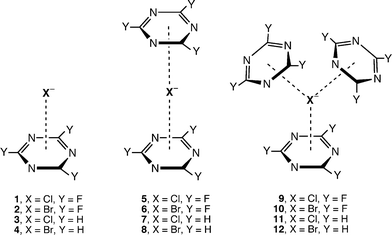 |
| | Fig. 12 Anion-π complexes of s-triazine and trifluoro-1,3,5-triazine with multiple interactions per anion. (Reproduced with permission29 from P. M. Deyà et al., J. Phys. Chem. A, 2005, 109, 9341. Copyright 2005 the American Chemical Society.) | |
Interplay between anion-π, cation-π and π–π interactions
Two elegant studies by Deyà et al.,30,31 performed at the MP219 level of theory, relate important information vis-à-vis the interplay between the anion-π, cation-π and π–π interactions. In the first study, systems with the same aromatic ring, i.e., 1,3,5-trifluorobenzene (Fig. 13a, b; X = F, Cl, Br; M = Li, Na) were considered,30 whereas in the latter study, the two aromatic rings were different (Fig. 13c).31 In the first study, it was shown that the distance r between the anion [X]– and the aromatic ring in 13–15 was shorter as compared to the simple anion-π complex, indicating that the π–π interaction strengthens the anion-π interaction.30 Moreover, the results showed that there was a highly synergetic effect between the interactions, i.e., the simultaneous formation of anion-π, π–π and cation-π interactions, e.g., complexes 16 and 17 are considerably more favorable than the independent establishment of the three interactions.30 Likewise, the results of the latter study show the cooperativity effects and the non-additivity of the energy values for these non-covalent interactions.31 For example, system 18 demonstrates the highest stabilization (non-additive) energy and the shortest π–π distance of 3.12 Å, most likely due to ion-induced polarization effects.31 Understanding the synergetic effect of these interactions is of great importance due to their ubiquitous presence in biological systems.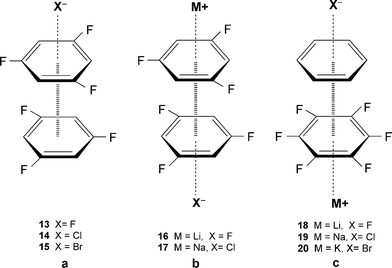 |
| | Fig. 13 Schematic representation of systems with multiple non-covalent interactions. (Part of Figure reproduced with permission30 from P. M. Deyà et al., ChemPhysChem, 2006, 7, 2487. Copyright 2006 Wiley-VCH Verlag GmbH & Co. KGaA.) (Part of Figure reproduced with permission31 from P. M. Deyà et al., New J. Chem., 2007, 31, 556. Copyright 2007 the Royal Society of Chemistry (RSC) on behalf of the Centre National de la Recherche Scientifique (CNRS).) | |
Experimental evidence of anion-π interactions
Structural evidence
CSD search. To further support the existence of anion-π interactions, Deyà et al.10 performed a search of the Cambridge Structural Database (CSD) in their pioneering study to unearth anion-π interactions in reported structures (not discussed, however, by the authors of the original reports) with the required criteria: “(1) the type of non-bonding contact was either intra or intermolecular; (2) the non-bonding contact was defined by distance criteria, i.e., less than the sum of van der Waals radii; (3) a hit was stored when a non-bonding contact existed between the interacting anion and all six carbon/nitrogen atoms of the aromatic ring and, (4) a negative charge was explicitly defined on the interacting atom.”12 The search for perfluorobenzene derivatives with electronegative atoms resulted in 1,944 hits with anion-π interactions present. The majority of the resulting structures had anions at angles θ close to 90° with respect to the aromatic rings (Fig. 4) and interaction distances r closer than the van der Waals contact.10 Crystallographic studies. Apart from the previously discussed theoretical studies (vide supra) and CSD search, the first X-ray crystal structures with recognized and unambiguous anion-π interactions were independently reported in 2004 by Meyer et al.32 and Reedijk et al.33 Meyer et al. reported a study of the electron deficient ligand hexakis(pyridin-2-yl)-[1,3,5]-triazine-2,4,6-triamine (L1; Fig. 14A) complexed with Cu(II) chloride to afford [(L1)2(CuCl)3][CuCl4][Cl] (Fig. 14B).32 The two L1 ligands are arranged in a nearly perfect face-to-face position (plane-to-plane distance ∼3.78 Å). A salient feature of this structure is the position of one of the charge-compensating [Cl]– ions, which establishes anion-π interactions with a triazine ring of L1. The contact distance to the ring centroid is 3.11 Å and the angle of the same contact is 88°, both of which are close to the theoretically predicted values by Mascal et al.11 and Deyà et al.,10 respectively.![(A) Schematic drawing of hexakis(pyridin-2-yl)-[1,3,5]-triazine-2,4,6-triamine (L1); (B) Front (left) and side (right) views of {[(L1)2(CuCl)3][Cl]}2+. Anion-to-triazine centroid contact 3.11 Å. Atom colors: C = grey, H = white, N = dark blue, Cu = light blue and Cl = yellow. Figure adapted from ref. 32.](/image/article/2008/CS/b614208g/b614208g-f14.gif) |
| | Fig. 14 (A) Schematic drawing of hexakis(pyridin-2-yl)-[1,3,5]-triazine-2,4,6-triamine (L1); (B) Front (left) and side (right) views of {[(L1)2(CuCl)3][Cl]}2+. Anion-to-triazine centroid contact 3.11 Å. Atom colors: C = grey, H = white, N = dark blue, Cu = light blue and Cl = yellow. Figure adapted from ref. 32. | |
Almost simultaneously, Reedijk et al. reported a remarkable supramolecular system involving the dendritic octadentate ligand N,N′,N″,N‴-tetrakis{2,4-bis(di-2-pyridylamino)-1,3,5-triazinyl}-1,4,8,11-tetraazacyclotetradecane (L2; Fig. 15A) coordinated to Cu(II) chloride to afford the tetranuclear complex [Cu4(L2)Cl4][Cl]4(H2O)13 (Fig. 15B).33 In this complex, 16 pyridyl rings of L2 are coordinated to four different Cu(II) ions, and two [Cl]– anions are encapsulated in the two cavities formed by the pyridyl rings (shortest anion-to-centroid distance ∼3.50 Å), as well as in close contact to the triazine rings (shortest distance 3.012 Å), thus establishing anion-π interactions (Fig. 15B).3,33 The latter study prompted Reedijk et al. to design new triazine-based ligands and relevant systems with anion-π interactions (primarily with nitrate ions). These studies were expertly detailed in a recent review by Reedijk et al.;3 two of the systems are briefly discussed in the section for reports that combine structural evidence with theoretical support (vide infra).
![(A) Schematic drawing of N,N′,N″,N‴-tetrakis{2,4-bis(di-2-pyridylamino)-1,3,5-triazinyl}-1,4,8,11-tetraazacyclotetradecane (L2). (B) Crystal structure of [Cu4(L2)Cl4][Cl]4(H2O)13 from two different perspectives. Atom colors: C = grey, H = white, N = dark blue, Cu = light blue and Cl = yellow. Figure adapted from ref. 33.](/image/article/2008/CS/b614208g/b614208g-f15.gif) |
| | Fig. 15 (A) Schematic drawing of N,N′,N″,N‴-tetrakis{2,4-bis(di-2-pyridylamino)-1,3,5-triazinyl}-1,4,8,11-tetraazacyclotetradecane (L2). (B) Crystal structure of [Cu4(L2)Cl4][Cl]4(H2O)13 from two different perspectives. Atom colors: C = grey, H = white, N = dark blue, Cu = light blue and Cl = yellow. Figure adapted from ref. 33. | |
A comprehensive study undertaken by our group resulted in the isolation of unprecedented macrocyclic complexes that displayed multiple anion-π contacts. Reactions of Ni(II) or Zn(II) ions with 3,6-bis(2′-pyridyl)-1,2,4,5-tetrazine (bptz) (Fig. 16A) in acetonitrile led to the formation of metallacyclophanes, which were templated by the anions present.34 The tetrahedral [BF4]– and [ClO4]– anions templated the formation of molecular squares, e.g., [Ni4(bptz)4(CH3CN)8⊂BF4][BF4]7 (Fig. 16B), whereas the octahedral [SbF6]– anion led to exclusive formation of the pentagon [Ni5(bptz)5(CH3CN)10⊂SbF6][SbF6]9 (Fig. 16C).34 Several studies indicate that the nuclearity of the cyclic product is dictated by the anion. Furthermore, in each case, the encapsulated anion resides in the void cavity of the macrocycle, establishing short contacts with the centroids of the electron deficient tetrazine rings (anion-π4 and anion-π5 systems for the molecular square and pentagon, respectively).34
![(A) Schematic drawing of bptz in the anti orientation; crystal structures of the (B) square [Ni4(bptz)4(CH3CN)8⊂BF4]7+ and (C) pentagon [Ni5(bptz)5(CH3CN)10⊂SbF6]9+. Atom colors: C = grey, H = white, N = dark blue, Ni = light blue, B = orange, F = green and Sb = yellow. Figure adapted from ref. 34.](/image/article/2008/CS/b614208g/b614208g-f16.gif) |
| | Fig. 16 (A) Schematic drawing of bptz in the anti orientation; crystal structures of the (B) square [Ni4(bptz)4(CH3CN)8⊂BF4]7+ and (C) pentagon [Ni5(bptz)5(CH3CN)10⊂SbF6]9+. Atom colors: C = grey, H = white, N = dark blue, Ni = light blue, B = orange, F = green and Sb = yellow. Figure adapted from ref. 34. | |
Square grid structures exhibiting anion-π interactions were also recently reported by Domasevitch et al.35 The new heteroaromatic derivative pyridazino[4,5-d]pyridazine (L3; Fig. 17A) was used to generate metal–organic frameworks, e.g., [Cu(H2O)2(L3)2][ClO4]2·4H2O (Fig. 17B). In this salt, the oxygen atoms of the enclosed perchlorate anions interact with the pyridazine rings.35 The same ligand L3 is present in the Cu(II) grid-type structure [Cu(µ-OH)(L3)][H2NSO3]·H2O, formed in the presence of sulfamate anions [H2NSO3]–, which establish ‘double’ anion-π interactions (through both the nitrogen and oxygen atoms) with the pyridazine rings (Fig. 17C).35
![(A) Schematic drawing of pyridazino[4,5-d]pyridazine (L3). (B) 2D square grid structure of [Cu(H2O)2(L3)2][ClO4]2·4H2O with each of the square meshes encapsulating two perchlorate anions with short O⋯π contacts. (C) Anion-π interactions through both N and O atoms in [Cu(µ-OH)(L3)][H2NSO3]·H2O. Atom colors: C = grey, O = red, Cl = yellow, Cu = light blue, N = blue, S = purple and H = white. Figure adapted from ref. 35.](/image/article/2008/CS/b614208g/b614208g-f17.gif) |
| | Fig. 17 (A) Schematic drawing of pyridazino[4,5-d]pyridazine (L3). (B) 2D square grid structure of [Cu(H2O)2(L3)2][ClO4]2·4H2O with each of the square meshes encapsulating two perchlorate anions with short O⋯π contacts. (C) Anion-π interactions through both N and O atoms in [Cu(µ-OH)(L3)][H2NSO3]·H2O. Atom colors: C = grey, O = red, Cl = yellow, Cu = light blue, N = blue, S = purple and H = white. Figure adapted from ref. 35. | |
Several structures of the π-electron deficient ligand HAT-(CN)6 (1,4,5,8,9,12-hexaazatriphenylene hexacarbonitrile; Fig. 18A), which combines high π-acidity due to the electron withdrawing cyano groups with high molecular polarizability (Fig. 18B),18 display anion-π interactions in the solid state.36,37 Kitagawa et al. reported the tetranuclear structure [Cu(dppFc*)]2{HAT-(CN)6}2][PF6]2 (dppFc* = 1,1′-bis(diphenylphosphino)octamethyl ferrocene), wherein anion-π interactions are established between the HAT-(CN)6 units and the [PF6]– ions due to the electron deficient character of the HAT-(CN)6 core, instead of intermolecular π-stacking of the non-coordinated CN groups (Fig. 18C; shortest anion F⋯central ring centroid distances 2.716 and 2.908 Å).36 Alternatively, the hexanuclear supramolecule [{Cu(dppFc)}3{HAT-(CN)6}][PF6]2, with a triangular shape, is formed when dppFc (dppFc = 1,1′-bis(diphenylphosphino) ferrocene) is used (Fig. 18D).36 Based on electrochemical studies, the latter copper complex contains the anion radical [HAT-(CN)6]˙– with the [PF6]– counterions centered over the planar [HAT-(CN)6]˙– (shortest anion F⋯central ring centroid distance 3.205 Å).36 Moreover, the cationic titanocene complexes [(Cp2Ti)3(µ3-HATNMe6)]n+ (n = 2, 3 or 4) of the HAT homolog, hexamethyl-1,6,7,12,13,18-hexaazatrinaphthylene (HATNMe6), exhibit anion-π interactions between the electron deficient central ring of the HATNMe6 moiety and the [PF]6– and [BF]4– counterions, with sandwiched [PF]6– anions between two cationic entities in the case of the [(Cp2Ti)3(µ3-HATNMe6)][PF6]2 complex.38
![(A) Schematic drawing of HAT-(CN)6. (B) ESP map (isodensity value 0.02 a.u.) of HAT-(CN)6 with ESP surface energies ranging from –16 (red) to 188 (blue) kcal mol–1 calculated by B3LYP/6-311+g(d,p). (C) [Cu(dppFc*)]2{HAT-(CN)6}2][PF6] (dppFc* = 1,1′-bis(diphenylphosphino)octamethyl ferrocene).36 (D) {[{Cu(dppFc)}3{HAT-(CN)6}][PF6]}+ (dppFc = 1,1′-bis(diphenylphosphino) ferrocene).36 (E) {([n-Bu4N][I])3[HAT-(CN)6]2}·3C6H6 looking down the c axis (the three iodide ions are distributed among four crystallographic positions).37 (F) Space-filling diagram of the repeat layers in ([n-Bu4N][I])3[HAT-(CN)6]2; the cations have been omitted for clarity.37 Atom colors: C = gray, H = white, N = dark blue, Cu = light blue, P = pink, F = green and Cl = yellow.](/image/article/2008/CS/b614208g/b614208g-f18.gif) |
| | Fig. 18 (A) Schematic drawing of HAT-(CN)6. (B) ESP map (isodensity value 0.02 a.u.) of HAT-(CN)6 with ESP surface energies ranging from –16 (red) to 188 (blue) kcal mol–1 calculated by B3LYP/6-311+g(d,p). (C) [Cu(dppFc*)]2{HAT-(CN)6}2][PF6] (dppFc* = 1,1′-bis(diphenylphosphino)octamethyl ferrocene).36 (D) {[{Cu(dppFc)}3{HAT-(CN)6}][PF6]}+ (dppFc = 1,1′-bis(diphenylphosphino) ferrocene).36 (E) {([n-Bu4N][I])3[HAT-(CN)6]2}·3C6H6 looking down the c axis (the three iodide ions are distributed among four crystallographic positions).37 (F) Space-filling diagram of the repeat layers in ([n-Bu4N][I])3[HAT-(CN)6]2; the cations have been omitted for clarity.37 Atom colors: C = gray, H = white, N = dark blue, Cu = light blue, P = pink, F = green and Cl = yellow. | |
Recently, our group co-crystallized [n-Bu4N][I] with HAT-(CN)6 to afford {([n-Bu4N][I])3[HAT-(CN)6]2}·3C6H6 (Fig. 18E), which exhibits charge transfer (CT) from the [I]– ions to the HAT-(CN)6 rings, as well as interesting anion-π interactions.37 In the solid-state, {([n-Bu4N][I])3[HAT-(CN)6]2}·3C6H6 consists of four different repeat layers (Fig. 18F); in layer 2 of this structure, the position of each disordered iodide ion is centered over the C–C bond of the NC–Cring positions of the HAT–(CN)6 molecule from layer 1, as well as directly under the same bonds of a HAT-(CN)6 molecule in layer 3. In layer 4, the fourth possible position of the disordered [I]– ions is positioned directly over the centroid of the central benzene ring of the HAT-(CN)6 molecule in layer 3 (3.8 Å from the centroid of the central ring and 3.6 Å from the HAT-(CN)6 central ring centroid in the next layer 1).37
Structural evidence with theoretical support
Deyà et al. collaborated with Saczewski to report a combined crystallographic and computational study on anion-π interactions involving cyanuric acid and its sulfur substituted derivatives (Fig. 8) in the presence of halides.25 The computational portion of the study is presented in the theoretical section (vide supra). To experimentally examine these cyanuric acid derivatives with halides, the thiocyanuric and dithiocyanuric acids with an ethylene ammonium arm attached to one of the triazine N atoms (L4 and L5, respectively; Fig. 19, left panel), were synthesized. In the halide salts of L4 and L5 (Fig. 19, right panel), anion-π interactions (3.18 and 3.28 Å for L4 and L5, respectively), as well as hydrogen bonds between the [Cl]– anion and the ammonium group of each ligand, result in robust structural motifs.25 In a similar vein, Mascal designed elegant novel cylindrophane-type highly selective fluoride receptors, both in the gas phase and in a water solvent model, based on π-electron deficient rings, e.g., s-triazine, cyanuric acid and boroxine (Fig. 5).20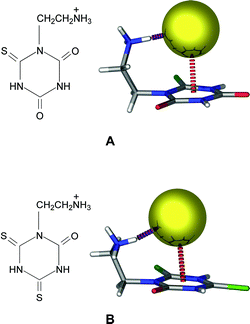 |
| | Fig. 19 Schematic drawing of the ligand and the crystal structure of its corresponding chloride complex for the ethylene ammonium derivative of (A) thiocyanuric acid (L4) and (B) dithiocyanuric acid (L5). Atom colors: C = grey, O = red, Cl = yellow, N = blue, S = green and H = white. Figure adapted from ref. 25. | |
A comprehensive crystallographic and theoretical study was undertaken by our group to probe the effect of anion-π interactions on the preferred structural motifs of the Ag(I) complexes obtained from the reaction of the Ag(I)X salts (X = [PF6]–, [AsF6]–, [SbF6]– and [BF4]–) with 3,6-bis(2′-pyridyl)-1,2,4,5-tetrazine (bptz; Fig. 20A) and 3,6-bis(2′-pyridyl)-1,2-pyridazine (bppn; Fig. 20B), which exhibit different π-acidities of their central rings (due to the four electron withdrawing nitrogen atoms on the central ring in bptz, compared to two in bppn; a significantly more electropositive area is observed in the bptz central tetrazine ring; see the ESP maps in Fig. 20).39 The bptz reactions led to polymeric, dinuclear and propeller-type species (Fig. 21A), depending on the anion, whereas the bppn reactions produced the grid-type structures [Ag4(bppn)4]4+ (Fig. 21B), regardless of the anion present. In the bppn structures, π–π stacking interactions are maximized, whereas multiple and shorter, and thus stronger, anion-π interactions between the anions and the tetrazine rings are established in the bptz complexes (shorter than those encountered in the bppn complexes by ∼0.2 Å).39 Furthermore, all the Ag(I) bptz complexes have more than one tetrazine ring π-contact per anion (e.g., in [Ag2(bptz)3][SbF6]2, each anion interacts with 3 tetrazine rings; Fig. 21A), whereas the bppn grids have only one pyridazine ring π-contact per anion (Fig. 21B). The multiple anion-π interactions per anion established in the case of the bptz complexes (as compared to one per anion in the bppn complexes) and the difference in the preferred structural motifs between the bptz and bppn structures are in accord with the higher π-acidic character of the bptz central tetrazine ring as compared to the more electron rich bppn pyridazine ring.39 It is notable that the structure of [Ag2(bptz)3][SbF6]2 exhibits the first crystallographic example of an anion-π6 system, i.e., a system having six anion-π interactions per participating [SbF6]– anion (Fig. 22).39 Another noteworthy point about this structure is that, apart from anion-π interactions, the central tetrazine ring of bptz participates in π–π stacking interactions with another tetrazine ring (Fig. 22). The cooperative effect between these non-bonding interactions has been theoretically investigated in elegant studies by Deyà et al.30,31
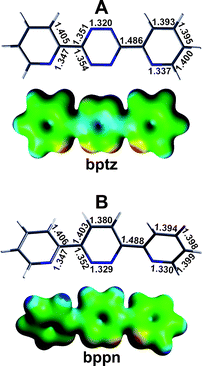 |
| | Fig. 20 BP86/TZP geometry optimization and ESP maps for (A) bptz and (B) bppn in the syn orientation. The ESP maps were generated with ADFView at an isodensity value of 0.02 and a color scale of 126 (blue) to – 63 (red) kcal mol–1. (Reproduced with permission39b from K. R. Dunbar et al., J. Am. Chem. Soc., 2006, 128, 5895. Copyright 2006 the American Chemical Society.) | |
![(A) A fragment of the [Ag2(bptz)3][SbF6]2 structure depicting three anion-π contacts between each [SbF6]– anion and the tetrazine rings. (B) The grid-type structure of [Ag4(bppn)4][SbF6]4 depicting the π–π (purple dashed lines) and anion-π interactions (red dashed lines). Figure adapted from ref. 39b.](/image/article/2008/CS/b614208g/b614208g-f21.gif) |
| | Fig. 21 (A) A fragment of the [Ag2(bptz)3][SbF6]2 structure depicting three anion-π contacts between each [SbF6]– anion and the tetrazine rings. (B) The grid-type structure of [Ag4(bppn)4][SbF6]4 depicting the π–π (purple dashed lines) and anion-π interactions (red dashed lines). Figure adapted from ref. 39b. | |
![Anion-π interactions between a [SbF6]– anion and six tetrazine rings in [Ag2(bptz)3][SbF6]2. F⋯centroid distance = 3.265(3) Å (red dashed lines), F⋯tetrazine plane distance = 2.844 Å. The π–π contacts (3.36 Å) are indicated with purple dashed lines. Figure reproduced with permission from ref. 39b. (Reproduced with permission39b from K. R. Dunbar et al., J. Am. Chem. Soc., 2006, 128, 5895. Copyright 2006 the American Chemical Society.)](/image/article/2008/CS/b614208g/b614208g-f22.gif) |
| | Fig. 22 Anion-π interactions between a [SbF6]– anion and six tetrazine rings in [Ag2(bptz)3][SbF6]2. F⋯centroid distance = 3.265(3) Å (red dashed lines), F⋯tetrazine plane distance = 2.844 Å. The π–π contacts (3.36 Å) are indicated with purple dashed lines. Figure reproduced with permission from ref. 39b. (Reproduced with permission39b from K. R. Dunbar et al., J. Am. Chem. Soc., 2006, 128, 5895. Copyright 2006 the American Chemical Society.) | |
The evidence gleaned from the solid state structures vis-à-vis the relative strength of the anion-π interactions for bptz and bppn, was corroborated by DFT single point energy calculations on several of their Ag(I) complexes. In Fig. 23, the ESP maps for [Ag2(bptz)2(CH3CN)2][AsF6]2 as (a) a dication and (b) a neutral species are shown. The dication (Fig. 23a) demonstrates high electropositive character (more blue) on the central tetrazine rings, which is greatly diminished in [Ag2(bptz)2(CH3CN)2][AsF6]2 (Fig. 23b) due to a flow of electron density from the [AsF6]– anions to the bptz π-acidic central rings and the establishment of favorable anion-π interactions between [AsF6]– and the tetrazine rings. In the singly-charged cation {[Ag2(bptz)2(CH3CN)2][AsF6]}+, the ring distal from the anion is clearly more electropositive than the ring in close proximity to the [AsF6]– anion (Fig. 24).
![ESP map from the BP86/TZP SPE calculations of (a) dication [Ag2(bptz)2(CH3CN)2]2+ with a color scale of 220 (blue) to –31 (red) kcal mol–1 and (b) neutral complex [Ag2(bptz)2(CH3CN)2][AsF6]2 with a color scale of 126 (blue) to –126 (red) kcal mol–1. The maps were generated with ADFView at a 0.02 isodensity value. (Reproduced with permission39b from K. R. Dunbar et al., J. Am. Chem. Soc., 2006, 123, 5895. Copyright 2006 the American Chemical Society.)](/image/article/2008/CS/b614208g/b614208g-f23.gif) |
| | Fig. 23 ESP map from the BP86/TZP SPE calculations of (a) dication [Ag2(bptz)2(CH3CN)2]2+ with a color scale of 220 (blue) to –31 (red) kcal mol–1 and (b) neutral complex [Ag2(bptz)2(CH3CN)2][AsF6]2 with a color scale of 126 (blue) to –126 (red) kcal mol–1. The maps were generated with ADFView at a 0.02 isodensity value. (Reproduced with permission39b from K. R. Dunbar et al., J. Am. Chem. Soc., 2006, 123, 5895. Copyright 2006 the American Chemical Society.) | |
![ESP map from the BP86/TZP SPE calculations for the singly-charged cation {[Ag2(bptz)2(CH3CN)2][AsF6]}+ with a color scale of 188 (blue) to –31 (red) kcal mol–1. The maps were generated with ADFView at a 0.02 isodensity value. (Reproduced with permission39b from K. R. Dunbar et al., J. Am. Chem. Soc., 2006, 128, 5895. Copyright 2006 the American Chemical Society.)](/image/article/2008/CS/b614208g/b614208g-f24.gif) |
| | Fig. 24 ESP map from the BP86/TZP SPE calculations for the singly-charged cation {[Ag2(bptz)2(CH3CN)2][AsF6]}+ with a color scale of 188 (blue) to –31 (red) kcal mol–1. The maps were generated with ADFView at a 0.02 isodensity value. (Reproduced with permission39b from K. R. Dunbar et al., J. Am. Chem. Soc., 2006, 128, 5895. Copyright 2006 the American Chemical Society.) | |
In the ESP maps of [Ag4(bppn)4][AsF6]4, the cationic unit demonstrates a small difference between the π-acidity of the central pyridazine ring and the outer pyridyl rings of bppn (Fig. 25a). Furthermore, the change in electropositive character of the bppn central rings in the cationic unit [Ag4(bppn)4]4+ is very small upon addition of the [AsF6]– anions, indicating negligible flow of electron density from the anion to the pyridazine ring in the neutral complex (Fig. 25b). These results concur with a more electron rich bppn central pyridazine ring, and thus much weaker anion-π interactions in the Ag(I)–bppn complexes as compared to those established in the Ag(I)–bptz complexes. In the formerly discussed studies of Ag(I)–bptz and Ag(I)–bppn complexes, polyatomic ions such as [BF4]–, [PF6]–, [AsF6]– and [SbF6]– were used to overcome solubility issues. Due to the lack of a theoretical investigation with such systems, we have also undertaken a comprehensive theoretical study of the anion-π interactions in a series of aromatic rings (e.g., benzene, pyridazine, triazine and tetrazine) with [BF4]– and [PF6]– ions; the results will be published soon. Recently, the results from the optimization of the trifluoro-s-triazine ring in the presence of complex anions at the MP219 level of theory were published by Deyà et al.40
![Electrostatic potential map from the BP86/TZP SPE calculations of the (a) cationic unit [Ag4(bppn)4]4+ with a color scale of 314 (blue) to –126 (red) kcal mol–1 and (b) neutral complex [Ag4(bppn)4][AsF6]4 with a color scale of 157 (blue) to –63 (red) kcal mol–1. The maps were generated with ADFView at a 0.02 isodensity value. The yellow areas on the anions are due to hydrogen bonding interactions between the anion fluorine atoms and the protons on the rings. (Reproduced with permission39b from K. R. Dunbar et al., J. Am. Chem. Soc., 2006, 128, 5895. Copyright 2006 the American Chemical Society.)](/image/article/2008/CS/b614208g/b614208g-f25.gif) |
| | Fig. 25 Electrostatic potential map from the BP86/TZP SPE calculations of the (a) cationic unit [Ag4(bppn)4]4+ with a color scale of 314 (blue) to –126 (red) kcal mol–1 and (b) neutral complex [Ag4(bppn)4][AsF6]4 with a color scale of 157 (blue) to –63 (red) kcal mol–1. The maps were generated with ADFView at a 0.02 isodensity value. The yellow areas on the anions are due to hydrogen bonding interactions between the anion fluorine atoms and the protons on the rings. (Reproduced with permission39b from K. R. Dunbar et al., J. Am. Chem. Soc., 2006, 128, 5895. Copyright 2006 the American Chemical Society.) | |
Another combined crystallographic and theoretical study by Reedijk et al. involved the ligand 2,4,6-tris(di-2-picolylamino)-[1,3,5]-triazine (L6; Fig. 26A).41 The ligand L6 reacts with Zn(NO3)·6H2O or Cu(NO3)·3H2O to afford [Zn3(L6)(NO3)6] or [Cu3(L6)(NO3)2(H2O)6][NO3]4, respectively. In these complexes, anion-π interactions are established between the triazine rings and the oxygen atoms of the nitrate ions (Fig. 26B). A notable point of the Cu(II) structure is that the triazine ring of L6 is simultaneously participating in two anion-π interactions, one on either side of the ring (centroid⋯O distances: 3.201 and 3.502 Å; Fig. 26B). The theoretical study of the nitrate-triazine ring-nitrate fragment at the MP219 level of theory resulted in an equilibrium distance of 3.2 Å between the tetrazine ring and the O atoms (Fig. 26C); the experimentally observed longer distance is attributed to the electron density donated to the tetrazine ring by the first nitrate ion, which makes the ring more electron rich and thus weakens the second nitrate-π interaction.3,41 Similar to those in Fig. 26C, ‘anion-π-sandwich’ and other notable anion-π-anion interactions were recently recorded in Co(II), Ni(II), Cd(II) 1-D coordination polymers of bis(2-pyrazylmethyl)sulfide and 2-benzylsulfanylmethylpyrazine (ligands with different π-acidities) in the presence of [NO3]– or [ClO4]– anions.42
![(A) Schematic drawing of 2,4,6-tris(di-2-picolylamino)-[1,3,5]-triazine (L6). (B) Crystal structure of [Cu3(L6)(NO3)2(H2O)6](NO3)4. (C) Theoretical model used for computational studies. Atom colors: O = red, N = dark blue, Cu = light blue, C = grey and H = white. Figure reproduced from ref. 41.](/image/article/2008/CS/b614208g/b614208g-f26.gif) |
| | Fig. 26 (A) Schematic drawing of 2,4,6-tris(di-2-picolylamino)-[1,3,5]-triazine (L6). (B) Crystal structure of [Cu3(L6)(NO3)2(H2O)6](NO3)4. (C) Theoretical model used for computational studies. Atom colors: O = red, N = dark blue, Cu = light blue, C = grey and H = white. Figure reproduced from ref. 41. | |
Further investigations on nitrate–triazine interactions were conducted by Reedijk et al. by reacting Cu(II) nitrate with the elaborate ligand N,N′,N″,N‴-tetrakis{2,4-bis(di-2-pyridylamino)-[1,3,5]-triazin-6-yl}triethylenetetraamine (L7; Fig. 27A).43 The obtained complex, [Cu4(L7)(NO3)4][NO3]4·12H2O (symmetry-related unit forming the tetranuclear complex shown in Fig. 27B), had a triazine ring simultaneously participating in π–π stacking and an anion-π interaction with the [NO3]– ion (Fig. 27c). This ‘dual’ interaction of the triazine ring, and the unusual position of the nitrate ion with respect to the triazine ring (with one oxygen atom pointing to one nitrogen atom of the ring), were theoretically analyzed by the authors.3,43
![(A) Schematic drawing of N,N′,N″,N‴-tetrakis{2,4-bis(dipyridylamino)-[1,3,5]-triazin-6-yl}triethylenetetraamine (L7). (B) Crystal structure of the unit {[Cu2(L7)(NO3)2][NO3]}+. (C) Model used for the theoretical studies. Atom colors: O = red, N = dark blue, Cu = light blue, C = gray and H = white. Figure adapted from ref. 43.](/image/article/2008/CS/b614208g/b614208g-f27.gif) |
| | Fig. 27 (A) Schematic drawing of N,N′,N″,N‴-tetrakis{2,4-bis(dipyridylamino)-[1,3,5]-triazin-6-yl}triethylenetetraamine (L7). (B) Crystal structure of the unit {[Cu2(L7)(NO3)2][NO3]}+. (C) Model used for the theoretical studies. Atom colors: O = red, N = dark blue, Cu = light blue, C = gray and H = white. Figure adapted from ref. 43. | |
Evidence of anion-π interactions in solution
Despite the mounting solid state evidence for anion-π interactions, few solution studies investigating anion-π contacts have emerged. The first study to recognize anion-π interactions in solution (although not named as such) was provided by Schneider et al., who reported weak but distinct attractive interactions between negative charges and the polarizable aryl parts of host–guest systems.8 In 2004, another solution study by Maeda et al.44 was performed on divalent metal (Ni(II), Pd(II) and Cu(II)) complexes of a tetrakis(pentafluorophenyl)-N-confused porphyrin (C6F5-NCP; L8), i.e., a porphyrin that has an α-β′ linked pyrrole ring, which thus possesses a reactive inner carbon and a peripheral nitrogen (Fig. 28). The metal complexes of L8 showed especially high association constants, Ka, for [F]– anions in CH2Cl2 (> 3 × 105 M–1), which were ascribed not only to the acidity of the NH peripheral group, but also to the anion-π interaction between the [F]– and the closest electron deficient fluorinated phenyl ring (Fig. 28). Evidence for this anion-π interaction in the Ni(II)–L8 complex was derived from the upfield shifts (∼2 ppm) of the 19F NMR resonances for the C6F5 ring near the N-confused pyrrole ring; the upfield shifts were attributed to the shielding effect due to the negative charge of the [F]– anion interacting with the C6F5 ring.44![Schematic drawing of an N-confused porphyrin (L8) interacting in solution with a halide anion [X]– through a hydrogen bond from the peripheral NH group on the pyrrole ring, as well as through an anion-π interaction with the nearest C6F5 ring. Figure adapted from ref. 44.](/image/article/2008/CS/b614208g/b614208g-f28.gif) |
| | Fig. 28 Schematic drawing of an N-confused porphyrin (L8) interacting in solution with a halide anion [X]– through a hydrogen bond from the peripheral NH group on the pyrrole ring, as well as through an anion-π interaction with the nearest C6F5 ring. Figure adapted from ref. 44. | |
Kochi et al. examined a series of neutral organic π-acceptors with electron deficient aromatic rings (Fig. 29a) in solution by UV/vis spectroscopy.45 Solutions of the tetraalkylammonium salts of [Cl]–, [Br]– or [I]– were slowly added to solutions of the depicted aromatic rings (Fig. 29a), which induced the appearance of new peaks in the spectra, indicative of the charge transfer (CT) character of the complexes, with appreciable formation constants and ratios [TCP/Cl–] = 1 : 1 to 4 : 1 (in Fig. 29b, the spectrum of tetracyanopyrazine (TCP), with incremental additions of [Pr4N][Br], is depicted).45 X-Ray structural analyses of the aforementioned systems with the halides (e.g., TCP with [Cl]–; Fig. 29c) revealed anion–ring contacts with distances shorter than the sum of the van der Waals radii. Regardless of the anion/ring ratio, close contacts (∼3.0 Å) exist between the halide anion and one or more aromatic rings, indicative of anion-π interactions originating from CT.45 The position of the anions over the periphery of the aromatic rings and the intense absorption bands in the visible region, however, firmly establish the CT character of these complexes, a finding that is corroborated by the recent study of Johnson et al.21
![(a) Schematic drawings of the aromatic systems studied by Kochi et al. (b) UV/vis spectra of a 5 mM tetracyanopyrazine (TCP) solution in acetonitrile upon the incremental additions of [Pr4N][Br]. Concentrations of [Br]–: line 1 = 0 mM, line 2 = 4.9 mM, line 3 =19 mM, line 4 = 46 mM, line 5 = 83 mM and line 6 = 208 mM. Inset: Mulliken dependence of the energy absorption band (νCT) on the reduction potential of the π-acceptor. (c) Crystal structure of TCP with [Cl]–. Atom colors: N = blue, C = grey and Cl = yellow. (Reproduced with permission45 from J. K. Kochi et al., Angew. Chem., Int. Ed., 2004, 43, 4650. Copyright 2004 Wiley-VCH Verlag GmbH & Co. KGaA.)](/image/article/2008/CS/b614208g/b614208g-f29.gif) |
| | Fig. 29 (a) Schematic drawings of the aromatic systems studied by Kochi et al. (b) UV/vis spectra of a 5 mM tetracyanopyrazine (TCP) solution in acetonitrile upon the incremental additions of [Pr4N][Br]. Concentrations of [Br]–: line 1 = 0 mM, line 2 = 4.9 mM, line 3 =19 mM, line 4 = 46 mM, line 5 = 83 mM and line 6 = 208 mM. Inset: Mulliken dependence of the energy absorption band (νCT) on the reduction potential of the π-acceptor. (c) Crystal structure of TCP with [Cl]–. Atom colors: N = blue, C = grey and Cl = yellow. (Reproduced with permission45 from J. K. Kochi et al., Angew. Chem., Int. Ed., 2004, 43, 4650. Copyright 2004 Wiley-VCH Verlag GmbH & Co. KGaA.) | |
A recent experimental study providing evidence of anion-π interactions in solution was reported by Johnson et al.46 Both ligands L9 and L10 (Fig. 30A), have the potential to establish hydrogen bonding between the NH group and the halide, but L9 has a significantly more electron deficient perfluorinated aryl ring as compared to L10. The comparative capacities of L9 and L10 to bind halides in solution were studied by 1H NMR titration experiments in CDCl3 in the presence of [n-BuN4]+ salts of [Cl]–, [Br]– and [I]–. For L9, the determined binding constants were large (Ka= 30, 20 and 34 M–1 for [Cl]–, [Br]– and [I]–, respectively), whereas for L10, the binding constants were too small (<1 M–1) to be measured appropriately by NMR spectroscopic methods.46 The theoretical geometry results at the HF23 level of theory (Fig. 30B) clearly show the [Cl]– ion positioned above the electron deficient C6F5 ring in the case of L9, as compared to the L10 complex. The latter results unambiguously confirm the presence of anion-π interactions for L9.46
![(A) Schematic drawings of L9 and L10. (B) HF/6-31+G* geometry optimization results of L9 and L10 in the presence of [Cl]– ions. (Reproduced with permission46 from D. W. Johnson et al., Chem. Commun., 2006, 506. Copyright 2006 the Royal Society of Chemistry (RSC) on behalf of the Centre National de la Recherche Scientifique (CNRS).)](/image/article/2008/CS/b614208g/b614208g-f30.gif) |
| | Fig. 30 (A) Schematic drawings of L9 and L10. (B) HF/6-31+G* geometry optimization results of L9 and L10 in the presence of [Cl]– ions. (Reproduced with permission46 from D. W. Johnson et al., Chem. Commun., 2006, 506. Copyright 2006 the Royal Society of Chemistry (RSC) on behalf of the Centre National de la Recherche Scientifique (CNRS).) | |
Systems with evidence of anion-π interactions both in the solid-state and in solution
Atwood et al. reported a series of organometallic calix[4]arene macrocyclic complexes that exhibited drastically altered host–guest behavior (enhancement of acidity for the interior of the cavity) due to capping of the calix[4]arene faces with transition metals (M = Rh, Ir or Ru), e.g., M = Ir and [BF4]– anions (Fig. 31). X-Ray crystallographic studies have shown that the tetrametallic species behave as anionic hosts, resulting in short anion⋯Ccalix contacts and appreciable anion binding constants (determined by 1H NMR titrations).47![X-Ray crystal structure of [{Ir(η5-C5Me5)}4(calix[4]arene)]6+ host with the [BF4]– anion in the π-acidic cavity. Atom colors: O = red, B = orange, Ir = light blue, F = green and C = grey. Short F⋯Ccalix contacts ∼2.9–3.0 Å. Figure adapted from ref. 47.](/image/article/2008/CS/b614208g/b614208g-f31.gif) |
| | Fig. 31 X-Ray crystal structure of [{Ir(η5-C5Me5)}4(calix[4]arene)]6+ host with the [BF4]– anion in the π-acidic cavity. Atom colors: O = red, B = orange, Ir = light blue, F = green and C = grey. Short F⋯Ccalix contacts ∼2.9–3.0 Å. Figure adapted from ref. 47. | |
In an analogous context, Fairchild and Holman reported an interesting system with evident anion-π interactions (in the solid state and in solution), wherein the six arene faces of Collet's cryptophane-E [(±)-E] were permetalated with [Cp*Ru(CH3CN)3]+ moieties to make the interior of the capsule π-acidic (Fig. 32A).48 Reaction of [(±)-E] with [Cp*Ru(CH3CN)3][CF3SO3] or [Cp*Ru(CH3CN)3][SbF6] resulted in the hexametalated capsule [(±)-2]6+with captured [CF3SO3]– or [SbF6]– ions inside the cavity (Fig. 32B). As indicated from the X-ray structural determinations, both anions are in close contact with the phenyl rings of the [(±)-2]6+ cryptophane. In the case of [(±)-2][CF3SO3]6, severe disorder precluded reliable determination of F⋯ring distances. The [(±)-2][SbF6]6 structure, however, revealed close anion F⋯centroid (3.08 Å) and F⋯C (2.97 Å) contacts. In solution, these cryptophanes accommodate anions in the cavities and show a preference for certain anions.48 The 19F NMR spectrum of [(±)-2][CF3SO3]6 in CD3NO2 exhibits two 19F resonances with a 1 : 5 (encapsulated : free) ratio (Fig. 32C, right spectrum). If [(±)-2][CF3SO3]6 is dissolved in CD3NO2 in the presence of [PF6]– ions, the triflate is exchanged for the smaller anion. This exchange does not take place with [BF4]– ions, most likely due to their smaller size, which precludes favorable anion-π contacts with the inner surfaces of the π-acidic phenyl rings.
![(A) Synthesis of (±)-{Cp*Ru)6(E)]6+ from (±)-cryptophane-E [(±)-E] and [Cp*Ru(CH3CN)3][X], where X = [CF3SO3]– or [PF6]–. (B) X-Ray crystal structures of {[(±)-2][CF3SO3]}5+ and {[(±)-2][SbF6]}5+. Atom colors: O = red, S = yellow, N = dark blue, Ru = light blue, F = green, C = grey and H = white. (C) 1H (left) and 19F (right) NMR spectra of [(±)-2][CF3SO3]6 at equilibrium in CD3NO2. (Reproduced with permission48 from K. T. Holman et al., J. Am. Chem. Soc., 2005, 127, 16364. Copyright 2005 the American Chemical Society.)](/image/article/2008/CS/b614208g/b614208g-f32.gif) |
| | Fig. 32 (A) Synthesis of (±)-{Cp*Ru)6(E)]6+ from (±)-cryptophane-E [(±)-E] and [Cp*Ru(CH3CN)3][X], where X = [CF3SO3]– or [PF6]–. (B) X-Ray crystal structures of {[(±)-2][CF3SO3]}5+ and {[(±)-2][SbF6]}5+. Atom colors: O = red, S = yellow, N = dark blue, Ru = light blue, F = green, C = grey and H = white. (C) 1H (left) and 19F (right) NMR spectra of [(±)-2][CF3SO3]6 at equilibrium in CD3NO2. (Reproduced with permission48 from K. T. Holman et al., J. Am. Chem. Soc., 2005, 127, 16364. Copyright 2005 the American Chemical Society.) | |
Importance and applications of anion-π interactions
The aforementioned theoretical and experimental studies indicate the rising interest of the scientific community in anion-π interactions, underscoring their significance. The design of efficient synthetic anion receptors or transporters is of prime importance for environmental (e.g., sensing, and removal of nitrate and phosphate ions from drinking water),3,18 biological and medicinal purposes (e.g., synthetic ion channels, membranes and pores),3,20,49 as well as catalysis.25 Among the other advantages, the neutrality of the receptors (aromatic rings), and the variety in size, geometry and shape of the anions, greatly improves the selectivity and directionality of anion-π interactions.13,18 Recent studies indicate that electron deficient π-rings are excellent candidates for the molecular recognition20,25,45,46,50 and transport51,52 of anions. In this respect, Mascal has proposed novel cylindrophane-type receptors based on π-electron deficient rings, e.g., triazine, cyanuric acid and boroxine (Fig. 33), which demonstrate a high level of selectivity for fluoride, both in the gas phase and in a water solvent model.20 Such receptors may find applications in both sensing and in the 19F labeling of targeted tracer probes in nuclear medicine.20 Moreover, controlled chloride ion sensing and selective recognition was recently achieved by synthesizing an artificial redox-active Fe-pseudocryptand with an isocyanuric platform (Fig. 34) that establishes favorable anion-π interactions with chloride ions.50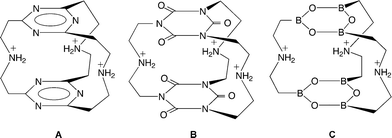 |
| | Fig. 33 Cylindrophane-type selective fluoride receptors based on π-electron deficient rings (A) 1,3,5-triazine, (B) cyanuric acid and (C) boroxine. Figure adapted from ref. 20. | |
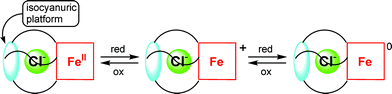 |
| | Fig. 34 The multi-step regulation of anion recognition by employing a redox-active Fe-pseudocryptand with an isocyanuric platform that generates a selective chloride binding cavity. Figure adapted from ref. 50. | |
In a recent remarkable investigation, Matile et al. synthesized and evaluated an unprecedented synthetic ion channel based on π-acidic oligo-(para-phenylene)-N,N-naphthalenediimide (O-NDI) rods as transmembrane chloride π-slides (Fig. 35B).51 The N,N-naphthalenediimide (NDI) building unit has a highly positive global quadrupole moment (Qzz = +19.4 B) and exhibits an electron deficient character at the center of the molecule (Fig. 35A). The O-NDI channel showed an unusual ion selectivity ([F]– > [Cl]– > [Br]– > [I]–), attributed to the existence of remarkably powerful anion-π interactions with the π-slide which compensate for the cost of ion dehydration.51 Such synthetic channels are of great interest in light of the importance of anion channels in diseases such as cystic fibrosis and other anion channelopathies.3,49
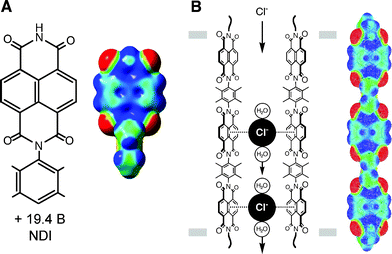 |
| | Fig. 35 (A) The N,N-naphthalenediimide unit (NDI) and its DFT-computed ESP map. (B) An oligo-(para-phenylene)-N,N-naphthalenediimide (O-NDI) rod as a chloride π-slide in lipid bilayer membranes and its DFT-computed ESP map. Blue areas: electron poor, red areas: electron rich. (Reproduced with permission51 from S. Matile et al., J. Am. Chem. Soc., 2006, 128, 14788. Copyright 2006 the American Chemical Society.) | |
In recent times, the bases of nucleic acids (especially when protonated), which are electron poor aromatic moieties, have been shown to establish anion-π interactions with various anions, as well as the lone pairs of electron rich molecules, e.g., H2O.3,53 The latter interactions have also been observed in proteins (especially with protonated histidine moieties), although to a lesser extent, due to the difficulty of reliably locating hydrogen atoms in the crystal structures of proteins, even at very high resolution.53 The recent finding that even non-electron deficient aromatic rings (e.g., the aromatic side groups of amino acids) can establish anion-π interactions under certain conditions,16–18 and the fact that 70% of enzyme substrates are anions,26,33 underscore the prime importance of further investigating the molecular basis of anion-π interactions and their hitherto unexplored consequences in biological systems.
Concluding remarks
This tutorial review provides an overview of the theoretical and experimental investigations that led to the recognition of the attractive, yet novel, anion-π interactions. During the initial phase of the relevant studies, basic principles were established and systems that exhibit anion-π interactions were unearthed. As anion-π interactions are gaining significant recognition from the scientific community and their pivotal role in many key chemical and biological processes is being appreciated, the challenge now lies in the design and development of highly selective anion receptors or transporters that will effectively accomplish various functions of prime environmental or biological importance.Acknowledgements
We would like to thank Professor M. Mascal and Professor S. Matile for kindly providing original figures from their work, as well as for helpful discussions. K. R. D. gratefully acknowledges the Robert A. Welch Foundation (A-1449) and the National Science Foundation (CHE-9906583). We would like to thank Mr Edward S. Funck and Mr Ian D. Giles for careful reading of this manuscript and helpful suggestions. We would also like to thank Dr L. M. Pérez for helpful discussions, as well as the Supercomputing Facility and Laboratory for Molecular Simulation at Texas A&M University for providing software and computer time.References
- J. Steed and J. L. Atwood, Supramolecular Chemistry, John Wiley, New York, 2001 Search PubMed.
- Special issue dedicated to Supramolecular Chemistry: Chem. Soc. Rev., 2007, 36(2), 125–440 Search PubMed.
- P. Gamez, T. J. Mooibroek, S. J. Teat and J. Reedijk, Acc. Chem. Res., 2007, 40, 435 CrossRef CAS and references therein.
- A. Bianchi and E. García-España, Thermodynamics of Anion Complexation in, Supramolecular Chemistry of Anions, ed. A. Bianchi, K. Bowman-James and E. García-España, Wiley-VCH, New York, 1997, pp. 217 Search PubMed.
- C. H. Park and H. E. Simmons, J. Am. Chem. Soc., 1968, 90, 2431 CrossRef CAS.
- P. D. Beer and P. A. Gale, Angew. Chem., Int. Ed., 2001, 40, 486 CrossRef and references therein.
- S. O. Kang, R. A. Begum and K. Bowman-James, Angew. Chem., Int. Ed., 2006, 250, 7882 CrossRef.
- H.-J. Schnieder, F. Werner and T. Blatter, J. Phys. Org. Chem., 1993, 6, 590 CrossRef CAS and references therein.
- I. Alkorta, I. Rozas and J. Elguero, J. Am. Chem. Soc., 2002, 124, 8593 CrossRef CAS.
- D. Quiñonero, C. Garau, C. Rotger, A. Frontera, P. Ballester, A. Costa and P. M. Deyà, Angew. Chem., Int. Ed., 2002, 41, 3389 CrossRef CAS.
- M. Mascal, A. Armstrong and M. D. Bartberger, J. Am. Chem. Soc., 2002, 124, 6274 CrossRef CAS.
- C. Garau, A. Frontera, D. Quiñonero, P. Ballester, A. Costa and P. M. Deyà, Anion-π Interactions, in Recent Research Developments in Chemical Physics, ed. S. G. Pandalai, Transworld Research Network, Kerala, India, 2004, vol. 5, pp. 227 Search PubMed and references therein.
- C. Garau, A. Frontera, D. Quiñonero, P. Ballester, A. Costa and P. M. Deyà, ChemPhysChem, 2003, 4, 1344 CrossRef CAS.
- C. Garau, A. Frontera, D. Quiñonero, P. Ballester, A. Costa and P. M. Deyà, J. Phys. Chem. A, 2004, 108, 9423 CrossRef CAS and references therein.
- D. Quiñonero, A. Frontera, D. Escudero, P. Ballester, A. Costa and P. M. Deyà, ChemPhysChem, 2007, 8, 1182 CrossRef CAS.
- C. Garau, D. Quiñonero, A. Frontera, P. Ballester, A. Costa and P. M. Deyà, New J. Chem., 2003, 27, 211 RSC.
- A. Clements and M. Lewis, J. Phys. Chem. A, 2006, 110, 12705 CrossRef CAS.
- C. Garau, A. Frontera, P. Ballester, D. Quiñonero, A. Costa and P. M. Deyà, Eur. J. Org. Chem., 2005, 179 CrossRef CAS.
- C. Möller and M. S. Plesset, Phys. Rev., 1934, 46, 618 CrossRef CAS; M. Head-Gordon, J. A. Pople and M. Frisch, J. Chem. Phys. Lett., 1988, 153, 503 Search PubMed.
- M. Mascal, Angew. Chem., Int. Ed., 2006, 45, 2890 CrossRef CAS and references therein.
- O. B. Berryman, V. S. Bryantsev, D. P. Stay, D. W. Johnson and B. P. Hay, J. Am. Chem. Soc., 2007, 129, 48 CrossRef CAS.
-
(a) R. F. W. Bader, Atoms in Molecules. A Quantum Theory, Clarendon, Oxford, UK, 1994 Search PubMed;
(b) R. F. W. Bader, Chem. Rev., 1991, 91, 893 CrossRef CAS.
-
(a) C. C. Roothan, Rev. Mod. Phys., 1951, 23, 69 CrossRef CAS;
(b) J. A. Pople and R. K. Nesbet, J. Chem. Phys., 1954, 22, 571 CAS;
(c) R. McWeeny and G. Dierksen, J. Chem. Phys., 1968, 49, 4852 CrossRef CAS.
- C. Garau, A. Frontera, D. Quiñonero, P. Ballester, A. Costa and P. M. Deyà, Chem. Phys. Lett., 2002, 359, 486 CrossRef CAS.
- A. Frontera, F. Saczewski, M. Gdaniec, E. Dziemidowicz-Borys, A. Kurland, P. M. Deyà, D. Quiñonero and C. Garau, Chem.–Eur. J., 2005, 11, 6560 CrossRef CAS and references therein.
- D. Kim, P. Tarakeshwar and K. S. Kim, J. Phys. Chem. A, 2004, 108, 1250 CrossRef CAS.
- C. Garau, A. Frontera, D. Quiñonero, P. Ballester, A. Costa and P. M. Deyà, Chem. Phys. Lett., 2004, 392, 85 CrossRef CAS.
- C. Garau, A. Frontera, D. Quiñonero, P. Ballester, A. Costa and P. M. Deyà, Chem. Phys. Lett., 2002, 399, 220.
- C. Garau, D. Quiñonero, A. Frontera, P. Ballester, A. Costa and P. M. Deyà, J. Phys. Chem. A, 2005, 109, 9341 CrossRef CAS.
- C. Garau, D. Quiñonero, A. Frontera, P. Ballester, A. Costa and P. M. Deyà, ChemPhysChem, 2006, 7, 2487 CrossRef.
- A. Frontera, D. Quiñonero, A. Costa, P. Ballester and P. M. Deyà, New J. Chem., 2007, 31, 556 RSC.
- S. Demeshko, S. Dechert and F. Meyer, J. Am. Chem. Soc., 2004, 126, 4508 CrossRef CAS.
- P. de Hoog, P. Gamez, I. Mutikainen, U. Turpeinen and J. Reedijk, Angew. Chem., Int. Ed., 2004, 43, 5815 CrossRef CAS.
-
(a) C. S. Campos-Fernández, R. Clérac and K. R. Dunbar, Angew. Chem., Int. Ed., 1999, 38, 3477 CrossRef CAS;
(b) C. S. Campos-Fernandez, R. Clérac, J. M. Koomen, D. H. Russell and K. R. Dunbar, J. Am. Chem. Soc., 2001, 123, 773 CrossRef CAS;
(c) C. S. Campos-Fernández, B. L. Schottel, H. T. Chifotides, J. K. Bera, J. Bacsa, J. M. Koomen, D. H. Russell and K. R. Dunbar, J. Am. Chem. Soc., 2005, 127, 12909 CrossRef CAS.
- I. A. Gural'skiy, P. V. Solntsev, H. Krautscheid and K. V. Domasevitch, Chem. Commun., 2006, 4808 RSC.
- S. Furkawa, T. Okubo, S. Masaoka, D. Tanaka, H. Chang and S. Kitagawa, Angew. Chem., Int. Ed., 2005, 44, 2700 CrossRef CAS.
- P. S. Szalay, J. R. Galán-Mascarós, B. L. Schottel, J. Bacsa, L. M. Pérez, A. S. Ichimura, A. Chouai and K. R. Dunbar, J. Cluster Sci., 2004, 15, 503 CrossRef CAS.
- I. M. Piglosiewicz, R. Beckhaus, G. Wittstock, W. Saak and D. Haase, Inorg. Chem., 2007, 46, 7610 CrossRef CAS.
-
(a) B. L. Schottel, J. Bacsa and K. R. Dunbar, Chem. Commun., 2005, 46 RSC;
(b) B. L. Schottel, H. T. Chifotides, M. Shatruk, A. Chouai, J. Bacsa, L. M. Pérez and K. R. Dunbar, J. Am. Chem. Soc., 2006, 128, 5895 CrossRef CAS.
- C. Garau, D. Quiñonero, A. Frontera, D. Escudero, P. Ballester, A. Costa and P. M. Deyà, Chem. Phys. Lett., 2007, 438, 104 CrossRef CAS.
- P. U. Maheswari, B. Modec, A. Pevec, B. Kozlevcar, C. Massera, P. Gamez and J. Reedijk, Inorg. Chem., 2006, 45, 6637 CrossRef CAS.
- C. A. Black, L. R. Hanton and M. D. Spicer, Inorg. Chem., 2007, 46, 3669 CrossRef CAS.
- H. Casellas, C. Massera, F. Buda, P. Gamez and J. Reedijk, New J. Chem., 2006, 30, 1561 RSC.
-
(a) H. Maeda, A. Osuka and H. Furuta, J. Inclusion Phenom. Macrocyclic Chem., 2004, 49, 33 Search PubMed;
(b) H. Maeda and H. Furuta, J. Porphyrins Phthalocyanines, 2004, 8, 67 CrossRef CAS.
- Y. S. Rosokha, S. V. Lindeman, S. V. Rosokha and J. K. Kochi, Angew. Chem., Int. Ed., 2004, 43, 4650 CrossRef CAS.
- O. B. Berryman, F. Hof, M. J. Hynes and D. W. Johnson, Chem. Commun., 2006, 506 RSC.
- M. Staffilani, K. S. B. Hancock, J. W. Steed, K. T. Holman, J. L. Atwood, R. K. Juneja and R. S. Burkhalter, J. Am. Chem. Soc., 1997, 119, 6324 CrossRef CAS.
- R. M. Fairchild and K. T. Holman, J. Am. Chem. Soc., 2005, 127, 16364 CrossRef CAS.
- A. P. Davis, D. N. Sheppard and B. D. Smith, Chem. Soc. Rev., 2007, 36, 348 RSC.
- T. Nabeshima, S. Masubuchi, N. Taguchi, S. Akine, T. Saiki and S. Sato, Tetrahedron Lett., 2007, 48, 1595 CrossRef CAS.
- V. Gorteau, G. Bollot, J. Mareda, A. Perez-Velasco and S. Matile, J. Am. Chem. Soc., 2006, 128, 14788 CrossRef CAS.
- V. Gorteau, G. Bollot, J. Mareda and S. Matile, Org. Biomol. Chem., 2007, 5, 3000 RSC.
- M. Egli and S. Sarkhel, Acc. Chem. Res., 2007, 40, 197 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2008 |
Click here to see how this site uses Cookies. View our privacy policy here.