Dendrimeric phosphines in asymmetric catalysis
Anne-Marie
Caminade
*,
Paul
Servin
,
Régis
Laurent
and
Jean-Pierre
Majoral
*
Laboratoire de Chimie de Coordination du CNRS, 205 route de Narbonne, 31077 Toulouse Cedex 4, France. E-mail: caminade@lcc-toulouse.fr; majoral@lcc-toulouse.fr; Fax: +33 5 6155 3003; Tel: +33 5 6133 3125
First published on 3rd September 2007
Abstract
Dendrimers are hyperbranched nanosized and precisely defined molecules, attracting increasing attention each year due to their numerous properties in catalysis, materials science, and biology. This tutorial review concerns the use of dendrimers as catalysts and focuses more precisely on their properties as enantioselective catalysts. Emphasis is put on chiral phosphine complexes constituting the core or the end groups of various types of dendrimers. The effect of the location of the catalytic entities, the effect of the size (generation) and the nature of the dendritic skeleton on the enantiomeric excesses are discussed.
 Anne-Marie Caminade | Anne-Marie Caminade is Director of Researches First Class at the CNRS since 2003 and co-head of the research group “Molecular, Macromolecular and Supramolecular Chemistry” in Toulouse. After two PhDs (1984 and 1988 in Toulouse), and two Post-docs (Institut Français du Pétrole and in Pr. M. Veith's group, Saarbrücken, Germany), she has worked since 1985 at the CNRS, where she is Director of Researches since 1997. Her research interest is in main group elements. She developed several aspects of the chemistry of phosphorus, including low coordinated compounds, coordination of transition metals, and synthesis of macrocycles. Her current research interest is in the synthesis, reactivity and study of properties and applications of dendrimers and dendritic molecules. She is the co-author of 250 publications and 25 patents, and received in 2006 the Organic Chemistry Prize of the French Chemical Society. |
 Paul Servin | Paul Servin received his MSc in Chemistry from Lund University, Sweden, in 2003. After his graduation, he started to work for Akzo Nobel in the Netherlands. He is currently a PhD student (Marie Curie Research grant) working under the supervision of A.-M. Caminade and J.-P. Majoral at LCC/CNRS in France. His research interest is to make water-soluble catalytic dendrimers for industrial application. |
 Régis Laurent | Régis Laurent was born in Porvoo (Finland) and studied chemistry at the University of Toulouse (France) where he received his PhD in 1994 under the supervision of Pr. J. Dubac and Pr A. Laporterie, working on microwave activation in organic chemistry. After post-doctoral studies for the BASF Company at the University of Saarbrücken (Germany) in the group of M. Veith, and an assistant researcher-teacher position (ATER) at the University of Toulouse in the group of L. Gorrichon, he got in 1996 a position at the CNRS in the group of J.-P. Majoral in Toulouse. He is currently “Chargé de Recherches”. His research interests are in the synthesis of phosphorus-containing dendrimers and dendrons, their applications in catalysis, asymmetric catalysis, catalysis in water, and in the elaboration of dendronized chiral conjugated polymers. |
 Jean-Pierre Majoral | Jean-Pierre Majoral is Emeritus Director of Researches Exceptional Class at the CNRS. He received his PhD from the Université Paul Sabatier of Toulouse in 1972. In 1972–1973 he worked as a Post-doc with Prof. A. Katritzky (University of East Anglia, Norwich, UK). He became Directeur de Recherche at the Centre National de la Recherche Scientifique in Toulouse in 1978. His research interest is mainly focused on the use of main group elements, especially phosphorus, in different areas of chemistry. Presently, he is involved in the preparation and the properties of macromolecules such as dendrimers and hyperbranched polymers. Emphasis is also laid on the studies of interactions between heavier main group elements and group 4 elements (titanium, zirconium, hafnium) with applications in organic and organometallic chemistry. He is a member of the Polish Academy of Sciences and of the Academia Europaea. He is the author of more than 400 publications and 31 patents. |
Introduction
Despite more than one century of industrial uses of catalysts,1 catalysis remains a highly active research area in both academia and industry. In particular, molecular catalysis research is directed towards the discovery or optimization of catalysts that have improved efficiencies, higher enantioselectivities, longer lifetimes, tolerance of air and moisture, and/or easier separation, recovery and recycling. This search is still of paramount importance connected in particular to the recent interest in “green chemistry”2 and “atom economy”.3 To fulfil most of these criteria, new approaches continue to inspire basic researches. Efforts have primarily involved the grafting of catalysts to soluble polymers. However, such an approach also has several drawbacks, such as a low loading capacity and above all poor definition of the polymeric catalysts, inducing problems of reproducibility.On the other hand, a very special type of branched polymer called “dendrimers”4–6 has attracted considerable attention as soluble supports of catalytic entities. In contrast to classical polymers, dendrimers enable the elaboration of precisely controlled structures. Dendrimers have been at the forefront of polymer research for almost two decades as a consequence of their very exceptional characteristics compared to conventional polymers: (i) a precisely defined and monodispersed structure, due to their step-by-step repetitive synthesis (“generation” after “generation”†), (ii) a highly branched architecture which enables the interior and exterior parts to be distinguished, and (iii) a large number of terminal groups, which governs the interactions of dendrimers with their environment. These unique features of dendritic macromolecules have shifted the main focus of the research community towards the design of functional dendrimers for specific high-end applications, amongst which catalysis occupies an important place since the first example in 1994,7 as emphasized in particular by the number of reviews published in this field within the past two years.8–17
Attachment of catalytic entities to a dendrimer is generally expected to favour the separation (and the reuse) of the catalyst from the products, but also to give rise to certain properties that are unavailable otherwise, such as an enhanced stability through steric isolation, or cooperativity between catalytic entities through proximal confinement. Furthermore, the number of catalytic entities attached to the dendrimer as well as their location can be regulated, which can be of crucial importance for the catalytic performance of the system. The most straightforward approach concerns their grafting as end groups to the dendrimer surface. This approach was the first one described,7 and is the most developed because it can be carried out by using a single reaction from commercially available dendrimers, and affords a high loading in catalytic entities (for a recent example of a dramatic “dendritic effect” see ref. 18). Another type of location for the catalytic entities is at the core of a dendrimer, generally called a dendron in this case. The synthesis is the same as for classical dendrimers but applied to a suitably functionalized core, and the surface groups can be varied; the main drawback of this approach is the very low loading (a single catalytic site per dendrimer). However, it is also rather frequently used with the aim of observing the effect of confinement (Fig. 1).
 | ||
| Fig. 1 Most frequently encountered locations of catalytic entities in dendritic molecules: as end groups (left) or at the core of dendrons (right). | ||
An essential component of the research about catalysis in general concerns asymmetric catalysis, which has led to breakthroughs in chemical sciences, not only in laboratories but also in industry, particularly in the manufacture of pharmaceuticals and agrochemicals.19 This topic is believed to be essential for the technologies of the 21st century, and researchers have discovered the opportunity to combine dendrimers and asymmetric catalysis.9 The aim of this review is to present an emerging area of research, which concerns dendrimeric chiral phosphines and their use in asymmetric catalysis. Indeed, metallic complexes of chiral phosphines have found multiple uses in asymmetric catalysis, but relatively few examples are known up to now in the field of dendrimers.
Synthesis of dendrimeric chiral phosphines
As shown in Fig. 1, the catalytically active site(s) can be located either at the core or at the surface of the dendritic structure. The nature of the dendritic scaffolds used to support the phosphines is diversified, as well as the type of chiral phosphines grafted, thus it appears important to describe first the synthesis of the diverse dendrimers synthesized. The methods of synthesis of the two types of compounds differ, thus they will be presented separately, beginning with phosphines linked to the core of dendrons, since this was the first example described.20Phosphine(s) at the core of chiral dendrons
In 1994, Brunner and Fürst presented an expanded chelate phosphine bearing 8 chiral substituents (compound 1a), which can be considered as the first example of a first generation chiral dendron bearing phosphines at its core (Scheme 1).20 The synthesis was carried out by reacting a tetrachlorodiphosphine with lithiobenzene derivatives bearing chiral (–)-borneol. This method was then applied to other diphosphines and other substituents of the aromatic, affording compounds 1b and 1c.21 Interestingly, this method was compatible with the presence of acetals (compounds 1d, 1e), which were then converted to aldehydes, which are prone to react with a variety of primary amines bearing chiral substituents. The series of first generation dendrons 2a–2g, 2i, and 3a–3h were obtained in this way (Scheme 1).22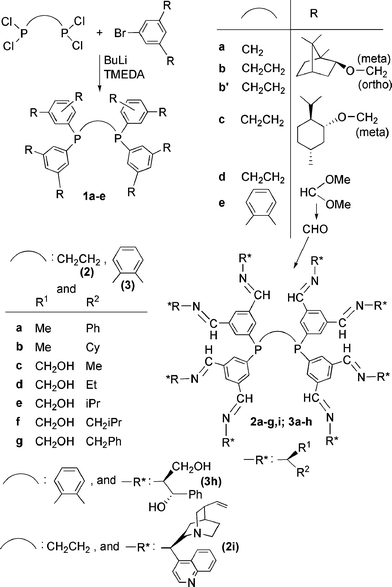 | ||
| Scheme 1 Synthesis of “expanded phosphines”. | ||
After this pioneering work, several other types of chiral dendrons possessing larger dendritic wedges and one or two phosphines at the core were described. In particular, Q. H. Fan et al.23–25 published a series of paper concerning a BINAP (2,2′-bis(diphenylphosphino)-1,1′-binaphthalene) ligand linked to the core of several generations of polyaryl ethers (“Fréchet” type dendrons).26 The reaction consisted of the attachment of dendritic wedges with a carboxylic acid at the focal point to 5,5′-diamino-BINAP (Scheme 2). The reaction was first carried out with dendrons where R = benzyl up to generation 3,23 then generation 425 (dendrons 4a-Gn, n = 1–4). It was also applied to dendrons possessing a variable number of alkyl chains to increase the solubility in organic solvents (dendrons 4b-Gn and 4c-Gn, n = 1–2).24 The same condensation method was also applied to 3,4-bis(diphenylphosphino)pyrrolidine to afford the series 5-Gn (n = 0–3) (Scheme 2).27
 | ||
| Scheme 2 Synthesis of dendritic BINAP and bis(diphenylphosphino)pyrrolidine. | ||
Another method was applied to the synthesis of polyether dendrons linked to a bridged biphenyl phosphine. In this case, the reaction occurred between the biphenol derivative of the protected (oxidized) phosphine and the dendritic wedge bearing a benzyl bromide at the focal point. In a second step, the phosphine oxides were reduced using HSiCl3. The series of dendritic compounds 6-Gn (n = 1–3) was obtained in this way (Scheme 3).28
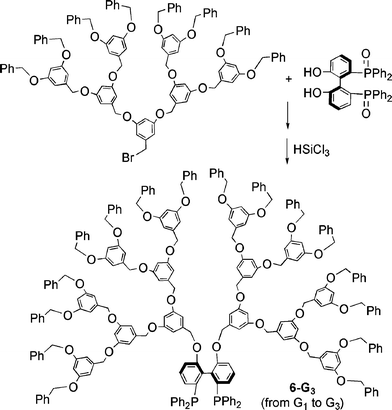 | ||
| Scheme 3 Synthesis of dendritic biphenyl phosphines. | ||
In an extension of this work, the same group reported recently the synthesis of dendritic chiral monodentate phosphoramidate ligands. The synthetic process again started from a polyether dendron bearing in this case an amino group at the focal point, which was first acetylated, then reduced to afford the secondary amine shown in Scheme 4. These dendrons were reacted with a chiral monophosphite, affording the series of dendrons 7-Gn (n = 0–2).29
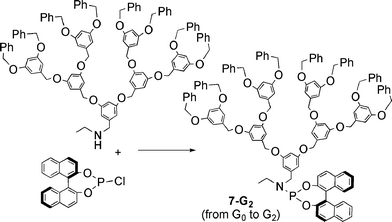 | ||
| Scheme 4 Synthesis of dendritic phosphoramidate ligand. | ||
Another example of a dendritic phosphoramidate compound was proposed by J. N. H. Reek, J. H. van Maarseveen et al.30 In this case, the nature of the dendritic wedge is totally different from the previous ones, since it is constituted of carbosilane derivatives. The synthetic process required several steps starting from a protected dicarbazole. The dendritic wedges were first grafted to the dicarbazole, the phenol groups were deprotected and reacted with P(NMe2)3 to afford compounds 8a,b-G3, but 8a-G3 rapidly decomposed (Scheme 5). This is the last example of phosphino derivatives linked to the core of chiral dendrons.
 | ||
| Scheme 5 Synthesis of phosphoramidate carbosilane dendrons. | ||
Phosphines as end groups of chiral dendrimers
In a series of papers starting in 1998, A. Togni et al. reported the first examples of dendrimers having chiral phosphino derivatives as end groups.31–33 In all cases, chiral ferrocenyldiphosphine ligands were linked to the surface of small dendrimers by a convergent process. These ligands were first connected to a dendron, which was then used to be grafted to various cores, such as benzene-1,3,5-tricarboxylic acid trichloride, adamantane-1,3,5,7-tetracarboxylic acid tetrachloride,31 or cyclo-tri- or tetra-phosphazene,32 affording the series of compounds 9a–c-Gn, 10a–c-Gn, 11a,d-Gn, and 12a,d-Gn, respectively, with n = 1 in most cases (Scheme 6). | ||
| Scheme 6 Synthesis of various types of dendrimers containing chiral ferrocenyldiphosphine ligands as end groups. | ||
Other series of dendrimers bearing chiral diphosphines as end groups were synthesized by L. Gade et al.34,353,4-Bis(diphenylphosphino)pyrrolidine was grafted using a strategy developed in polypeptide synthesis between amino-terminated dendrimers and carboxylic acid functionalized pyrrolidines. The well-known PPI (poly(propyleneimine))36–38dendrimers were used first,34 then the reaction was also applied to the well-known PAMAM (poly(amidoamine))39dendrimers to afford, in both cases from generations 0 to 4, dendrimers 13-Gn and 14-Gn, respectively (Scheme 7).
 | ||
| Scheme 7 Grafting of bis(diphenylphosphino)pyrrolidine to the surface of PPI and PAMAM dendrimers. | ||
We recently proposed the use of phosphorus-containing dendrimers40,41 for the synthesis of chiral dendrimers with monophosphines as end groups. The first dendrimer we described was built from a trifunctional core,40 and was synthesized in the last step by condensation reactions between the 24 aldehyde end groups of the third generation and (2S)-2-amino-1-(diphenylphosphinyl)-3-methylbutane, affording the chiral dendrimer15-G3 (Scheme 8).42
 | ||
| Scheme 8 Synthesis of a phosphorus dendrimer bearing chiral iminophosphines as end groups. | ||
The second type of phosphorus dendrimers with chiral phosphines as end groups was synthesized from a hexafunctional core (cyclotriphosphazene).41 Chiral ferrocenylphosphine-thioether ligands that have planar chirality were grafted by reaction of a phenol with the P(S)Cl2 end groups in basic conditions (caesium carbonate or sodium hydride). The series of dendrimers 16-Gn was synthesized from generation 1 to generation 4 (Scheme 9).43 Finally, a series of small generation carbosilane dendrimers with chiral phosphines as end groups was proposed by O. Rossell et al.44,45 The lithium derivatives of chiral phosphines protected by borane were reacted with the Si–Cl end groups of carbosilane dendrimers to afford compounds 17a-Gn (n = 0, 1) and 17b-G0 (Scheme 10). These compounds are unique examples up to now in which the chirality is directly on the phosphorus due to the unsymmetrical substitution of the phosphines.
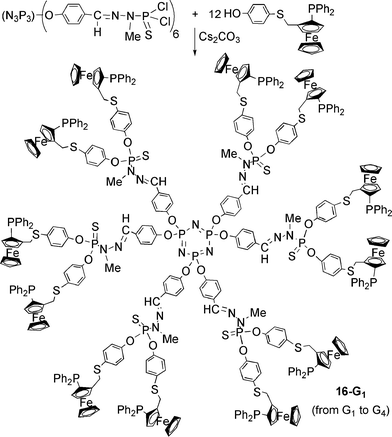 | ||
| Scheme 9 Grafting of chiral ferrocenylphosphines to a series of phosphorus dendrimers. | ||
 | ||
| Scheme 10 Grafting of chiral phosphines to carbosilane dendrimers. | ||
Catalytic properties of dendrimeric chiral phosphines
The dendrimeric phosphines shown in the first part of this review were all used for the complexation of transition metals, in particular rhodium and ruthenium derivatives. In most cases, the corresponding complexes were used as catalysts for the asymmetric hydrogenation of unsaturated bonds. Some palladium derivatives were also synthesized and mainly used for asymmetric allylations. In all cases, the percentage of catalyst is expressed in mol% of the number of catalytic sites, i.e. the number of end groups in the case of catalysts linked to the surface of the dendrimer.Asymmetric hydrogenations
The first examples of tentative enantioselective catalysis using dendrimeric phosphine complexes concerned the expanded phosphines shown in Scheme 1. The complexes obtained by the in situ de-dimerization of [Rh(COD)Cl]2 (COD = cyclooctadiene) were used for the catalyzed hydrogenation of α-N-acetamidocinnamic acid in methanol at 25 °C under H2 pressure (Scheme 11, eqn (1)). Hydrogenation occurred easily, but enantioselectivity was elusive in all cases (entries 1–5, Table 1). It was shown that the reaction rate depended on the steric hindrance close to catalytic centers (compare conditions for 1b and 1b′, entries 2 and 3, Table 1),21 but no difference could be observed between flexible and rigid ligands.22 | ||
| Scheme 11 Various types of asymmetric hydrogenation reactions carried out with dendritic catalysts. Conditions and results are given in Table 1. | ||
| Entry | Catalyst | Reaction | Conditions | Conv. (%) | ee (%) | Ref. |
|---|---|---|---|---|---|---|
| a 100% conversion in 24 h. b Recovered by precipitation and filtration. c Isolated yields. | ||||||
| 1 | 1a + [Rh(COD)Cl]2 | Eqn (1a) | 2 mol% cat., 20 bar H2, MeOH, rt, 20 h | 100 | 2 (R) | 20 |
| 2 | 1b + [Rh(COD)Cl]2 | Eqn (1a) | 3 bar H2, MeOH, rt, 2 min | 100 | Very low | 21 |
| 3 | 1b′ + [Rh(COD)Cl]2 | Eqn (1a) | 30 bar H2, MeOH, rt, 20 min | 100 | Very low | 21 |
| 4 | 2a–2g + [Rh(COD)Cl]2 | Eqn (1a) | 1 mol% cat. 30 bar H2, MeOH, rt, 20 h | 100 | 0–6 | 22 |
| 5 | 3a–3g + [Rh(COD)Cl]2 | Eqn (1a) | 1 mol% cat., 30 bar H2, MeOH, rt, 20 h | 100 | 0–6 | 22 |
| 6 | 4a-G1 + [Ru(p-cymene)Cl2]2 | Eqn (2b) | 0.8 mol% cat., 80 atm H2, 1.5 NEt3, MeOH–toluene, 2 h | 10.4a | 91.8 (R) | 23 |
| 7 | 4a-G2 + [Ru(p-cymene)Cl2]2 | Eqn (2b) | 0.8 mol% cat., 80 atm H2, 1.5 NEt3, MeOH–toluene, 2 h | 13.2a | 92.6 (R) | 23 |
| 8 | 4a-G3 + [Ru(p-cymene)Cl2]2 | Eqn (2b) | 0.8 mol% cat., 80 atm H2, 1.5 NEt3, MeOH–toluene, 2 h | 34.3a | 91.6 (R) | 23 |
| 9 | 4a-G3 + [Ru(p-cymene)Cl2]2 | Eqn (2b) | 0.8 mol% cat., 80 atm H2, 1.5 NEt3, MeOH–toluene, 5 h | 69.3a | 91.6 (R) | 23 |
| Recycled (1)b | Eqn (2b) | 0.8 mol% cat., 80 atm H2, 1.5 NEt3, MeOH–toluene, 5 h | 67.3 | 91.4 (R) | 23 | |
| Recycled (2)b | Eqn (2b) | 0.8 mol% cat., 80 atm H2, 1.5 NEt3, MeOH–toluene, 5 h | 68.9 | 91.8 (R) | 23 | |
| Recycled (3)b | Eqn (2b) | 0.8 mol% cat., 80 atm H2, 1.5 NEt3, MeOH–toluene, 5 h | 66.6 | 90.9 (R) | 23 | |
| 10 | 4b-G1 + [RuCl2(benzene)]2 | Eqn (2a) | 1 mol% cat., 80 atm H2, 1.5 NEt3, EtOH–hexane, 4 h | 100 | 88 (R) | 24 |
| 11 | 4b-G2 + [RuCl2(benzene)]2 | Eqn (2a) | 1 mol% cat., 80 atm H2, 1.5 NEt3, EtOH–hexane, 4 h | 100 | 87 (R) | 24 |
| 12 | 4b-G1 + [RuCl2(benzene)]2 | Eqn (2b) | 1 mol% cat., 80 atm H2, 1.5 NEt3, EtOH–hexane, 4 h | 100 | 90 (R) | 24 |
| 13 | 4b-G2 + [RuCl2(benzene)]2 | Eqn (2b) | 1 mol% cat., 80 atm H2, 1.5 NEt3, EtOH–hexane, 4 h | 100 | 90 (R) | 24 |
| 14 | 4b-G2 + [RuCl2(benzene)]2 | Eqn (2b) | 1 mol% cat., 80 atm H2, 1.5 NEt3, EtOH–hexane, 2 h | 94 | 90 (R) | 24 |
| Recycled (1)b | Eqn (2b) | 1 mol% cat., 80 atm H2, 1.5 NEt3, EtOH–hexane, 2 h | 93 | 90 (R) | 24 | |
| Recycled (2)b | Eqn (2b) | 1 mol% cat., 80 atm H2, 1.5 NEt3, EtOH–hexane, 2 h | 93 | 89 (R) | 24 | |
| Recycled (3)b | Eqn (2b) | 1 mol% cat., 80 atm H2, 1.5 NEt3, EtOH–hexane, 2 h | 91 | 89 (R) | 24 | |
| 15 | 4c-G1 + [RuCl2(benzene)]2 | Eqn (2a) | 1 mol% cat., 80 atm H2, 1.5 NEt3, EtOH–hexane, 4 h | 100 | 88 (R) | 24 |
| 16 | 4c-G2 + [RuCl2(benzene)]2 | Eqn (2a) | 1 mol% cat., 80 atm H2, 1.5 NEt3, EtOH–hexane, 4 h | 100 | 84 (R) | 24 |
| 17 | 4c-G1 + [RuCl2(benzene)]2 | Eqn (2b) | 1 mol% cat., 80 atm H2, 1.5 NEt3, EtOH–hexane, 4 h | 100 | 90 (R) | 24 |
| 18 | 4c-G2 + [RuCl2(benzene)]2 | Eqn (2b) | 1 mol% cat., 80 atm H2, 1.5 NEt3, EtOH–hexane, 4 h | 100 | 89 (R) | 24 |
| 19 | 4c-G1 + [RuCl2(benzene)]2 | Eqn (2b) | 1 mol% cat., 80 atm H2, 1.5 NEt3, EtOH–hexane–0.01H2O, 2.5 h | 38 | 80 (R) | 24 |
| 20 | 4a-G2 + [Ir(COD)Cl]2 | Eqn (3a) | 1.1 mol% cat., 45 atm H2, 11% I2, THF, rt, 20 h | >95 | 89 (S) | 25 |
| 21 | 4a-G2 + [Ir(COD)Cl]2 | Eqn (3a) | 1.1 mol% cat., 45 atm H2, 11% I2, THF–MeOH, rt, 20 h | 70 | 88 (S) | 25 |
| 22 | 4a-G2 + [Ir(COD)Cl]2 | Eqn (3a) | 1.1 mol% cat., 45 atm H2, without I2, THF, rt, 20 h | <5 | Nd | 25 |
| 23 | 4a-G2 + [Ir(COD)Cl]2 | Eqn (3a) | 0.002 mol% cat., 45 atm H2, 1.25% I2, THF, rt, 48 h | 86 | 88 (S) | 25 |
| 24 | 4a-G1 + [Ir(COD)Cl]2 | Eqn (3a) | 0.01 mol% cat., 45 atm H2, 1.25% I2, THF, rt, 5 h | 50 | 89 (S) | 25 |
| 25 | 4a-G2 + [Ir(COD)Cl]2 | Eqn (3a) | 0.01 mol% cat., 45 atm H2, 1.25% I2, THF, rt, 5 h | 75 | 89 (S) | 25 |
| 26 | 4a-G3 + [Ir(COD)Cl]2 | Eqn (3a) | 0.01 mol% cat., 45 atm H2, 1.25% I2, THF, rt, 5 h | 79 | 88 (S) | 25 |
| 27 | 4a-G4 + [Ir(COD)Cl]2 | Eqn (3a) | 0.01 mol% cat., 45 atm H2, 1.25% I2, THF, rt, 5 h | >95 | 87 (S) | 25 |
| 28 | 4a-G2 + [Ir(COD)Cl]2 | Eqn (3a) | 0.25 mol% cat., 45 atm H2, 5% I2, THF, rt, 1.5 h | >95 | 90 (S) | 25 |
| 29 | 4a-G2 + [Ir(COD)Cl]2 | Eqn (3b) or (3c) | 0.25 mol% cat., 45 atm H2, 5% I2, THF, rt, 1.5 h | >95 | 89 (S) | 25 |
| 30 | 4a-G2 + [Ir(COD)Cl]2 | Eqn (3d) | 0.25 mol% cat., 45 atm H2, 5% I2, THF, rt, 1.5 h | >95 | 84 (S) | 25 |
| 31 | 4a-G2 + [Ir(COD)Cl]2 | Eqn (3e) | 0.25 mol% cat., 45 atm H2, 5% I2, THF, rt, 1.5 h | 83 | 82 (R) | 25 |
| 32 | 4a-G2 + [Ir(COD)Cl]2 | Eqn (3f) or (3g) | 0.25 mol% cat., 45 atm H2, 5% I2, THF, rt, 1.5 h | >95 | 92 (R) | 25 |
| 33 | 4a-G2 + [Ir(COD)Cl]2 | Eqn (3h) | 0.25 mol% cat., 45 atm H2, 5% I2, THF, rt, 1.5 h | 77 | 76 (R) | 25 |
| 34 | 4a-G2 + [Ir(COD)Cl]2 | Eqn (3i) | 0.25 mol% cat., 45 atm H2, 5% I2, THF, rt, 1.5 h | 87 | 89 (S) | 25 |
| 35 | 4a-G2 + [Ir(COD)Cl]2 | Eqn (3j) | 0.25 mol% cat., 45 atm H2, 5% I2, THF, rt, 1.5 h | 77 | 87 (S) | 25 |
| 36 | 4a-G2 + [Ir(COD)Cl]2 | Eqn (3k) | 0.25 mol% cat., 45 atm H2, 5% I2, THF, rt, 1.5 h | >95 | 87 (S) | 25 |
| 37 | 4a-G3 + [Ir(COD)Cl]2 | Eqn (3a) | 0.1 mol% cat., 45 atm H2, 0.5% I2, THF, 25–30 °C, 8 h | >95 | 87 (S) | 25 |
| Recycled (2)b | Eqn (3a) | 0.1 mol% cat., 45 atm H2, 0.5% I2, THF, 25–30 °C, 8 h | >95 | 86 (S) | 25 | |
| Recycled (5)b | Eqn (3a) | 0.1 mol% cat., 45 atm H2, 0.5% I2, THF, 25–30 °C, 32 h | 80 | 85 (S) | 25 | |
| 38 | 5a-G1 + [Ru(COD)2]BF4 | Eqn (1a) | 0.125 mol% cat., 60 atm H2, MeOH–toluene, rt, 1 h | 91 | 96.8 (S) | 27 |
| 39 | 5a-G2 + [Ru(COD)2]BF4 | Eqn (1a) | 0.125 mol% cat., 60 atm H2, MeOH–toluene, rt, 1 h | 93 | 96.9 (S) | 27 |
| 40 | 5a-G3 + [Ru(COD)2]BF4 | Eqn (1a) | 0.125 mol% cat., 60 atm H2, MeOH–toluene, rt, 1 h | 79 | 96.9 (S) | 27 |
| 41 | 5a-G4 + [Ru(COD)2]BF4 | Eqn (1a) | 0.125 mol% cat., 60 atm H2, MeOH–toluene, rt, 6 h | 20 | 97.0 (S) | 27 |
| 42 | 5b-G1 + [Ru(COD)2]BF4 | Eqn (1a) | 0.125 mol% cat., 60 atm H2, MeOH–toluene, rt, 1 h | 96 | 96.8 (S) | 27 |
| 43 | 5b-G2 + [Ru(COD)2]BF4 | Eqn (1a) | 0.125 mol% cat., 60 atm H2, MeOH–toluene, rt, 1 h | 86 | 95.1 (S) | 27 |
| 44 | 5b-G3 + [Ru(COD)2]BF4 | Eqn (1a) | 0.125 mol% cat., 60 atm H2, MeOH–toluene, rt, 1 h | 59 | 94.6 (S) | 27 |
| 45 | 5a-G3 + [Ru(COD)2]BF4 | Eqn (1a) | 1 mol% cat., 60 atm H2, MeOH–toluene, rt, 0.5 h | 94 | 97.5 (S) | 27 |
| Recycled (1)b | Eqn (1a) | 1 mol% cat., 60 atm H2, MeOH–toluene, rt, 0.5 h | 81 | 97.7 (S) | 27 | |
| Recycled (2)b | Eqn (1a) | 1 mol% cat., 60 atm H2, MeOH–toluene, rt, 0.5 h | 55 | 97.7 (S) | 27 | |
| 46 | 6-G1 + [RuCl2(benzene)]2 | Eqn (4a) | 0.5 mol% cat., 40 atm H2, EtOH–CH2Cl2, 60 °C, 24 h | 100 | 92.0 | 28 |
| 47 | 6-G2 + [RuCl2(benzene)]2 | Eqn (4a) | 0.5 mol% cat., 40 atm H2, EtOH–CH2Cl2, 60 °C, 24 h | 100 | 86.6 | 28 |
| 48 | 6-G3 + [RuCl2(benzene)]2 | Eqn (4a) | 0.5 mol% cat., 40 atm H2, EtOH–CH2Cl2, 60 °C, 24 h | 100 | 91.3 | 28 |
| 49 | 6-G1 + [RuCl2(benzene)]2 | Eqn (4b) | 0.5 mol% cat., 40 atm H2, EtOH–CH2Cl2, 60 °C, 24 h | 100 | 81.1 | 28 |
| 50 | 6-G2 + [RuCl2(benzene)]2 | Eqn (4b) | 0.5 mol% cat., 40 atm H2, EtOH–CH2Cl2, 60 °C, 24 h | 100 | 77.9 | 28 |
| 51 | 6-G3 + [RuCl2(benzene)]2 | Eqn (4b) | 0.5 mol% cat., 40 atm H2, EtOH–CH2Cl2, 60 °C, 24 h | 100 | 79.6 | 28 |
| 52 | 6-G1 + [RuCl2(benzene)]2 | Eqn (4e) | 0.5 mol% cat., 40 atm H2, EtOH–CH2Cl2, 60 °C, 24 h | 100 | 94.1 | 28 |
| 53 | 6-G2 + [RuCl2(benzene)]2 | Eqn (4e) | 0.5 mol% cat., 40 atm H2, EtOH–CH2Cl2, 60 °C, 24 h | 100 | 89.1 | 28 |
| 54 | 6-G3 + [RuCl2(benzene)]2 | Eqn (4e) | 0.5 mol% cat., 40 atm H2, EtOH–CH2Cl2, 60 °C, 24 h | 100 | 88.4 | 28 |
| 55 | 7-G0 + [Rh(COD)2]BF4 | Eqn (1b) | 0.1 mol% cat., 10 atm H2, CH2Cl2, rt, 12 min | 95.8 | 97.3 (S) | 29 |
| 56 | 7-G1 + [Rh(COD)2]BF4 | Eqn (1b) | 0.1 mol% cat., 10 atm H2, CH2Cl2, rt, 12 min | 97.0 | 97.5 (S) | 29 |
| 57 | 7-G2 + [Rh(COD)2]BF4 | Eqn (1b) | 0.1 mol% cat., 10 atm H2, CH2Cl2, rt, 12 min | 74.0 | 97.3 (S) | 29 |
| 58 | 7-G0 + [Rh(COD)2]BF4 | Eqn (1d) | 1 mol% cat., 20 atm H2, CH2Cl2, rt | 100 | 96.0 (S) | 29 |
| 59 | 7-G1 + [Rh(COD)2]BF4 | Eqn (1d) | 1 mol% cat., 20 atm H2, CH2Cl2, rt | 100 | 97.3 (S) | 29 |
| 60 | 7-G0 + [Rh(COD)2]BF4 | Eqn (1k) | 1 mol% cat., 20 atm H2, CH2Cl2, rt | 100 | 95.6 (S) | 29 |
| 61 | 7-G1 + [Rh(COD)2]BF4 | Eqn (1k) | 1 mol% cat., 20 atm H2, CH2Cl2, rt | 100 | 94.6 (S) | 29 |
| 62 | 7-G0 + [Rh(COD)2]BF4 | Eqn (5) | 1 mol% cat., 20 atm H2, CH2Cl2, rt | 100 | 97.7 (R) | 29 |
| 63 | 7-G1 + [Rh(COD)2]BF4 | Eqn (5) | 1 mol% cat., 20 atm H2, CH2Cl2, rt | 100 | 97.0 (R) | 29 |
| 64 | 8b-G3 + [Rh(COD)2]BF4 | Eqn (1b) | 1 mol% cat. (2.2 ligand/Rh), 5 bar H2, CH2Cl2, rt, 2.5 h | 100 | 95 | 30 |
| 65 | 9a-G0 + [Rh(COD)2]BF4 | Eqn (5) | 1 mol% cat., 1 bar H2, MeOH, rt, 20 min | 83c | 98.6 | 33 |
| 66 | 9a-G1 + [Rh(COD)2]BF4 | Eqn (5) | 1 mol% cat., 1 bar H2, MeOH, rt, 20 min | 86c | 97.9 | 33 |
| 67 | 9b-G0 + [Rh(COD)2]BF4 | Eqn (5) | 1 mol% cat., 1 bar H2, MeOH, rt, 20 min | 78c | 98.7 | 33 |
| 68 | 9b-G1 + [Rh(COD)2]BF4 | Eqn (5) | 1 mol% cat., 1 bar H2, MeOH, rt, 20 min | 88c | 98.1 | 33 |
| 69 | 9b-G2 + [Rh(COD)2]BF4 | Eqn (5) | 1 mol% cat., 1 bar H2, MeOH, rt, 20 min | 86c | 98.2 | 33 |
| 70 | 10a-G0 + [Rh(COD)2]BF4 | Eqn (5) | 1 mol% cat., 1 bar H2, MeOH, rt, 20 min | 74c | 98.7 | 33 |
| 71 | 10a-G1 + [Rh(COD)2]BF4 | Eqn (5) | 1 mol% cat., 1 bar H2, MeOH, rt, 20 min | 70c | 98.0 | 33 |
| 72 | 10b-G1,G2 + [Rh(COD)2]BF4 | Eqn (5) | 1 mol% cat., 1 bar H2, MeOH, rt, 20 min | 78c | 98.0 | 33 |
| 73 | 13-G0,G1 + [Rh(COD)2]BF4 | Eqn (1b) | 0.25 mol% cat., 30 bar H2, MeOH, 25 °C, 200 min | 100 | ∼93 | 34 |
| 74 | 13-G2,G3 + [Rh(COD)2]BF4 | Eqn (1b) | 0.25 mol% cat., 30 bar H2, MeOH, 25 °C, 200 min | 100 | ∼91 | 34 |
| 75 | 13-G4 + [Rh(COD)2]BF4 | Eqn (1b) | 0.25 mol% cat., 30 bar H2, MeOH, 25 °C, 200 min | ∼60 | ∼88 | 34 |
| 76 | 13-G0 + [Rh(COD)2]BF4 | Eqn (5) | 0.25 mol% cat., 30 bar H2, MeOH, 25 °C, 150 min | 100 | ∼74 | 34 |
| 77 | 13-G1 + [Rh(COD)2]BF4 | Eqn (5) | 0.25 mol% cat., 30 bar H2, MeOH, 25 °C, 150 min | 100 | ∼68 | 34 |
| 78 | 13-G2,G3 + [Rh(COD)2]BF4 | Eqn (5) | 0.25 mol% cat., 30 bar H2, MeOH, 25 °C, 150 min | ∼97 | ∼61 | 34 |
| 79 | 13-G4 + [Rh(COD)2]BF4 | Eqn (5) | 0.25 mol% cat., 30 bar H2, MeOH, 25 °C, 150 min | ∼90 | ∼60 | 34 |
| 80 | 17a-G0 + [Rh(COD)Cl]2 | Eqn (5) | 0.2 mol% cat., 10 bar H2, THF, 20 °C, 2 h | 94.4 | ∼0 | 45 |
| 81 | 17a-G1 + [Rh(COD)Cl]2 | Eqn (5) | 0.2 mol% cat., 10 bar H2, THF, 20 °C, 2 h | 68.6 | ∼0 | 45 |
| 82 | 17b-G0 + [Rh(COD)Cl]2 | Eqn (5) | 0.2 mol% cat., 10 bar H2, THF, 20 °C, 2 h | 64 | ∼0 | 45 |
On the contrary, all compounds shown in Scheme 2 were successfully used as ligands for enantioselective hydrogenations. Compounds 4a-Gn (n = 1–3) were reacted in situ with [Ru(p-cymene)Cl2]2 and used for the asymmetric hydrogenation of 2-[p-(2-methylpropyl)phenyl]acrylic acid, leading to ibuprofen (Scheme 11, eqn (2)). All these dendritic catalysts showed higher ee values than the parent BINAP complex. The rate of reaction increased when using higher generation catalysts (compare entries 6–8, Table 1). Furthermore, the third generation (4a-G3) could be recovered by precipitation and reused for at least three cycles with the same activity and enantioselectivity (entry 9).23 Very moderate TOF (turn over frequency) values were obtained (6.5–21.4 h–1). The presence of alkyl chains as end groups of dendrons 4b-Gn and 4c-Gn (n = 1, 2) made their metal complexes exclusively soluble in hydrocarbons; the use of a miscible ethanol–hexane (1 : 1) mixture allowed complete conversions in 4 h with high enantioselectivity for the asymmetric hydrogenation of 2-arylacrylic acids (entries 10–13 and 15–18, Table 1). Upon completion of the reaction, addition of a small amount of water (2.5%) induced phase separation and provided facile catalyst recovery and reuse, with only a very slight loss of enantioselectivity (entry 14). However, water must be avoided during the catalytic process, as illustrated by the drop in conversion and ee in entry 19.24
The series of compounds 4-Gn were also able to complex in situ[Ir(COD)Cl]2, and the corresponding complexes were used for the asymmetric hydrogenation of quinaldine and quinoline derivatives (Scheme 11, eqn (3)). The complex of 4a-G2 was used for the first tests, illustrating the influence of the solvent (much lower activity in the presence of methanol, compare entries 20 and 21), the necessity to have iodine as an additive (compare entries 20 and 22), and the extremely high efficiency even with a very low ratio of catalyst (entry 23). The dendrimer generation also has an effect on the catalyst performance, which increases gradually with increasing dendrimer generation from 4a-G1 to 4a-G4 (entries 24 to 27). In view of these excellent results, the Ir complex of 4a-G2 was used for the asymmetric hydrogenation of a series of quinoline derivatives with good to excellent enantioselectivity (entries 28 to 36). The recyclability of the third generation was also tested with the Ir complex of 4a-G3. This catalyst was reused at least 5 times with a similar enantioselectivity, but with a lower activity (entry 37, Table 1). Extremely high TONs (turn over numbers) were obtained, up to 43![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 with 4a-G2.25
000 with 4a-G2.25
The in situ reaction of dendrons 5-Gn with [Rh(COD)2]BF4 affords a series of complexes used for the asymmetric hydrogenation of α-N-acetamidocinnamic acid. Contrary to the previous reports using compounds 2 and 3 (entries 1–5)20–22 enantioselectivity is high with the Rh complexes of 5a-Gn. However, the reaction rate decreased with increasing generation, and the catalyst almost lost its activity when going from generation 3 to generation 4 (entries 38–41). Presuming that such phenomenon might be due to changes in conformation inducing the encapsulation of the core, the back-folded dendrimers 5b-Gn were used in the same catalytic experiments. The first generation had almost the same efficiency, but an important loss in activity was observed for the third generation, confirming the importance of the steric hindrance (entries 43–45, Table 1).27
The asymmetric hydrogenation of a series of β-keto esters was carried out using the dendritic complexes obtained by the in situ complexation of [RuCl2(benzene)]2 by the dendrons 6-Gn (Scheme 11, eqn (4)). The enantioselectivity slightly decreased on going from the non-dendritic ligand to generation 1 and to further generations. Selected examples are given in Table 1 (entries 46–54); the case of eqn (4c) is almost identical to (4b), and (4d) to (4e).28
A series of α-dehydroamino acid esters was hydrogenated asymmetrically (Scheme 11, eqn (1)) by the rhodium complexes of dendrons 7-Gn, obtained by reaction with [Rh(COD)2]BF4. In the case of 2-acetamido cinnamate (eqn (1b)), even very low catalyst loading rapidly gave high yields and high enantioselectivities (entries 55–57), albeit 7-G2 gave a slightly lower reaction rate. Excellent enantioselectivities were also achieved for all the other α-dehydroamino acid esters, which were better or comparable to those obtained using the analogous non-dendritic catalyst. The reaction is compatible with several types of substituents, and it was shown that meta- or para-substituents (entries 58–59) on the phenyl group gave slightly higher ee values as compared to ortho-substituents (entries 60–61, Table 1). These catalysts were also used for the asymmetric hydrogenation of dimethyl itaconate (Scheme 11, eqn (5)) with very good enantioselectivities (entries 62–63).29 The last example of the use of dendrons for asymmetric hydrogenation was provided by the complex of 8b-G3 obtained in situ by reaction with [Rh(COD)2]BF4 at a ligand/Rh ratio of 2.2; it also allowed the hydrogenation of type eqn (1b) with high ee (entry 64, Table 1).30
The Rh complexes of the dendritic chiral ferrocenyl ligands shown in Scheme 6 were used for the asymmetric hydrogenation of dimethyl itaconate (Scheme 11, eqn (5)), constituting the first example of phosphino dendritic end groups used for asymmetric catalysis. In first attempts, the [Rh(COD)2]BF4 complexes of 9a-Gn and 10a-Gn (n = 0, 1) were used and afforded very good enantioselectivities.31 Analogous results were obtained with 11a-G1 and 12a-G1.32 These results were gathered and expanded in another paper,33 also affording information about the use of 9b-Gn and 10b-Gn (n = 0–2). Similar results were obtained in all cases (Table 1, entries 65–72).
The series of compounds 13-Gn and 14-Gn (Scheme 7), could have allowed an interesting comparison of their catalytic properties with the dendrons 5a,b-Gn, since they possess the same type of chiral phosphines in different locations, but a different metal was complexed, hampering such comparison. After complexation of [Rh(COD)2]BF4 by the series 13-Gn, the resulting complexes were used for the asymmetric hydrogenation of methylacetamidocinnamate (Scheme 11, eqn (1b)), and of dimethyl itaconate (eqn (5)). In both cases, a decrease in the catalytic activity was observed for the highest generations, as well as a decrease in the selectivity. Such phenomena, which might be due to decreased accessibility to the catalytic sites, were observed with the fourth generation in the first case (entries 73–75, Table 1), and from the second generation in the second case (entries 76–79).34
To conclude this part about asymmetric hydrogenation, despite the numerous examples of successful use of dendritic catalysts described above, some poor results may occur, as already illustrated by the expanded phosphines 1, 2, and 3.20–22 Analogous poor results were obtained with the carbosilane dendrimers 17a,b-Gn. Their complexes with [Rh(COD)Cl]2 used in the hydrogenation of dimethyl itaconate (Scheme 11, eqn (5)) did not afford any enantiomeric excess, and an important decrease in activity was observed even for the first generation (entries 80–82, Table 1).45
Miscellaneous enantioselective reactions
Besides classical hydrogenations using H2, other types of catalyzed reactions finally result in hydrogenation of unsaturated bonds. The first example concerned the hydrosilylation of acetophenone with diphenylsilane. The reaction was carried out in THF, with 0.25 mol% of the in situ generated catalysts (2 or 3 + [Rh(COD)Cl]2). In all cases, the enantiomeric excesses were poor, but the ethylene-bridged ligands gave slightly higher optical inductions (2a: 2.8% ee (R); 2b: 10.3% ee (S); 2e: 18.1% ee (S)) than the analogous rigid phenylene-bridged ligands (3a: 2.4% ee (R); 3b: 2.6% ee (R); 3e: 0% ee).22Acetophenone was also used recently for a transfer hydrogenation from 2-propanol in the presence of tBuOK, using 0.5 mol% of the catalyst obtained by complexation of compounds 17-Gn with [RuCl2(p-cymene)]2. The percentage conversion after 6 h at 82 °C was ∼75% with 17a-G0 and 60% with 17b-G0, but the enantiomeric excesses were not satisfactory (about 15%).45Allylation reactions using dendritic catalysts are the second most studied type of enantioselective reactions, after hydrogenations. The first mention of these reactions concerned the enantioselective allylation (5–11% ee) of 1,5-dimethylbarbituric acid with allyl acetate using the expanded phosphines of types 2 and 3, but no details were given.22 Much better results were obtained with several types of dendrimers bearing Pd complexes as end groups. The first example concerned the phosphino ferrocenes 9-Gn and 10-Gn reacted in situ with [Pd(dba)2]. The enantioselectivities afforded by the zeroth and first generations for the allylic alkylation shown in Scheme 12 (eqn (6a)) were relatively good (entries 1–7 in Table 2), but the second generations were not soluble enough.33
 | ||
| Scheme 12 Enantioselective allylations using dendritic catalysts. Conditions and results are given in Table 2. | ||
| Entry | Catalyst | Reaction | Conv. (%) | ee (%) | Ref. |
|---|---|---|---|---|---|
| a 1 mol% cat., BSA (N,O-bis(trimethylsilyl)acetamide), CH2Cl2, rt, 20 h. b 0.3 mol% cat., DMSO, 45 °C. c 2.5 mol% cat., BSA, CH2Cl2, rt, 24 h. d 2 mol% cat., BSA, CH2Cl2, rt, 3 h. e Isolated yields. | |||||
| 1 | 9a-G0 + [Pd(dba)2]a | Eqn (6a) | 92e | 90 (S) | 33 |
| 2 | 9a-G1 + [Pd(dba)2]a | Eqn (6a) | 92e | 89 (S) | 33 |
| 3 | 9b-G0 + [Pd(dba)2]a | Eqn (6a) | Nd | 85 (S) | 33 |
| 4 | 9b-G1 + [Pd(dba)2]a | Eqn (6a) | 85e | 90 (S) | 33 |
| 5 | 10a-G0 + [Pd(dba)2]a | Eqn (6a) | 96e | 91 (S) | 33 |
| 6 | 10a-G1 + [Pd(dba)2]a | Eqn (6a) | 91e | 90 (S) | 33 |
| 7 | 10b-G1 + [Pd(dba)2]a | Eqn (6a) | 96e | 90 (S) | 33 |
| 8 | 13-G0 + [PdCl2(NCPh)2]b | Eqn (7a) | ∼18 | 35 | |
| 9 | 13-G1 + [PdCl2(NCPh)2]b | Eqn (7a) | ∼30 | 35 | |
| 10 | 13-G2,G3,G4 + [PdCl2(NCPh)2]b | Eqn (7a) | ∼42 | 35 | |
| 11 | 14-G0 + [PdCl2(NCPh)2]b | Eqn (7a) | ∼32 | 35 | |
| 12 | 14-G1 + [PdCl2(NCPh)2]b | Eqn (7a) | 50 | 35 | |
| 13 | 14-G2 + [PdCl2(NCPh)2]b | Eqn (7a) | 57 | 35 | |
| 14 | 14-G3 + [PdCl2(NCPh)2]b | Eqn (7a) | 62 | 35 | |
| 15 | 14-G4 + [PdCl2(NCPh)2]b | Eqn (7a) | 69 | 35 | |
| 16 | 15-G3 + [PdCl(allyl)]2 + KOAcc | Eqn (6a) | 100 | 90 (R) | 42 |
| Recycled (1) | Eqn (6a) | 100 | 82 (R) | 42 | |
| Recycled (2) | Eqn (6a) | 92 | 82 (R) | 42 | |
| 17 | 15-G3 + [PdCl(allyl)]2 + LiOAcc | Eqn (6a) | 100 | 91 (R) | 42 |
| Recycled (1) | Eqn (6a) | 100 | 85 (R) | 42 | |
| Recycled (2) | Eqn (6a) | 98 | 84 (R) | 42 | |
| 18 | 15-G3 + [PdCl(allyl)]2 + LiOAcc | Eqn (6b) | 100 | 95 (R) | 42 |
| Recycled (1) | Eqn (6b) | 97 | 94 (R) | 42 | |
| Recycled (2) | Eqn (6b) | 90 | 92 (R) | 42 | |
| 19 | 16-G1 + [PdCl(allyl)]2 + LiOAcd | Eqn (6b) | 100 | 93 (R) | 43 |
| 20 | 16-G2 + [PdCl(allyl)]2 + LiOAcd | Eqn (6b) | 100 | 92 (R) | 43 |
| 21 | 16-G3 + [PdCl(allyl)]2 + LiOAcd | Eqn (6b) | 100 | 90 (R) | 43 |
| 22 | 16-G4 + [PdCl(allyl)]2 + LiOAcd | Eqn (6b) | 100 | 91 (R) | 43 |
The other series of phosphino ferrocene derivatives 16-Gn, reacted in situ with [PdCl(allyl)]2, gave even better results in terms of enantioselectivity for allylic alkylations (Scheme 12, eqn (6b)), with almost no difference depending on the generation, and no problem of solubility (entries 19–22, Table 2). However, attempted reuse of these catalysts was unsuccessful, due to a significant decrease in efficiency and enantioselectivity.43 On the contrary, dendrimer15-G3 also reacted with [PdCl(allyl)]2, was easily recovered by precipitation and reused at least two times. This compound allowed determination of the influence of the co-catalyst (LiOAc better than KOAc, compare entries 16 and 17, Table 2) and of the substituent R (diphenyl-2-propenyl pivalate (Scheme 12, eqn (6b)) better than diphenyl-2-propenyl acetate (eqn (6a)) (compare entries 17 and 18, Table 2).42
Another type of allylation was carried out using the dendritic complexes obtained by reaction of dendrimers 13-Gn and 14-Gn with [PdCl2(NCPh)2]. A strong dependence on the type of dendrimer (PPI or PAMAM) and the generation was observed for the allylic amination shown in Scheme 12, eqn (7). In the case of PPI dendrimers (13-Gn), an increase in the enantioselectivity was observed at the beginning (entries 8–9, Table 2), but a plateau is rapidly attained (entry 10). In contrast, the PAMAM dendrimers (14-Gn) gave better results and a continuous increase in the enantioselectivity from the zeroth to the fourth generation (entries 11–15, Table 2). All these dendritic catalysts gave better results than the corresponding monomeric catalysts.35
Finally, two other types of enantioselective catalyses using chiral dendritic phosphino complexes were proposed. The first one was the hydroboration of styrene, using the rhodium complexes of dendrimers 9-Gn and 10-Gn, obtained by reaction with [Rh(COD)2]BF4. In this case, in addition to enantiomers, regioisomers (branched (B) or linear (L)) were obtained (Scheme 13, eqn (8)). In all cases, the regioselectivity for the branched isomer is high, from 89 : 11 (entry 9, Table 3) to 98 : 2 (entry 6, Table 3) and better for the lowest generations, but the enantioselectivities for the branched product is modest (entries 1–9, Table 3), and also generally better for the lowest generations.33
 | ||
| Scheme 13 Regio- and enantioselective hydroboration and hydrovinylation of styrene. | ||
| Entry | Catalyst | Reaction | Conv. (%) | Selectivity | ee (%) | Ref. |
|---|---|---|---|---|---|---|
| a 1 mol% cat., THF, rt, 5–13 h. b Isolated yields. c Branched : linear selectivity. d Obtained from reaction of 17-Gn with [Pd(2-MeC3H4)Cl]2 then with AgBF4 (or NaBARF (NaB[3,5-(CF3)2C6H3]4)) in situ; 0.2 mol% cat., CH2Cl2, 15 bar styrene, 25 °C, 2 h. e % Selectivity for 3-phenyl-1-butene with respect to co-dimers. f idem footnote d, but after 6 h. | ||||||
| 1 | 9a-G0 + [Rh(COD)2]BF4a | Eqn (8) | 77b | 95 : 5c | 64 (S) | 33 |
| 2 | 9a-G1 + [Rh(COD)2]BF4a | Eqn (8) | 64b | 90 : 10c | 60 (S) | 33 |
| 3 | 9b-G0 + [Rh(COD)2]BF4a | Eqn (8) | 87b | 96 : 4c | 68 (S) | 33 |
| 4 | 9b-G1 + [Rh(COD)2]BF4a | Eqn (8) | 71b | 91 : 9c | 61 (S) | 33 |
| 5 | 9b-G2 + [Rh(COD)2]BF4a | Eqn (8) | 57b | 97 : 3c | 67 (S) | 33 |
| 6 | 10a-G0 + [Rh(COD)2]BF4a | Eqn (8) | 86b | 98 : 2c | 68 (S) | 33 |
| 7 | 10a-G1 + [Rh(COD)2]BF4a | Eqn (8) | 71b | 92 : 8c | 61 (S) | 33 |
| 8 | 10b-G1 + [Rh(COD)2]BF4a | Eqn (8) | 63b | 95 : 5c | 67 (S) | 33 |
| 9 | 10b-G2 + [Rh(COD)2]BF4a | Eqn (8) | 97b | 89 : 11c | 64 (S) | 33 |
| 10 | 17a-G0[Pd(2-MeC3H4)]BF4d | Eqn (9) | 31.3 | 97.4%e | 63 (S) | 44 |
| 11 | 17a-G0[Pd(2-MeC3H4)]BF4f | Eqn (9) | 55.7 | 77.3%e | 56 (S) | 44 |
| 12 | 17a-G1[Pd(2-MeC3H4)]BF4d | Eqn (9) | 34.0 | 95.6%e | 65 (S) | 44 |
| 13 | 17a-G1[Pd(2-MeC3H4)]BF4f | Eqn (9) | 53.8 | 77.9%e | 58 (S) | 44 |
| 14 | 17a-G0[Pd(2-MeC3H4)]BARFd | Eqn (9) | 29.5 | 98.5%e | 75 (S) | 44 |
| 15 | 17a-G0[Pd(2-MeC3H4)]BARFf | Eqn (9) | 58.4 | 92.0%e | 75 (S) | 44 |
| 16 | 17a-G1[Pd(2-MeC3H4)]BARFd | Eqn (9) | 27.4 | 98.6%e | 79 (S) | 44 |
The asymmetric hydrovinylation of styrene was carried out using the Pd complexes of the carbosilane dendrimers 17a-Gn, obtained by reacting first with [Pd(2-MeC3H4)Cl]2, then with AgBF4 or NaB[3,5-(CF3)2C6H3]4 (NaBARF) as a halide extractor. At moderate conversions, the selectivity for 3-phenyl-1-butene was excellent, and the enantiomeric excesses were good both for the zeroth and first generations (entries 10, 12, Table 3). At higher conversions, the selectivity decreased dramatically, and the enantiomeric excess was reduced, in the case of BF4 as counter ion (entries 11, 13, Table 3). However, changing BF4 to the BARF counter ion had a dramatic influence on the results: an important increase in both the selectivity and the enantiomeric excess was observed, even for high conversions (entries 14–16, Table 3). Compound 17b-G0 was also tested in these reactions, but it gave very poor results for the enantioselectivity, even if the TOF is better (524 to 1425 h–1) than for 17a-Gn (45 to 85 h–1).44
Conclusions
This review has emphasized the diversity of locations and structures of phosphino dendritic catalysts usable for asymmetric catalyses, but it is difficult to deduce rules from this diversity. Indeed, even if it has been shown that the internal skeleton has an influence on the enantioselectivity (see results obtained with 13-Gn and 14-Gn),35 no data exist to date to determine the role of the catalytic site(s) location (core or surface). However, the quantity of matter needed for a given number of catalytic sites is much lower when these sites are located on the surface. Comparison of the properties of dendritic catalysts in terms of efficiency and enantioselectivity often give results analogous to those of monomeric catalysts, but a few poorer results, as well as spectacular improvements in some cases also have been reported. Furthermore, in most cases it is possible to recover and reuse a dendritic catalyst; this possibility allows both to reduce the cost and to have purer products, not contaminated by the catalysts. The first aspect is important when sophisticated ligands such as dendrimers are concerned; the second aspect is particularly important for pharmaceuticals. Finally, it must be emphasized that the number and types of enantioselective catalyses studied using phosphino dendritic complexes are very limited, thus there is plenty of room for new research in this field.Acknowledgements
Thanks are due to the European Community through the contract MRTN-CT-2003-503864 (AQUACHEM) for financial support (in particular grant to P. S.). This work was also supported by the CNRS.References
- P. Sabatier and J. B. Senderens, C. R. Hebd. Seances Acad. Sci., 1899, 128, 1173 Search PubMed.
- M. Poliakoff, J. M. Fitzpatrick, T. R. Farren and P. T. Anastas, Science, 2002, 297, 807 CrossRef CAS.
- B. Trost, Angew. Chem., Int. Ed. Engl., 1995, 34, 259 CrossRef CAS.
- J. M. J. Fréchet and D. A. Tomalia, Dendrimers and other dendritic polymers, John Wiley and Sons, Chichester, 2001 Search PubMed.
- G. R. Newkome, C. N. Moorefield and F. Vögtle, Dendrimers and dendrons. Concepts, syntheses, applications, Wiley VCH, Weinheim, 2001 Search PubMed.
- J. P. Majoral and A. M. Caminade, Chem. Rev., 1999, 99, 845 CrossRef CAS.
- G. Van Koten, D. M. Grove, P. Wijkens, P. W. van Leeuwen, J. C. de Wilde, A. W. van der Made and J. W. C. Knapen, Nature, 1994, 372, 659 CrossRef CAS.
- B. Helms and J. M. J. Fréchet, Adv. Synth. Catal., 2006, 348, 1125 CrossRef.
- J. K. Kassube and L. H. Gade, Top. Organomet. Chem., 2006, 20, 61 CAS.
- J. P. Majoral, A. M. Caminade and R. Laurent, ACS Symp. Ser., 2006, 928, 230 CAS.
- D. Méry and D. Astruc, Coord. Chem. Rev., 2006, 250, 1965 CrossRef CAS.
- J. N. H. Reek, S. Arevalo, R. Van Heerbeek, P. C. J. Kamer and P. W. N. M. Van Leeuwen, Adv. Catal., 2006, 49, 71.
- D. Astruc, K. Heuze, S. Gatard, D. Mery, S. Nlate and L. Plaut, Adv. Synth. Catal., 2005, 347, 329 CrossRef CAS.
- A. M. Caminade and J. P. Majoral, Coord. Chem. Rev., 2005, 249, 1917 CrossRef CAS.
- A. Dahan and M. Portnoy, J. Polym. Sci., Part A: Polym. Chem., 2005, 43, 235 CrossRef CAS.
- C. Liang and J. M. J. Fréchet, Prog. Polym. Sci., 2005, 30, 385 CrossRef CAS.
- R. van de Coevering, R. J. M. Klein Gebbink and G. van Koten, Prog. Polym. Sci., 2005, 30, 474 CrossRef CAS.
- A. Ouali, R. Laurent, A. M. Caminade, J. P. Majoral and M. Taillefer, J. Am. Chem. Soc., 2006, 128, 15990 CrossRef CAS.
- See for example: (a) R. Noyori, Asymmetric Catalysis in Organic Synthesis, Wiley, New York, 1994 Search PubMed; (b) E. N. Jacobsen, A. Pfaltz and H. Yamamoto, Comprehensive Asymmetric Catalysis, Springer, Berlin, 1999 Search PubMed; (c) I. Ojima, Catalytic Asymmetric Synthesis, Wiley, New York, 2000 Search PubMed; (d) R. Noyori, Angew. Chem., Int. Ed., 2002, 41, 2008 CrossRef CAS.
- H. Brunner and J. Fürst, Tetrahedron, 1994, 50, 4303 CrossRef CAS.
- H. Brunner, J. Organomet. Chem., 1995, 500, 39 CrossRef CAS.
- H. Brunner, M. Janura and S. Stefaniak, Synthesis, 1998, 1742 CrossRef CAS.
- Q. H. Fan, Y. M. Chen, X. M. Chen, D. Z. Jiang, F. Xi and A. S. C. Chan, Chem. Commun., 2000, 789 RSC.
- G. J. Deng, Q. H. Fan, X. M. Chen, D. S. Liu and A. S. C. Chan, Chem. Commun., 2002, 1570 RSC.
- Z. J. Wang, G. J. Deng, Y. M. Li, Y. M. He, W. J. Tang and Q. H. Fan, Org. Lett., 2007, 9, 1243 CrossRef CAS.
- C. J. Hawker and J. M. J. Fréchet, J. Am. Chem. Soc., 1990, 112, 7638 CrossRef CAS.
- B. Yi, Q. H. Fan, G. J. Deng, Y. M. Li, L. Q. Qiu and A. S. C. Chan, Org. Lett., 2004, 6, 1361 CrossRef CAS.
- G. J. Deng, G. R. Li, L. Y. Zhu, H. F. Zhou, Y. M. He, Q. H. Fan and Z. G. Shuai, J. Mol. Catal. A: Chem., 2006, 244, 118 CrossRef CAS.
- W. J. Tang, Y. Y. Huang, Y. M. He and Q. H. Fan, Tetrahedron: Asymmetry, 2006, 17, 536 CrossRef CAS.
- P. N. M. Botman, A. Amore, R. van Heerbeek, J. W. Back, H. Hiemstra, J. N. H. Reek and J. H. van Maarseveen, Tetrahedron Lett., 2004, 45, 5999 CrossRef CAS.
- C. Köllner, B. Pugin and A. Togni, J. Am. Chem. Soc., 1998, 120, 10274 CrossRef.
- R. Schneider, C. Köllner, I. Weber and A. Togni, Chem. Commun., 1999, 2415 RSC.
- C. Köllner and A. Togni, Can. J. Chem., 2001, 79, 1762 CrossRef CAS.
- G. D. Engel and L. H. Gade, Chem.–Eur. J., 2002, 8, 4319 CrossRef CAS.
- Y. Ribourdouille, G. D. Engel, M. Richard-Plouet and L. H. Gade, Chem. Commun., 2003, 1228 RSC.
- E. Buhleier, W. Wehner and F. Vögtle, Synthesis, 1978, 155 CrossRef CAS.
- C. Wörner and R. Mühlhaupt, Angew. Chem., Int. Ed. Engl., 1993, 32, 1306 CrossRef.
- E. M. M. de Brabander-van den Berg and E. W. Meijer, Angew. Chem., Int. Ed. Engl., 1993, 32, 1308 CrossRef.
- D. A. Tomalia, H. Baker, J. Dewald, M. Hall, C. Kallos, S. Martin, J. Roeck, J. Ryder and P. Smith, Polym. J. (Tokyo), 1985, 17, 117 Search PubMed.
- N. Launay, A. M. Caminade, R. Lahana and J. P. Majoral, Angew. Chem., Int. Ed. Engl., 1994, 33, 1589 CrossRef.
- N. Launay, A. M. Caminade and J. P. Majoral, J. Organomet. Chem., 1997, 529, 51 CrossRef CAS.
- R. Laurent, A. M. Caminade and J. P. Majoral, Tetrahedron Lett., 2005, 46, 6503 CrossRef CAS.
- L. Routaboul, S. Vincendeau, C. O. Turrin, A. M. Caminade, J. P. Majoral, J. C. Daran and E. Manoury, J. Organomet. Chem., 2007, 692, 1064 CrossRef CAS.
- L. I. Rodriguez, O. Rossell, M. Seco, A. Grabulosa, G. Muller and M. Rocamora, Organometallics, 2006, 25, 1368 CrossRef CAS.
- L. I. Rodriguez, O. Rossell, M. Seco and G. Muller, J. Organomet. Chem., 2007, 692, 851 CrossRef CAS.
Footnote |
| † A new “generation” is created each time the number of end groups is multiplied, most frequently by two or by three. |
| This journal is © The Royal Society of Chemistry 2008 |