The structure of protonated acetone and its dimer: infrared photodissociation spectroscopy from 800 to 4000 cm−1†
Gary E.
Douberly
,
Allen M.
Ricks
,
Brian W.
Ticknor
and
Michael A.
Duncan
*
Department of Chemistry, University of Georgia, Athens, GA, USA. E-mail: maduncan@uga.edu; Fax: 706-542-1234; Tel: 706-542-1998
First published on 22nd November 2007
Abstract
The infrared spectra of protonated acetone and the proton bound acetone dimer are obtained revealing vibrational resonances associated with the shared proton motions, which are in agreement with the predictions from ab initio, MP2, harmonic frequency calculations.
Proton-bound dimers are prototypes for studying strong ionic hydrogen bonds of the form R–O⋯H+⋯O–R′. Ionic hydrogen bonding motifs of this type play important roles in biological processes, such as proton transport across cell membranes.1 To obtain a microscopic understanding of these processes, a detailed characterization of the 3-D proton transfer potential is required. Structural studies of gas phase proton-bound dimers have been reported using infrared spectroscopic techniques capable of probing the vibrational resonances associated with the shared proton motion.2–7 The shared proton resonances are sensitive probes of the structure of the O⋯H+⋯O bond and the broad proton transfer potentials associated with these systems.7 Here, we employ a broadly tunable, table top infrared laser system, along with the method of rare gas tagging,8 to characterize the structure of protonated acetone and its proton bound dimer.
The structure of protonated acetone was first investigated in superacid matrices with NMR9 and FTIR10 spectroscopy. Chemical shifts of the proton bound to the oxygen lone pair revealed a diminished sp2 hybridization of the oxygen as a result of electron donation from the carbonyl group to the proton. Several studies of proton-bound clusters of acetone have been reported using mass spectrometers.11–15 Both electron impact13 and photoionization11 of neutral acetone clusters produced ionic complexes of the form H+(C3H6O)n. Multiple isomers of H+(C3H6O)2 have been implicated in many of these studies, although with conflicting results.14 Most recently, a combined mass spectrometry and ab initio study15 supported the existence of the enolic protonated diacetone alcohol molecule, along with the proton-bound acetone dimer. Nevertheless, the dimer was found to be the global minimum structure, and the enol species was found to be less stable by 7.4 kcal mol−1. Spectroscopy of H+(C3H6O)2 in isolation has not been reported.
The details of our experiment are given elsewhere.8,16 We synthesize H+(C3H6O)n and H+(C3H6O)nArmclusters in a pulsed supersonic expansion, electric discharge source. The gas mixture of H2 (10%) and Ar contains acetone at its ambient vapor pressure. Mass selection of specific clusters is achieved with a specially designed reflectron time-of-flight mass spectrometer. Photofragmentation of the mass-selected cations is achieved by overlapping the ion packet with the output of a tunable (700 to 4400 cm−1) infrared OPO/OPA/AgGaSe2 laser. Given the large binding energies of H+(C3H6O)n ions,12 single-photon IR photodissociation (IRPD) spectra are obtained by monitoring the loss of a weakly attached Ar atom from the complexes. IR intensities may be biased in this method because of the energy-dependent dissociation yield, but band positions are expected to match the absorption spectrum.
Fig. 1 shows the IRPD spectrum of H+(C3H6O)Ar, along with the ab initio (MP2/6-311++g(d,p))17 harmonic frequency calculations (scaled by a factor of 0.96). The S/N in the 1000–2000 cm−1 region reflects the lower laser power available here. The structure of the complex, with the proton bound to the oxygen lone pair, is shown as the inset. The predicted binding energy of Ar to H+(C3H6O) is only ∼500 cm−1, and IRPD bands are observed between 1150 and 3500 cm−1 in excellent agreement with theory. The COH+ bend transition is found at 1155 cm−1 while the carbonyl (CO) stretch is at 1580 cm−1. Therefore, protonation of acetone leads to a 150 cm−1CO stretch red shift, which can be compared to a recent study of the acetone CO stretch upon complexation with Mg+, Al+, and Ca+ ions.18 In these metal acetone complexes, the interactions are mostly electrostatic, leading to smaller (50–100 cm−1) red shifts. The features in the IRPD spectrum near 2915 cm−1 are assigned to the methyl stretching vibrations, and the OH+ stretch transition is located at 3378 cm−1. From the ab initio results, the change in the spectrum is predicted to be small upon addition of Ar. The Ar is bound on the proton, resulting in a 0.002 Å increase in the OH bond length, along with a predicted OH+ stretch redshift of 59 cm−1. It is interesting to compare the OH+ stretch redshift to the analogous shift for the H+(CO2)Ar complex,16,19 which was experimentally determined to be 702 cm−1. The more substantial redshift for H+(CO2)Ar is due to the larger (0.03 Å) increase in the OH bond length upon Ar complexation. The different Ar induced geometric changes predicted are simply due to the differences in proton affinity (PA)20 between Ar and CO2 (ΔPA = 117 kJ mol−1), in comparison to Ar and acetone (ΔPA = 443 kJ mol−1). Johnson and coworkers have shown that there is a monotonic increase in the frequency of the shared proton stretch (νsp) in protonated binary complexes as ΔPA increases.7 The νsp transitions in homogeneous dimers are found near 1000 cm−1. As the proton becomes more unevenly shared in heterogeneous complexes, νsp shifts towards higher frequencies. Indeed, the OH stretch, νsp, transitions for H+(CO2)Ar and H+(C3H6O)Ar are in excellent agreement with the ΔPA(νsp) trend reported previously.7
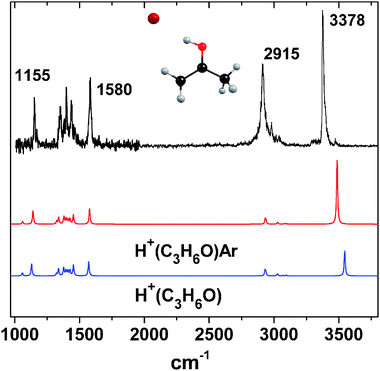 | ||
| Fig. 1 IRPD spectrum of the H+(C3H6O)Ar complex (corrected for laser power), along with ab initio calculations for protonated acetone with (red) and without (blue) Ar. | ||
The IRPD spectrum of the H+(C3H6O)2Ar species is shown in Fig. 2. Several bands are observed below 1800 cm−1, and there are no bands in the region of the protonated monomer OH+ stretch (3378 cm−1). As noted above, a previous mass spectrometry study supported the existence of the enolic protonated diacetone alcohol molecule, in addition to the proton-bound dimer.15 However, there are two intense OH stretch transitions of the alcohol predicted at 3650 and 1560 cm−1 that are missing in the IRPD spectrum, indicating that the enolic species is not efficiently produced under our experimental conditions. A comparison of the ab initioproton-bound dimer spectra with (red) and without (ESI†) argon reveals little difference, except for a 46 cm−1 red shift of one band near 1300 cm−1. This is in contrast to the predictions for the previously reported H+(CO2)2 dimer,16 which exhibited an Ar induced 230 cm−1 blue shift of the shared proton vibration parallel to the OH+O axis, (νsp(∥)). The origin of this effect is the reduction in symmetry of the OH+O bonding motif in the H+(CO2)2 dimer, with the proton becoming more unevenly shared, leading to the blue shift. Complexation with Ar, however, does not change the OH+ bond lengths in the proton-bound acetone dimer because the Ar is bound directly on top of the proton, preserving the essentially Ci symmetry of the core dimer ion. As a result, the perturbation due to Ar is small. Given the small binding energy (175 cm−1) to H+(C3H6O)2, Ar provides an ideal leaving group for studying the dimer with IRPD.
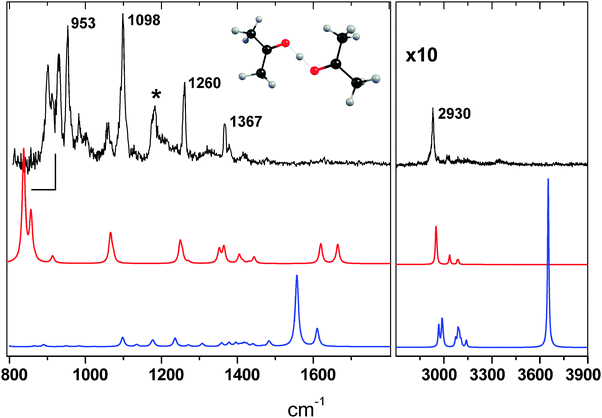 | ||
| Fig. 2 IRPD spectrum of the H+(C3H6O)2Ar complex (normalized to laser power), along with ab initio calculations for the proton bound dimer Ar complex (red) and the protonated diacetone alcohol isomer (blue). | ||
The equal sharing of the proton between the oxygen lone pairs leads to a ∼2700 cm−1 redshift of the OH+ stretch compared to the monomer, although there are five dimer modes involving νsp(∥). As observed for the H+(CO2)2 dimer, the ab initio normal mode vibrations associated with the shared proton motions in H+(C3H6O)2 include the motions of both acetone units, such as methyl bending and CCC stretching. A detailed assignment of the ab initio and experimental spectra are given in the ESI,† and we highlight here the νsp(∥, ⊥) transitions of the proton bound dimer. The frequencies of the shared proton vibrations are given in Table 1. The IRPD bands at 1367, 1260, and 1098 cm−1 are all associated with shared proton motions. The 1367 cm−1 and the smaller peak 15 cm−1 to the blue are assigned to in-plane (COH+OC) νsp(⊥) transitions. The band at 1260 cm−1 is in good agreement with the predicted frequencies of both an out-of-plane νsp(⊥) and an in-plane νsp(⊥) mode. The out-of-plane νsp(⊥) corresponds to the band mentioned above that is red shifted by the Ar. This seems reasonable, given the proton oscillates out of the plane and into the Ar, which likely leads to an increased binding of the Ar in the excited vibrational state and hence a red shift. The in-plane νsp(⊥) at this frequency involves the in-phase CCC asymmetric stretching of both acetones. The most intense band at 1098 cm−1, along with the weaker band at 1057 cm−1, are in the vicinity of the predicted frequencies of two bands associated with in- or out-of-phase methyl bending, along with some νsp(∥) character. One of these two transitions is predicted to be three times as intense as the other, perhaps accounting for the relative intensities of the two IRPD bands.
| ν sp(∥) | ν sp(⊥)b oop | ν sp(⊥) ip | |
|---|---|---|---|
| a Frequencies are scaled by 0.96, and the units are in cm−1 (km mol−1 for IR intensity). b Notation: ip (in-plane), oop (out-of-plane). | |||
| Ab initio | 1065 (130), 838 (930), 631 (3300), 454 (720), 337 (990) | 1255 (66), 857 (390) | 1249 (170), 1365 (140) |
| IRPD | 1098, 900, 930 | 1260, 930, 953 | 1367, 1260 |
As is apparent in Fig. 2, above 1020 cm−1, the H+(C3H6O)2Ar harmonic frequency calculation is in good agreement with the IRPD spectrum, except for the band centered at 1185 cm−1 (marked with *). In addition, the two carbonyl (CO) stretch bands predicted at 1620 and 1660 cm−1 appear only as a broad, weak feature centered near 1630 cm−1.
There are four intense H+(C3H6O)2 (>1000 km mol−1) transitions with strong νsp(∥) character predicted at 333, 451, 626, and 836 cm−1, which are all shifted by less than 5 cm−1 by complexation with Ar. Although the band at 1185 cm−1 does not match any features in the calculated spectrum, it seems reasonable to assign this transition to the overtone of the νsp(∥) transition predicted at 626 cm−1, in part due to the calculated intensity of the fundamental (3400 km mol−1). With the current setup, we are capable of probing the fundamental νsp(∥) band predicted at 836 cm−1 that involves the simultaneous out of phase symmetric CCC stretch and out of phase methyl rocking motions. While there are no transitions observed between 700 and 900 cm−1, a group of intense bands occurs in the IRPD spectrum between 900 and 950 cm−1. We note here that there are six other normal modes predicted in this region that correspond to methyl bending and CCC stretching motions, although with much smaller oscillator strengths. We therefore assign the structure between 900 and 950 cm−1 to the brightνsp(∥) transition that is apparently anharmonically coupled to the other dark modes of the complex also predicted in this region. Splittings and intensity sharing have also been observed in the proton-bound dimers of H2O5 and CO2,16 along with other R–O⋯H+⋯O–R′ systems,7 and is attributed to the extensive coupling of the shared proton motions to the molecular framework. In contrast to the transitions above 1020 cm−1, the νsp(∥) band origin is not predicted well with the harmonic frequency calculation, which is perhaps not surprising given the anharmonic nature of the proton transfer potential in systems of this type.5 In conclusion, the IRPD spectrum indicates only the presence of the lowest energy proton bound acetone dimer. Furthermore, the vibrational modes associated with shared proton motions involve significant bending and stretching of the extended molecular framework.
Acknowledgements
We gratefully acknowledge support for this work from the National Science Foundation (grant no. CHE-0551202).References
- C. A. Wraight, Biochim. Biophys. Acta, 2006, 1757, 886–912 CAS.
- K. R. Asmis, N. L. Pivonka, G. Santambrogio, M. Brummer, C. Kaposta, D. M. Neumark and L. Wöste, Science, 2003, 299, 1375 CrossRef CAS.
- D. T. Moore, J. Oomens, L. Van der Meer, G. Von Helden, G. Meijer, J. Valle, A. G. Marshall and J. R. Eyler, ChemPhysChem, 2004, 5, 740 CrossRef CAS.
- J. M. Headrick, E. G. Diken, R. S. Walters, N. I. Hammer, R. A. Christie, J. Cui, E. M. Myshakin, M. A. Duncan, M. A Johnson and K. D. Jordan, Science, 2005, 308, 1765 CrossRef CAS.
- N. I. Hammer, E. G. Diken, J. R. Roscioli, M. A. Johnson, E. M. Myshakin, K. D. Jordan, A. B. McCoy, X. Huang, J. M. Bowman and S. Carter, J. Chem. Phys., 2005, 122, 244301 CrossRef.
- (a) T. D. Fridgen, L. MacAleese, P. Maitre, T. B. McMahon, P. Boissel and J. Lemaire, Phys. Chem. Chem. Phys., 2005, 7, 2747 RSC; (b) T. D. Fridgen, L. MacAleese, T. B. McMahon, J. Lemaire and P. Maitre, Phys. Chem. Chem. Phys., 2006, 8, 955 RSC.
- J. R. Roscioli, L. R. McCunn and M. A. Johnson, Science, 2007, 316, 249 CrossRef CAS.
- (a) W. H. Robertson and M. A. Johnson, Annu. Rev. Phys. Chem., 2003, 54, 173 CrossRef CAS; (b) M. A. Duncan, Intl. Rev. Phys. Chem., 2003, 22, 407 Search PubMed.
- (a) M. Brookhart, G. C. Levy and S. Winstein, J. Am. Chem. Soc., 1967, 89, 1735–1737 CrossRef CAS; (b) G. A. Olah, A. L. Berrier and G. K. Surya Prakash, J. Am. Chem. Soc., 1982, 104, 2373–2376 CrossRef CAS.
- P. V. Huong and G. Noel, Spectrochim. Acta, Part A, 1976, 32A, 831–835 CrossRef CAS.
- Z. Luczynski and H. Wincel, Int. J. Mass Spectrom. Ion Phys., 1977, 23, 37–44 CrossRef CAS.
- Y. K. Lau, P. P. S. Saluja and P. Kebarle, J. Am. Chem. Soc., 1980, 102, 7429–7433 CrossRef CAS.
- A. J. Stace and A. K. Shukla, J. Phys. Chem., 1982, 86, 865–867 CrossRef CAS.
- L. S. Santos, R. Catharino and M. N. Eberlin, J. Mass Spectrom. Lett., 2005, 40, 127–128 Search PubMed.
- K. Norrman, T. I. Solling and T. B. McMahon, J. Mass Spectrom., 2005, 40, 1076–1087 CrossRef CAS.
- G. E. Douberly, A. M. Ricks, B. W. Ticknor and M. A. Duncan, J. Phys. Chem. A, 2007, in press Search PubMed.
- M. W. Schmidt, K. K. Baldridge, J. A. Boatz, S. T. Elbert, M. S. Gordon, J. H. Jensen, S. Koseki, N. Matsunaga, K. A. Nguyen, S. Su, T. L. Windus, M. Dupuis and J. A. Montgomery, J. Comput. Chem., 1993, 14, 1347 CrossRef CAS.
- J. Velasquez, E. D. Pillai, P. D. Carnegie and M. A. Duncan, J. Phys. Chem. A, 2006, 110, 2325 CrossRef CAS.
- O. Dopfer, R. V. Olkhov, D. Roth and J. P. Maier, Chem. Phys. Lett., 1998, 296, 585 CrossRef CAS.
- E. P. Hunter and S. G. Lias, “Proton Affinity Evaluation,”NIST Chemistry WebBook, NIST Standard Reference Database Number 69, National Institute of Standards and Technology, Gaithersburg, MD, (http:∥webbook.nist.gov), 2005.
Footnote |
| † Electronic supplementary information (ESI) available: Full details of the ab initio calculations, including structures, energetics, vibrational frequencies and intensities. See DOI: 10.1039/b716165d |
| This journal is © the Owner Societies 2008 |