Evidence of a water layer in solid-contact polymeric ion sensors†
Roland
De Marco
*a,
Jean-Pierre
Veder
a,
Graeme
Clarke
a,
Andrew
Nelson
b,
Kathryn
Prince
b,
Ernö
Pretsch
c and
Eric
Bakker
a
aNanochemistry Research Institute, Department of Applied Chemistry, Curtin University of Technology, GPO Box U1987, Perth, Western Australia, 6845, Australia. E-mail: r.demarco@curtin.edu.au; Fax: +61 (0)8![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 92667322; Tel: +61 (0)92662602
92667322; Tel: +61 (0)92662602
bAustralian Nuclear Science and Technology Organization (ANSTO), PMB 1, Menai, New South Wales, 2234, Australia
cInst. Biogechem. & Pollutant Dynam., ETH Zürich, CHN F 16, CH-8092, Zürich, Switzerland
First published on 19th October 2007
Abstract
This paper presents the very first direct structural evidence for the formation of a 100 ± 10 Å water layer in coated-wire polymeric-membrane ion-selective electrodes (ISEs).
Solid-state ion-selective electrodes (ISEs) utilizing a polymeric sensing membrane have shown great promise for use in mainstream analytical chemistry. Such solid-state polymeric ISEs do not require an inner filling solution (normally required in liquid-contact ISEs) and are suited superbly for miniaturization as coated-wire electrodes (CWEs).1,2 The CWEs also exhibit rapid response times and a long life out of solution,2 which are desirable qualities for any ion sensor. Small sensors enable novel applications in systems that contain minute amounts of analyte, such as intracellular measurements3–5 where improvements can be made on existing technologies, and in lab-on-a-chip technologies, where a considerable reduction in the size of components can lead to advantages over traditional analytical techniques.6–9
Miniaturized polymeric ion sensors provide exciting opportunities for the application of these concentration-sensitive devices in the detection of ultratrace amounts of total analyte (e.g. attomole quantities),10 which may also be coupled to novel bioassays utilizing nanoparticle-tagged biomolecules.11,12
The underlying problem associated with solid-state polymeric ISEs or CWEs is their susceptibility to a decline in stability and their relatively mediocre detection limits and selectivities over lengthened periods of time in solution. The degradation in sensor response has recently been ascribed to the existence of a water layer between the membrane and the electrode substrate, which behaves unintentionally as an electrolyte reservoir that re-equilibrates on each and every change in sample composition.13,14 In recent times, the presence of a hydrophobic conductive polymer or redox mediator, acting as a redox-buffering underlayer, has been shown to be important for the ion-to-electron transduction process,15,16 noting that this ion-to-electron transduction concept had been established with silver sulfide solid-state sensors in early work by Rhodes and Buck.17 Accordingly, inhibition of the deterioration in sensitivity and stability of all solid-state polymeric ISEs15,16,18–20 is due to an elimination of the water reservoir that tends to form at the electrode-substrate/sensing-membrane interface. Furthermore, the solid contact allows the elimination of the liquid contact utilized in conventional polymeric membrane ISEs, obviating spurious ion fluxes and the need to control and optimize the filling solution in the liquid contact to achieve low detection-limit sensors.21–23
Neutron reflectometry (NR) is an excellent surface-analysis technique for investigating in situ the nanostructures of surfaces, interfaces and thin films, and can be used to monitor the development of water layers at the solid-contact/ion-sensing-membrane interface of CWE ISEs.24 With NR, it is necessary to prepare molecularly thin and atomically flat films (tens to hundreds of Å in thickness and a few Å root mean square (rms) in roughness), so as to enable sufficient reflectivity (via reduced roughness) and to minimize the number of interference fringes arising from the coherent neutron scattering from successive layers of polymeric material (through diminished thickness) in the thin film. If the prerequisites are satisfied then it is possible to derive atomic-scale information about the physical nature of thin films. Theoretical modelling of the NR reflectivity data for a 120-Å-thick water layer beneath a 200-Å-thick ISE layer (see Fig. S1 in the supplementary information†) reveals that, for perfectly resolved and atomically flat layers, the water layer produces a series of interference fringes that are interspersed with the corresponding interference fringes for the ISE film, thereby confirming that NR can be used to detect water layers in CWEs.
In this communication, the authors present the very first structural and spectroscopic evidence, as deduced using NR, electrochemical impedance spectroscopy (EIS), and secondary-ion mass spectrometry (SIMS), for the formation of water layers at the metal substrate/membrane interface of CWE Ag+ and Ca2+ ISEs, noting that these ISEs were chosen as model systems in this study due to their excellent response attributes.
Fig. 1a presents a snap-shot of EIS complex-plane impedance plots as a function of exposure for a CWEAg+ ISE soaking in 10−3 M AgNO3 for 48 hours. The data reveals a high-frequency semi-circle that is ascribable to the bulk membrane resistance coupled with a spurious resistance such as the contact resistance between the membrane and the gold-substrate electrode. From Fig. 1b, it is evident that, during conditioning, the bulk membrane resistance–contact resistance rises sharply until approximately 3 hours, and diminishes gradually until about 20 hours, at which point it stabilizes. The initial increase in resistance has been explained by ion trapping whereby hydrophilic ionic impurities are dissolved into water nanodroplets in the membrane, creating electrical discontinuities.25 The EIS trend after 3 hours may be interpreted in terms of the establishment of a water layer comprising both water and transported ions, and the wet ISE membrane/gold interface ultimately yields a lower contact resistance manifested as a diminished bulk membrane resistance–contact resistance at high frequency. Significantly, a conventional liquid-contact polymeric-membrane ISE shows the initial increase in resistance, but not the eventual decline in resistance that is consistent with the presence of an interfacial water layer.
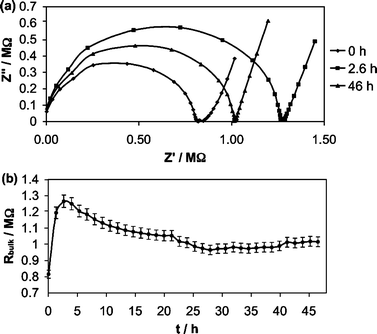 | ||
| Fig. 1 EIS data for a gold CWE Ca2+ ISE conditioned in 10−3 M CaCl2 over a prolonged period: (a) complex-plane impedance plot; (b) bulk membrane resistance as a function of time. | ||
X-Ray reflectometry (XR) was used to determine the thickness and integrity of the spin-cast ISE films, so as to ascertain their suitability in subsequent NR measurements. Accordingly, Fig. 2 shows the XR curve for a molecularly thin Ag+-selective plasticized PVC film, as measured in air without conditioning. Fitting of the reflectivity data to a single-layer model yielded a PVC film thickness of 217 Å and a surface roughness of 11 Å rms. The blurring of the interference fringes in the X-ray reflectivity data at Q values above 0.25 Å−1 is indicative of slight roughening of the polymer coating, as the atomically smooth silicon wafer possesses a surface roughness of a few Å rms. Subsequent work on unplasticized copolymer ISEs (unpublished findings) has shown that this surface roughening of a spin-cast film is due to the high volatility of the tetrahydrofuran (THF) casting solvent, which promotes the deposition of a non-uniform film. Nevertheless, the XR data show that the THF spin-cast film is adequate for measurements on the NR instrument used in this study that possesses a Q range from 0 to 0.25 Å−1.
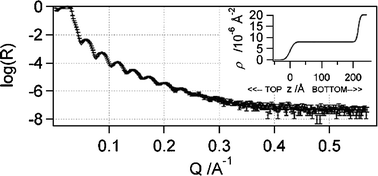 | ||
| Fig. 2 XR reflectivity curve in air for a molecularly thin spin-cast film of Ag+ ISE deposited onto atomically smooth silicon. Note, R is the neutron reflectivity, Q is the magnitude of the neutron-scattering vector, and ρ is the scattering-length density (SLD). | ||
Note that electrochemical studies on molecularly thin films like the one proposed in the present study are experimentally viable, as a previous potentiometric study of a valinomycin-based bilayer membrane revealed a classical Nernstian response to potassium over a wide dynamic range.26
Unfortunately, the NR profile of the PVC film in air cannot be contrasted with the ones in the electrolyte, as the disparate contrasts in neutron-scattering-length density (NSLD) of the polymer against air as well as D2O or D2O–H2O or H2O cannot be compared. Furthermore, as the NSLDs for air and PVC are too similar, there is insufficient neutron-scattering contrast to meaningfully extract information from this featureless NR profile.
The in situ NR data for the PVC Ag+-selective film (see Fig. 3 and 4 for the reflectivity and NSLD curves respectively) required a fitting of two physically dissimilar films with roughening factors. This can be explained by the scattering of neutrons from the polydisperse nanodroplets of water in the membrane, and the presence of an interior film (100–120 Å thick) possessing a NSLD akin to a composite of water and polydisperse nanodroplets of organic liquid (e.g.membrane plasticizer exuded into the water layer).25 Furthermore, the NSLD values for the water-infused materials (i.e.water layer with exuded plasticizer and polymer with water nanodroplets) varied in accordance with changes in the contrast in NSLD values for D2O to D2O–H2O to H2O. This is strong evidence for the reliability of the NR fits. Also noteworthy is the fact that the XR thickness of the original polymer film is congruent with the NR fitted data for the electrolyte-treated samples. Unfortunately, the NR results for the Ag+ ISE bathed in 10−3 M AgNO3 in D2O, a mixture of D2O–H2O, along with H2O (see the reflectivity curves in Fig. 3a–c and NSLD curves in Fig. 4a–c), together with the NR best-fit parameters (see Table S1 in the supplementary information†), revealed a smearing of the interference fringes. Similar to the interpretation of the XR data, this can be explained by a roughening of the films, along with an apparent loss of contrast in NSLD values. Clearly, the blurring at the interface between the PVC and water layer, as evidenced in the NSLD profiles, is ascribable to roughening of the films due to neutron scattering from the polydisperse water inclusions in the PVC layer and polydisperse plasticizer nanodroplets in the water layer (see Fig. 5). Furthermore, the fitted data for the films provided apparent NSLDs for the PVC film akin to a composite of the plasticized PVC film and inclusions of water, and a water layer consistent with the presence of exuded organic material (i.e. plasticizer) from the membrane. This is symbolic of the permeation of water nanodroplets into the PVC film and plasticizer into the water layer; well-known phenomena that have been studied extensively by various researchers25,27–31 where it was shown that the water droplets are nanoscaled.27 This phenomenon is responsible for the polymer’s NSLD value approaching that of the aqueous electrolyte, as well as the observed smearing of the interference fringes due to roughening as neutrons scatter off the polydisperse water and/or plasticizer nanodroplets along with the surrounding polymer matrix and/or water layer. Also, the H2O NR data (see Fig. 3c) showed signs of a time dependency over the duration of the NR measurement (i.e., about 20 hours), as evidenced by a slight increase in reflectivity between Q values of 0.175 and 0.225 Å−1. This time dependency is responsible for the slight deviation between the fitted and experimental reflectivity data at higher Q values.
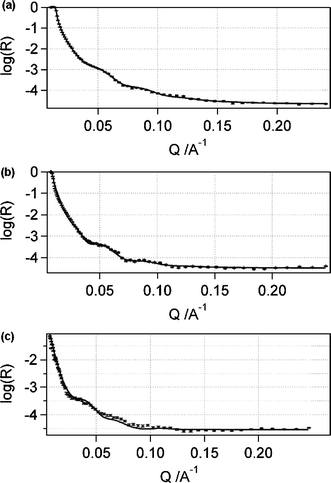 | ||
| Fig. 3 NR reflectivity curves for a molecularly thin spin-cast film of Ag+-selective membrane deposited onto an atomically smooth silicon wafer in 10−3 M AgNO3 in (a) D2O, (b) D2O–H2O and (c) H2O. | ||
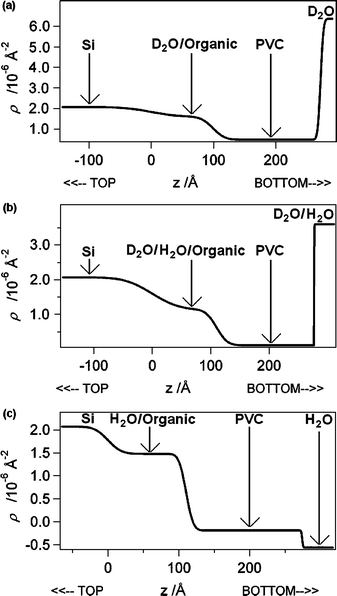 | ||
| Fig. 4 NR NSLD curves for a molecularly thin spin-cast film of Ag+ ISE deposited onto an atomically smooth silicon wafer in 10−3 M AgNO3 in (a) D2O, (b) D2O–H2O and (c) H2O. | ||
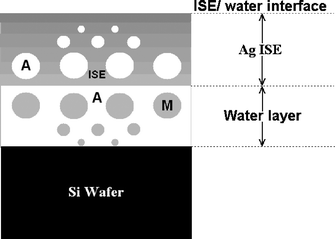 | ||
| Fig. 5 Schematic diagram depicting the physical structure in a CWE ISE membrane, noting the formation of a water layer comprising exuded plasticizer (i.e. M) and a membrane phase saturated with polydisperse water (i.e. A). | ||
Following conditioning of CWE Ca2+ ISEs in separate solutions of 0.1 M Ca(NO3)2, 0.1 M CaCl2 and 0.1 M NaCl with subsequent peeling of the ISE membrane to expose the underlying Au-substrate electrode, SIMS depth profiling revealed a surplus of cation (i.e.Ca2+ and Na+) and anion (i.e.Cl− and NO3−), as compared to the control Au sample, symbolizing the transportation of water and ions to the metallic-electrode/membrane interface (see Table 1 for average 133CsNM+ counts for static SIMS profiles of these substrates). Note the following important points: (1) the control Au sample comprised a CWE that had not been exposed to electrolyte; (2) the SIMS intensities represented raw ion counts without consideration of elemental sensitivity factors; and (3) the relatively high experimental uncertainty of the SIMS ion counts (i.e. 10–20% deviation) did not permit a comparison of the relative permeation rates of ions through the ISE membrane. Clearly, the water layer acts as a reservoir for membrane-transported ions, and provides a source of uncontrollable and spurious ion fluxes that manifest themselves in the same way as transmembrane fluxes do in the non-optimized filling solutions of liquid-contact polymeric ISEs.14 Without doubt, this is a critical problem that must be resolved if solid-contact ISEs are to be used widely in analytical chemistry.
| Sample | 133Cs23Na+ | 133Cs14N+ | 133Cs35Cl+ | 133Cs40Ca+ |
|---|---|---|---|---|
| Au (N = 4) | 4059 (61) | 527 (22) | 129 (48) | 4787 (45) |
| Au PVC Ca(NO3)2 (N = 4) | — | 1627 (8) | — | 22298 (21) |
| Au PVC CaCl2 (N = 4) | — | — | 1729 (5) | 30357 (10) |
| Au PVC NaCl (N = 3) | 29785 (49) |
— | 1366 (20) | — |
Fig. 5 presents a schematic diagram of a polymeric CWE ISE, particularly as it relates to the Ag+ ISE investigated in the in situ NR study of the evolution of water layers in CWE ISEs. In the model, the presence of polydisperse water nanodroplets is proposed, as suggested elsewhere by Liet al.30 and supported strongly in this work by the NSLD data and smearing of the interference fringes for the fitted-polymer ISE film in the NR reflectivity data. Furthermore, the interfacial water layer is infused by polydisperse nanodroplets of the membrane plasticizer, as evidenced by an intermediate NSLD for water and the organic plasticizer, and this phenomenon has been observed by other investigators25,31 who noted membrane exudation of plasticizer into the electrolyte, so the plasticizer is also expected to migrate into the water layer of the CWE ISE.
Conclusions
This work has provided the first experimental evidence for the formation of a water layer in CWE ISEs, as depicted in Fig. 5. Significantly, this undesirable water layer acts as an intermediate electrolyte reservoir manifesting itself in the same way as a non-optimized filling solution in a liquid-contact ISE, thereby providing spurious ion fluxes that degrade the detection limit and selectivity of CWE ISEs.The outcome of this research has important ramifications for solid-contact polymeric ISEs; it is necessary to develop strategies to eliminate the formation of a detrimental water layer at the electrode-substrate/ISE-membrane interface if the superior response characteristics of all-solid-state polymeric ISEs are to be achieved.
Acknowledgements
The authors acknowledge the financial support of the Australian Research Council (LX0454397 and DP0665400), Australian Institute of Nuclear Science and Engineering, National Institutes of Health (EB002189) and Swiss National Foundation. We also thank Mr Armand Atanacio and Dr Tracey Hanley at ANSTO for assistance with the SIMS and XR research respectively.References
- R. W. Cattrall and I. C. Hamilton, Ion-Sel. Electrodes Rev., 1984, 6, 125–172 Search PubMed.
- R. W. Cattrall and H. Freiser, Anal. Chem., 1971, 43, 1905–1906 CrossRef CAS.
- Z. M. Hu, T. Buhrer, M. Muller, B. Rusterholz, M. Rouilly and W. Simon, Anal. Chem., 1989, 61, 574–576 CrossRef CAS.
- R. Steiner, M. Oehme, D. Ammann and W. Simon, Anal. Chem., 1979, 51, 351–353 CrossRef CAS.
- F. F. Lanter, D. Erne, D. Ammann and W. Simon, Anal. Chem., 1980, 52, 2400–2402 CrossRef CAS.
- G. H. Sanders and A. Manz, J. Assoc. Lab. Automation, 2000, 5, 40–45 Search PubMed.
- A. Manz, N. Graber and H. M. Widmer, Sens. Actuators, B, 1990, B1, 244–248 CrossRef.
- W. Ehrfeld, Electrochim. Acta, 2003, 48, 2857–2868 CrossRef CAS.
- P. Dittrich and A. Manz, Nat. Rev. Drug Discovery, 2006, 5, 210–218 CrossRef CAS.
- A. Malon, T. Vigassy, E. Bakker and E. Pretsch, J. Am. Chem. Soc., 2006, 128, 8154–8155 CrossRef CAS.
- R. Thurer, T. Vigassy, M. Hirayama, J. Wang, E. Bakker and E. Pretsch, Anal. Chem., 2007, 79, 5107–5110 CrossRef.
- K. Y. Chumbimuni-Torres, Z. Dai, N. Rubinova, Y. Xiang, E. Pretsch, J. Wang and E. Bakker, J. Am. Chem. Soc., 2006, 128, 13676–13677 CrossRef CAS.
- E. Bakker and E. Pretsch, TrAC, Trends Anal. Chem., 2005, 24, 199–207 CrossRef CAS.
- M. Fibbioli, W. E. Morf, M. Badertscher, N. F. de Rooj and E. Pretsch, Electroanalysis, 2000, 12, 1286–1292 CrossRef CAS.
- J. Bobacka, Anal. Chem., 1999, 71, 4932–4937 CrossRef CAS.
- A. Michalska, Anal. Bioanal. Chem., 2006, 384, 391–406 CAS.
- R. K. Rhodes and R. P. Buck, Anal. Chim. Acta, 1979, 110, 185–196 CrossRef CAS.
- J. Bobacka, Electroanalysis, 2006, 18, 7–18 CrossRef CAS.
- J. Migdalski, T. Blaz and A. Lewenstam, Anal. Chim. Acta, 1996, 322, 141–149 CrossRef.
- J. Bobacka, A. Ivaska and A. Lewenstam, Electroanalysis, 2003, 15, 366–374 CrossRef CAS.
- K. Y. Chumbimuni-Torres, N. Rubinova, A. Radu, L. T. Kubota and E. Bakker, Anal. Chem., 2006, 78, 1318–1322 CrossRef CAS.
- J. Sutter and E. Pretsch, Electroanalysis, 2006, 18, 19–25 CrossRef CAS.
- J. Sutter, A. Radu, S. Peper, E. Bakker and E. Pretsch, Anal. Chim. Acta, 2004, 523, 53–59 CrossRef CAS.
- A. S. Brown, S. A. Holt, P. M. Saville and J. W. White, Aust. J. Phys., 1997, 50, 391–405 Search PubMed.
- G. Horvai, E. Graf, K. K. Toth, E. Pungor and R. Buck, Anal. Chem., 1986, 58, 2735–2740 CrossRef CAS.
- H. Minami, N. Sato, M. Sugawara and Y. Umezawa, Anal. Sci., 1991, 7, 853–862 CrossRef CAS.
- A. D. C. Chan and D. J. Harrison, Anal. Chem., 1993, 65, 32–36 CrossRef CAS.
- X. Li, S. Petrovic and D. J. Harrison, Sens. Actuators, B, 1990, B1, 275–280 CrossRef CAS.
- Z. Li, X. Li, M. Rothmaier and D. J. Harrison, Anal. Chem., 1996, 68, 1726–1734 CrossRef CAS.
- Z. Li, X. Li, S. Petrovic and D. J. Harrison, Anal. Chem., 1996, 68, 1717–1725 CrossRef CAS.
- D. Messadi and J. M. Vergnaud, J. Appl. Polym. Sci., 1982, 27, 3945–3955 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, neutron reflectometry (NR) cell design, theoretically modelled NR data for PVC with and without a water layer, and NR best-fit data. See DOI: 10.1039/b714248j |
| This journal is © the Owner Societies 2008 |