Dynamic imaging of molecules using high order harmonic generation
Jon P.
Marangos
*,
Sarah
Baker
,
Nathaniel
Kajumba
,
Joseph S.
Robinson
,
John W. G.
Tisch
and
Ricardo
Torres
Blackett Laboratory, Imperial College London, Prince Consort Road, South Kensington, London, UK SW7 2BZ. E-mail: j.marangos@imperial.ac.uk
First published on 14th November 2007
Abstract
We review recent progress towards imaging the electronic wavefunctions and nuclear dynamics of small molecules using the high order harmonics emitted when a molecule experiences an intense laser field. We illustrate that the essence of high harmonic emission is contained in the recombination amplitude between the continuum portion of the electronic wavefunction, that is formed through field ionization and which is accelerated and driven back to recollide in the laser field, and the bound electronic state. We review for the non-specialist some recent experimental and theoretical work dealing with high harmonic generation (HHG) in molecules. Particular attention is paid to two types of experiment recently performed in our group. The first of these types of experiment is the measurement of signatures of molecular electronic structure using HHG from molecules with a fixed orientation in space. The second is the use of HHG to track extremely fast proton rearrangement following ionization in light molecules, using the intrinsic temporal variation of the recolliding electron energy to extract these dynamics from measurements of the high harmonics.
 Jon P. Marangos Jon P. Marangos | Jon Marangos (Lockyer Professor of Physics, Imperial College) is the Head of the Quantum Optics and Laser Science group in the Blackett Laboratory at Imperial College and the Director of the Blackett Laboratory Laser Consortium. Since gaining his PhD in 1986 he has carried out research in laser spectroscopy, non-linear optics, laser-matter interactions and coherent control at Imperial College and through collaborations in Europe, Japan and North America. His current work is mainly in the areas of ultra-fast strong field phenomena and attosecond science. |
 John W. G. Tisch John W. G. Tisch | John Tisch is Reader in Physics working in the Quantum Optics and Laser Science group within the Physics Department at Imperial College London. His research interests are ultrafast laser physics and attosecond science. He gained his Ph.D. degree in 1995 at Imperial College and has also worked at the ETH in Zürich. |
Introduction
It has recently been demonstrated that it is possible to use high order harmonic generation (HHG) from molecules in a strong laser field to image molecular structure and dynamics with simultaneously sub-angstrom spatial resolution and sub-femtosecond temporal resolution. These measurements are enabled by several recent technical advances; the availability of high intensity light pulses in the near IR with pulse duration down to just a few optical cycles, improved theoretical understanding that provides a path to calculate strong field processes even from non-trivial molecular structures, and the new possibilities to fix the orientation of molecules in space using a variety of laser alignment techniques most notably non-adiabatic alignment.The aim of this review will be to describe techniques for ultra-fast measurement of molecular structure and dynamics that are based upon HHG and our understanding of the electron dynamics driven by a laser pulse within an optical cycle, and the role of molecular structure. We will concentrate upon the results of our own research but will also cover results from colleagues in other laboratories where appropriate. The intention is to explain these techniques to scientists not previously working in strong laser fields/non-linear optics and particularly to researchers working in the area of chemical physics. The more specialist reader may wish also to consult a technically more detailed recent review on the subject.1
High harmonics are generated when an atom or molecule is exposed to an intense laser field and the resulting coherent radiation extends to photon energies that can easily exceed 100 eV. To understand the process of high harmonic generation (HHG) we must appreciate that it is closely tied to the driven electron dynamics in a strong laser field. A consequence of this connection to the dynamics in the field is the ultra-fast (few hundred attoseconds) duration of extreme ultraviolet (XUV) harmonics that are emitted. This property of HHG radiation has in the last few years found application in a variety of attosecond domain measurements. Whilst the high photon energy and short duration of radiation are a consequence of the influence of the laser field on the ionized electron, the amplitude of emission depends upon properties of the atomic or molecular system. Thus the HHG signal carries time resolved information about the atomic or molecular structure that over the last few years we have started to understand how to interpret.
1.1 Electron dynamics driven by a strong laser field
We begin by reviewing the key ideas behind high order harmonic generation. This field is rooted in non-linear optics and laser physics so in what follows we will attempt to stress the physically important ideas and avoid technical detail (a more technical review of general HHG can be found in ref. 2 and of molecular HHG in ref. 1).If the laser field is strong (intensity >1014W cm−2) and linearly polarized, HHG can be understood using a semi-classical model proposed in 1993.3,4 In the semi-classical picture the process separates into three distinct steps. First the laser field ionizes the atom or molecule through field ionization, the dominant mechanism for this is quantum tunneling through the field suppressed potential barrier. The tunnel ionization liberates some electron amplitude from the binding potential and so launches an electron wavepacket into the continuum. Tunnel ionization occurs with high probability only near the peak of the sinusoidally varying electric field amplitude (Fig. 1) due to the exponential dependence of the tunneling rate on the factor  5–7 (where Ip is the ionization potential of the bound state and E(t) the electric field amplitude, both in atomic units). The exponential dependence of the tunneling rate ensures that (a) the highest energy electronic states are field ionized first, and (b) the ionization is confined in time to ∼± 200 attoseconds (or <1/10th of an optical cycle) around the cycle peak.
5–7 (where Ip is the ionization potential of the bound state and E(t) the electric field amplitude, both in atomic units). The exponential dependence of the tunneling rate ensures that (a) the highest energy electronic states are field ionized first, and (b) the ionization is confined in time to ∼± 200 attoseconds (or <1/10th of an optical cycle) around the cycle peak.
 | ||
| Fig. 1 The electron trajectory displacement (vertical scale) and return energy (darker = higher return energy) within the trajectories launched by ionization in the optical half-cycle peaking at t = 0. The laser electric field as a function of time is shown as a dashed line (800 nm, 2 × 1014W cm−2), whilst the trajectory associated with the highest electron energy is indicated by a dashed white line. | ||
In the next step, the electron wavepacket moves in response to the laser field: first being accelerated away from the parent ion, and then returning at some later time on the order of half an optical cycle period (typically 0.5–1.5 fs for a laser field at 800 nm) as the laser field reverses direction. A characteristic of the strong field limit is the large amplitude of the excursion of the ionized electron from the core, this may be many times the dimensions of the molecule and will take the electron to regions of space where the Coulomb interaction with the ion core is weak. During this process the electron wavepacket gains energy from the laser field. More subtly, the various momentum components within the electron wavepacket gain a phase that depends upon the timing of the ionization event, which determines the laser field they subsequently experience.
The evolution of the electron wavepacket in the laser field can essentially be treated as classical motion of a charge in an oscillating electric field. The details of the motion are then determined by the phase of the field when the electron was ionized, the field frequency and amplitude, with the following boundary conditions: that it appears at the parent ion with an initial zero momentum,8,9 and that it must return to the parent ion in order for recombination to occur. Solution of the classical equations for an electron moving in an oscillating electric field with the above constraints shows that only those electrons that tunnel ionize in the optical cycle shortly after the peak, i.e. while the field is falling may return (Fig. 1), those arriving in the continuum before the peak, i.e. while the field is rising, never return and so they directly ionize.
The third step is a recombination process of the electron wavepacket with its parent ion in which the energy gained by the electron in the field is emitted as a high energy photon, whilst the electron makes a transition back to the ground state of the system. The spectrum of emitted radiation extends up to a cut-off energy set by the maximum possible electron return energy, which from classical considerations is 3.17Up + Ip (where Up is the ponderomotive (quiver) energy of a free electron in the field and Ip the ionization potential of the state). For laser intensities typically used for HHG ((1–5) × 1014W cm−2) the cut-off photon energies for a most molecules will lie in the range ∼30–110 eV.
Electrons that are born into the continuum between the peak of the electric field and about 1/20th of a cycle follow the so called “long trajectories”, travelling far from the core before the electric field reverses direction (Fig. 1). Electrons launched into the continuum between 1/20th of a cycle and the zero-crossing of the electric field follow “short trajectories”, that is, their path in the continuum is short since the electric field reverses direction relatively quickly following their birth. For electrons born at 1/20th of a cycle after the peak the short and long trajectories converge and have the highest energy upon return, giving rise to the highest energy photon emission (the harmonic cut-off). Within each “class” of trajectories, the energy of the colliding wavepacket varies depending on its time of birth:9,10e.g. for electrons following shorter trajectories, those which follow the very shortest paths return with relatively low energy since they have experienced little acceleration by the field. Long trajectories have the opposite dependence, with the electrons spending longer in the field returning with smaller energy. Some long trajectories give rise to multiple electron returns but their role in HHG is usually insignificant.
1.2 Recolliding electrons and HHG emission
For electrons following the short trajectories there exists a direct relationship between the electron return time, and the energy of the harmonic photon emitted: successively higher orders of harmonics are generated at longer time delays. There is in effect an encoding between the return time and the frequency in the emitted spectrum through the energy chirp (i.e. variation with time) of the electron wave packet. This property of HHG is fundamental to the new technique demonstrated in our work, since it allows a range of pump–probe delays to be accessed by analysis of a single harmonic spectrum (this will be discussed further in section 3).It is useful to examine the properties of the returning electron. These are determined by the peak intensity (field) of the pulse and the carrier wave frequency as these set the scale of the ponderomotive energy; Up = e2E02/4meω2 (where E0 is the electric field amplitude and ω the angular frequency of the field). The return electron kinetic energy will range from a few eV for the very lowest energy electrons up to 3.17Up for the highest returns. This should be added to the ground state binding energy, Ip, gained from the Coulomb field as the electron recombines to obtain the energy of the photon emitted. For an 800 nm field focused to an intensity of 5 × 1014W cm−2, Up = 32 eV and so the highest return energy is slightly above 100 eV. Assuming that the electrons returning in this field have a kinetic energy in the range 10 to 100 eV, then the de Broglie wavelength of the electron wave packet spans the range from 3.9 × 10−10 m (for 10 eV electrons) down to 1.22 × 10−10 m (for 100 eV electrons); the latter wavelength is close to the size of a typical diatomic molecule. At this same intensity a field of 1.6 μm will have return energies up to 400 eV so the shortest de Broglie wavelength in the wavepacket will be 0.61 × 10−10 m i.e. close to the size of a hydrogen atom.
It is the small electron de Broglie wavelength (rather than the much longer wavelength of the corresponding emitted XUV radiation) that makes the HHG process a sensitive probe of molecular structure. The recombination amplitude for the process is sensitive to structural details within the molecule down to a scale comparable to the de Broglie wavelength. The situation is akin to an electron diffraction measurement, but with the emitted electromagnetic field carrying the structural information rather than the elastically scattered electrons.
For the electrons that return, the recombination channel is only one possible outcome of the many events that might occur in the re-encounter with the parent ion core. The electron returns with sufficient kinetic energy to excite a range of inelastic scattering processes, such as recombination to any of the excited states or excitation of a second electron either to a bound state or the continuum. The electron may also elastically scatter.
Why is it then that we can isolate much of what is going on in the strong field process given that there are so many potential outcomes? The heart of the answer is that in order for the generated HHG field to be large enough to be observable the contributions from all atoms/molecules in the sample should add coherently. In part this requires that (in the language of non-linear optics) the generated field is phase matched i.e. that it propagates in phase with the material dipole that drives it. On a microscopic scale it requires that the driven dipoles each have identical phase, which can only be satisfied if they begin and end with the same state i.e. usually the ground state of the neutral atom or molecule. This means the macroscopic HHG signal is derived only from those molecules that have undergone the 3-step process that begins and ends in the ground state. In effect this is a powerful sift that makes HHG blind to the myriad of other strong field processes that may occur, and instead gives the signal a unique specificity that allows us to say precisely what type of process the observed XUV radiation arose from.
Imaging techniques based upon HHG have considerable potential as they may offer a new route to directly retrieving molecular structure. HHG emission from a properly prepared medium yields a strong signal that can be measured reliably in a single shot. Moreover, the timescale of these measurements is very fast, which makes them commensurate with the fast time-scale of non-adiabatic alignment. The ionization/recollison process at the heart of HHG takes place upon a sub-optical cycle timescale, which presents the opportunity to use these measurements to probe some of the fastest processes in molecules, as was recently demonstrated using the chirp encoding of the electron recollision to image proton rearrangement in a molecule following ionization.11 The ultimate ambition is to use the technique of HHG structural imaging to make “movies” of molecular structure changing during a chemical reaction for which temporal resolution approaching a few femtoseconds is required.
2. Structural imaging using HHG from aligned molecules
2.1 Influence of molecular ground state wavefunctions on HHG signals
Much progress has been reported in recent years towards using the XUV pulses emitted in the HHG process in atoms, which can approach durations as short as 100 attoseconds, to make attosecond time domain measurements.10,12–16 The HHG signal obtained directly from a molecular sample provides an alternative way to make attosecond domain measurements of molecular structure. The recollision of the electron is a brief event, taking much less than an optical period, and occurs at a well synchronized moment with respect to the ionization, and with an appreciable momentum (returning with kinetic energy in the 10–100 eV range for typical fields). As a consequence of these properties the recollision electron can be used as a probe in several ways. For instance, Corkum and colleagues first pointed out that the ionization event forms correlated electron/nuclear wavepackets in a molecule; these can be used to determine vibrational or dissociation dynamics from measurements of the kinetic energy released when the molecules are collisionally ionized by the returning electron. In principle, the elastically scattered electron momentum distribution carries structural information through the interferences between the amplitudes arising from different scattering centers (electron diffraction), a process termed laser induced electron diffraction. Recently there have been advances both theoretically17–21 and experimentally22 towards realizing this concept.It is, however, the emission of harmonics from a molecular target of aligned molecules that has been shown most successfully to carry structural information about the molecule with the potential for a very high temporal resolution.23–28 This is possible because the HHG amplitude is dependent upon the dipole amplitude for the transition between the continuum state and the ground state. As the return electron energy is in the range of 10–100 eV, the de Broglie wavelengths in the wave packet span the spatial scales of interest in the ground state wavefunction and so the dipole is sensitive to the electronic structure of the molecular ground state.24
For a linearly polarized strong field with electric field vector in the z direction the harmonic spectrum from a single molecule is given by S(ω) ∼ω4|D(ω)|2, where D(ω) is the Fourier transform of the time-dependent dipole moment D(t) = 〈Ψ(t)|−ez|Ψ(t)〉 and Ψ(t) is the time-dependent electron wave function evolving in the combined molecular and laser field potentials. When the wave-function Ψ(t) is written in terms of a superposition of bound and continuum8 parts Ψ(t) = a0(t)Ψo + ac(t)Ψc the time-dependent dipole takes the simple (length gauge) form:
 | (1) |
Numerical treatments25,26,29,30 of HHG have been carried out by solving the time-dependent Schrödinger equation for the simple molecules H2+ and H2. It was found that as the angle θ between laser polarization and the internuclear axis was varied, for a given harmonic order a characteristic minimum of the calculated dipole amplitude and modulation of the phase appeared around a specific angle.25 By comparison with a simple plane-wave model for the returning electron it was argued that this was due to interference between the dipole amplitudes from the two atomic centers in the molecule when they were separated by half the de Broglie wavelength λB of the returning electron.26 This idea is closely analogous to laser induced electron diffraction. The measurement of these recombination interference signatures for certain values of k will give almost directly the internuclear separation.26,27 The conditions for interference were found to correspond to the simple conditions for constructive and destructive interference, respectively:26,28
 | (2) |
In the strong field limit the so called strong field approximation (SFA) is frequently applied. The major assumptions in SFA are that: (a) there is no influence of the laser field upon the molecular bound state (and only the ground state is considered); (b) the continuum state is dressed only by the strong laser field (i.e. a Volkov state) and is not influenced by the molecular potential. In this approximation the continuum states are assumed to be plane waves exp(ik·r). Based upon a derivation from the full SFA formulation8 it has been argued that for a molecule with well defined alignment it is possible to extract the molecular state wave function Ψ0 from measurements of the harmonic spectrum.24 In this simplified picture, adopted by Itatani and co-workers,24 the complex integrals that must be evaluated for all electron trajectories are factored into a simple expression for the harmonic amplitude. For z-polarized light a recollision wavepacket can be written as the sum (integral) over the possible momenta of plane waves:
 | (3) |
 | (4) |
2.2 Determination of the structure of small molecules using HHG
We now examine the first steps towards implementation of the ideas outlined above. There are some severe challenges to the practical application of the HHG imaging techniques to a real molecule. Some of these are technical; for instance achieving high enough sample densities of well aligned molecules in the sample. There are also matters of principle regarding the interpretation of HHG. For instance, the validity of the assumption that the HHG can be adequately described within a strong field approximation (SFA) model, which is an essential simplifying assumption to permit straightforward structural retrieval from the measurements.To make any measurement the orientation of the molecules must first be fixed in space. Early experiments employing adiabatic alignment showed the modulation of the HHG yield as a result of partial alignment in the molecular ensemble.23 More recently this technique was employed to study the alignment dependence of lower order HHG (9th–17th) with angle in the CO2 molecule.31 These experiments demonstrated a modulation of the HHG yield as a function of the presence of alignment,32 but technical problems (amplitude noise on the aligning field) appear to have limited the extent of alignment and so the depth of modulation.
More recent work using the impulsive alignment technique33 has shown much larger alignment dependent modulations of HHG, indicative of higher degrees of alignment, with the added benefit that the alignment is field free at the rotational revivals and sub-revivals.34,35 Several reports have been made where high order harmonic generation from an ensemble of spatially aligned linear molecules24,27,28,34 has been measured using the impulsive method. In this technique the molecular alignment is controlled by an initial ultra-fast laser pulse (duration ∼100 fs and intensity ∼1013W cm−2), which excites a rotational wavepacket (coherent superposition of rotational states) in the molecules.33,36,37 The rotational wavepacket exhibits strong molecular axis alignment at a regular period corresponding to rotational revivals, half-revivals and quarter revivals. For a rotational period T these occur at delays of T, T/2 and T/4 (and integer multiples thereof). A second higher intensity ultra-fast laser pulse then produces high harmonic emission from the molecules when they are close to the maximum degree of alignment. By varying the polarization angle of this laser field with respect to the alignment field polarization the HHG emission intensity for different angular distributions can be measured (see Fig. 2).
 | ||
| Fig. 2 (a) Schematic layout of the experiment to measure HHG spectra from laser aligned molecules. The blue and red lines represent the aligning and harmonic generating beams, respectively. (b) Distribution of molecular orientations in an aligned sample probed at different angles θ. | ||
The simultaneous observation of the ionization yield along with the intensity of the soft X-ray harmonic emission by Kanai et al.27 allowed the efficiency of the first (ionization) step of the process to be separated from the efficiency of the final step when the electron wave recombines to the initial molecular state. In their measurements CO2 was found to have a minimum in the harmonic emission when the ionization was maximum. Increased ionization was accompanied by maximum harmonic emission in the cases of O2 and N2, but in the case of CO2 this was more than offset by the reduced efficiency of the recombination step of the process. The parts of the molecular electronic state of CO2 located near the two oxygen atoms make equal but opposite contributions to the X-ray emission, due to the anti-symmetric nature of the πg HOMO. The suppression of the harmonic emission occurred when the two oxygen atoms in the molecule, which are a distance of 0.232 nm apart, are separated by exactly one electron wavelength in the direction of the electron travel (laser polarization). The soft X-ray emission amplitude from each of the two oxygen atoms is then exactly out of phase, which leads to destructive interference in the total emission. We have measured CO2 and O2 in an impulsively aligned sample, but go further by measuring the HHG spectrum to much higher harmonic orders (Fig. 3) and by confirming the essential role of two-center interference, not only in CO2, but also providing evidence for this process in O2.
 | ||
| Fig. 3 HHG spectra from CO2 measured for an isotropic sample (red line) and a non-adiabatically aligned sample (black line). The strong suppression of the signal for the aligned case extending from the 25th to 41st harmonics is an evidence of two-center interference. | ||
The group at NRC, Ottawa made a series of harmonic spectrum measurements for a sample of aligned N2 molecules over a range of alignment angles θ from 0 to 90° (Fig. 4). They also measured the harmonic spectrum from an atomic reference (Ar) that has the same ionization potential as N2. By assuming that the electron wavepacket amplitude a(k) will be the same for N2 and Ar they were able to use eqn (4) to divide through the measured harmonic spectra of the molecule by the reference spectrum and, using the (known) dipole matrix for Ar, determine the normalized dipole matrix amplitude 〈exp(ikz)|z |Ψ0〉 of N2 for each alignment angle. After making a few realistic assumptions about the phase variation of the dipole with θ they were then able to tomographically reconstruct Ψ0 for the molecule using a Radon transform method (as used in medical tomography). The retrieved wavefunction was in fair agreement with the calculated HOMO for this molecule.24
 | ||
| Fig. 4 HHG spectra recorded for N2 aligned at different angles between 0 and 90° relative to the polarization axis of the laser. The HHG spectrum from argon, used as a reference, is also shown. Reprinted by permission from Macmillan Publishers Ltd.: Nature, 432, 867, copyright 2004. | ||
2.3 Steps toward HHG imaging of small organic molecules
We wish now to address the issue of scalability of the molecular imaging methods to bigger systems. There have been some studies devoted to HHG in randomly oriented organic molecules,38–40 but quantitative studies in aligned systems have been confined to simple diatomic and triatomic linear molecules. There are a number of obstacles to investigating larger molecules. For imaging to be efficient a large enough number of harmonic orders must be produced, thus providing a sufficient spread of momenta to allow a complete characterization of the orbital in momentum space. Increasing the molecular size, however, generally reduces the ionization potential, thus bringing the saturation of ionization down to lower laser intensities, with the consequence that the cut-off (highest energy) harmonic will be at relatively low orders. Besides, laser intensities must be kept low enough and the pulses be short enough to prevent molecular fragmentation, which is also more likely in larger molecules. These obstacles can be partially overcome by the use of shorter laser pulses, as was the case for the work considered here. A better solution in the future will be to use a longer wavelength laser (>800 nm) with a short pulse duration (<10 fs), which extends the cut-off harmonic at a given intensity to higher energies due to the λ2 scaling of the ponderomotive energy with the laser wavelength.The mathematical steps in structure retrieval from HHG are based upon the strong field approximation (SFA), i.e. both the influence of the laser field upon the bound state and the influence of the Coulomb potential upon the continuum electron states are neglected. The SFA lacks translational and gauge invariance41 and the limitations derived from this problem are expected to be more significant for extended molecular systems. Even in those cases where a small molecule was addressed, the assumption that the wave function can be properly described by a single electron orbital has needed to be modified.42 It is now clear that what is retrieved via tomographic reconstruction is inevitably a signature of a multi-electron wave function. It is suspected that multi-electron processes driven by the laser field may become more significant with increasing molecular size as, for example, the polarization of the ionic core by the laser field will modify the field ionization.43,44
The first studies of HHG from aligned molecules were restricted to simple diatomic and triatomic linear systems. We recently looked at the HHG from significantly more complex aligned molecules, namely the conjugated organic molecules acetylene (HC![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CH), and allene (H2C
CH), and allene (H2C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CCH2).45
CCH2).45
The experiment was performed using the 10 Hz ASTRA Laser Facility at the Rutherford Appleton Laboratory, UK. This system provided laser pulses of 14 fs duration and ∼0.5 mJ at 800 nm after a hollow fibre pulse compressor. Approximately 80% of the beam energy was split off before the hollow fibre and sent through a delay line in order to produce a pulse to induce non-adiabatic alignment, correctly synchronized with the harmonic generating field. The beams were recombined with a normal incidence beam splitter and were focused by a 40 cm focal length off-axis parabolic mirror into a pulsed gas jet. The alignment beam had an energy of 200 μJ and a pulse duration of 60 fs, giving an estimated on target intensity of 3 × 1013W cm−2, which was insufficient to cause any detectable HHG. The harmonic generating beam had an on target intensity of ∼1.5 × 1014W cm−2. The short duration of the harmonic generating pulse is very important for accurate structural retrieval since it prevents a significant realignment of the molecules at the time of harmonic emission and avoids saturation of the ionization.
The stagnation pressure of the gases was 2 bar, and the nozzle diameter was 100 μm. The temperature of the gas in the interaction region was estimated to be less that 100 K, low enough to ensure a significant degree of molecular alignment. A halfwave plate in the aligning beam controlled the angle between the aligning and harmonic generating (driving) fields. The harmonics produced in the gas jet were spectrally dispersed by a flat field spectrometer and detected with a microchannel plate detector fitted with a phosphor screen. A CCD camera was used to image the phosphor screen. Harmonics from the 17th to the 27th order were detected in acetylene and allene.
The harmonic spectra were recorded for different time delays between the aligning and driving pulses while maintaining their polarizations parallel. Spectra were also recorded in the absence of an aligning field under otherwise identical conditions. When correctly calibrated, these spectra were used to extract the signal ratio between the aligned and isotropic samples.
The measured spectra were simulated using the approach described by Lewenstein et al.8 following the assumptions of the single active electron and strong field approximations. This goes beyond the simplified factorized expression (eqn (4)) by considering systematically the contributions from all viable electron trajectories that may contribute. The HOMO orbitals were calculated as a superposition of gaussian like functions with the GAMESS-UK package46 using the 321-G basis set. Keeping the electric field in the z direction, the different orientations of the molecules were introduced by applying the rotation matrix, as defined in ref. 47, on the spatial coordinates of the orbitals.
The angular dependence of the harmonic yield from acetylene and allene was measured for delays between the aligning and driving pulses corresponding to the maximum molecular alignment (along the laser field) or maximum anti-alignment (located preferentially in the plane perpendicular to the aligning field) in the rotational revival. The harmonic signal was then recorded as a function of the angle between the aligning and probe polarization directions. The results shown in Fig. 5 are the ratio of the measured signal for each angle to that obtained under otherwise identical conditions in a randomly aligned sample (i.e. with the alignment pulse blocked).
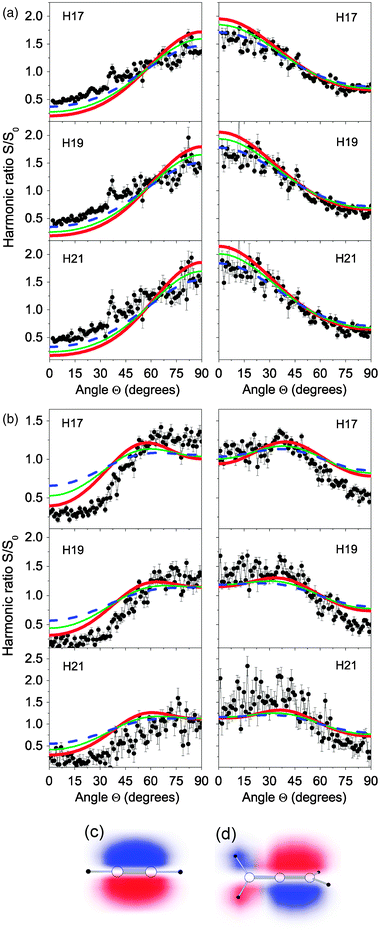 | ||
| Fig. 5 (a) Harmonic ratio between aligned and isotropic samples of acetylene molecules as a function of the angle between the aligning and driving field polarizations measured at the time of maximum alignment (left column) and maximum anti-alignment (right column). The red thick solid, green thin solid and blue dashed curves show the calculated ratio in a sample at 20, 50 and 100 K, respectively. (b) Idem for allene. (c) Representation of one of the two degenerate orbitals comprising the HOMO of acetylene, different colours indicate different signs of the wavefunction. (d) Idem for allene. | ||
In acetylene and allene the HOMO is dominated by bonding πu orbitals between the carbon atoms (see Fig. 5), though the allene molecule has additional contributions to the HOMO from electron amplitude located at the H atoms on the end of the molecule. These wavefunction differences are reflected in the angular dependence of the HHG yield that is different for the two molecules. The theoretical simulations agree well with the observed angular dependence of the harmonic ratio both qualitatively and quantitatively. The calculated data for 20, 50 and 100 K are plotted in the figures. The modulation of the calculated signal with the sampling angle depends upon the degree of alignment in the molecular ensemble, as expected. In spite of the temperature uncertainty, the calculations reproduce the measured ratios and, more importantly, the subtle difference observed in allene that typifies its orbital structure.
Studying the dependence of the harmonic yield with the alignment angle during the aligned transient states (revivals) of acetylene and allene, we have observed a clear signature of their HOMO structures and found good agreement with our single active electron SFA calculation,45 which takes into account the distribution of molecular orientations in the aligned sample.
This result demonstrates that signatures of the electronic wavefunction can be found in the HHG signal in these aligned organic molecules and that the SFA provides a fairly robust means to model the measured data. Complete retrieval of the wavefunction would also be possible if a wider range of harmonic orders were measured (e.g. by using a longer wavelength laser as discussed above).
2.4 Unique features of HHG imaging and future challenges
The tomographic reconstruction of the HOMO orbital of N2 from the scans of HHG spectra for different alignment angles,24 and the observation of interference dips in harmonic spectra from CO2 molecules,27,28 are the first steps towards techniques with wider applicability. The full analysis of the reconstruction of structure by this pair of closely related techniques for these two simple molecules is still being tackled. For instance, the concept of tomographic reconstruction of a single electron wave function (the HOMO of N2) as determined in ref. 24, rather than the physically more appropriate multi-electron wavefunction of the real molecule has been examined. Recently a more sophisticated calculation (including exchange effects) has shown the reconstructed wave function to be still closer to the appropriate multi-electron wave function than single electron wavefunction of N2.42 At the time of writing, the reconstruction measurements have only been published for N2 but progress towards performing these for other molecules is being made. For two-center interference measurements it is not yet clear how general the two- or multi-center interference signatures will be, and whether their observation will be confined only to molecules where the wavefunction can be accurately described as a simple linear combination of atomic orbitals centered at the atomic locations.An important issue for both techniques, and for variants built upon this basic idea of using HHG for imaging, is the extent to which the SFA is an applicable theory for structural reconstruction. The great simplicity of SFA in reducing the HHG emission to a simple mapping of the angle dependence of the transition dipole moment is very attractive and powerful. SFA, however, neglects the Coulombic binding potential in the continuum states and the laser field is ignored in the bound states, therefore compromising gauge invariance and the accuracy of the dipole moment calculations. Alternative, more accurate, approaches lose the simple relationship between HHG emission and dipole matrix element and so do not lend themselves immediately to the retrieval of molecular structure. There is clearly much work still to be carried out in this area both by the theorist, for instance to examine what can be done to preserve the simple ideas offered by SFA in a more accurate theoretical framework,41,48 and by the experimenter to learn how to do these experiments in a way that reduces the reconstruction difficulties, for instance by ensuring that a large recollision momentum is achieved to improve the accuracy of the SFA.
We would argue that despite these difficulties there are considerable prospects to use these techniques as time resolved probes. Even if there are uncertainties in the retrieval, there are certain situations where large structural changes are happening quickly, accompanied perhaps by symmetry changes in the electronic wavefunction, and so a time resolved method (even if it is not exact) will provide enormous new insight in chemical physics. The incorporation of HHG probe techniques into a pump–probe scenario is envisaged. In fact two pumps—one to induce alignment and a second to investigate some photochemical change—are required. The recording of HHG spectra at known molecular axis alignment and with variable delay with respect to the second probe should suffice to provide a new class of ultra-fast measurements that can follow structural changes. Short laser pulses (of 10 fs or less) in the second pump and probe will be required to achieve high temporal resolution, but these are now becoming available over a wide range of wavelengths. The position in the spectrum of the interference dip minimum can be followed in time through such a measurement, and through this the temporal evolution of the internuclear separation will be determined.
3. Measuring molecular dynamics on a sub-cycle timescale
3.1 Chirp encoded electron recollision imaging
For many molecules the nuclei are massive (typically >104me) and undergo negligible motion on the timescale of the active electron ionization–recollision involved in HHG (∼1 fs). Nevertheless we must anticipate that the electrons remaining in the cation will undergo a variety of processes connected to the sudden removal of a valence electron and the presence of the laser field. We speculate below on whether we can gain insight into these very fast electronic dynamics from measuring the HHG emission.If the nuclei are protons their motion cannot be ignored during the HHG process. In this very important case, significant motion of the nuclei can occur in a few femtoseconds if the ionization process places the proton on a steep part of the ionic potential energy surface (as will often be the case). The motion of the protons will then significantly change the harmonic emission.49 We will describe how the frequency–time encoding of the harmonic spectrum can be used to measure the ultra-fast rearrangement of protons in a molecule in the few femtoseconds following the event of ionization in a strong field. This is especially interesting as it is not possible to see this very fast motion by any conventional means.
In the cases of molecular structure determination using HHG that were considered in section 2, it can be reasonably assumed that the atomic nuclei were too massive to move appreciably during the electron return time. That said, recent work has shown that if a pump probe configuration is employed (in which the first pulse creates vibrational modes in a spherical molecule such as SF6), then the harmonic signal generated by the probe will modulate as a function of delay time. It has been noted that the Fourier transform of this modulation includes signatures of all the active Raman modes, indicating that the HHG signal is sensitive to the small changes in internuclear separation (∼0.01 Å) caused by the vibrational motion.50 This is a new and highly sensitive technique, for detection of vibrational excitation in molecules.
The intrinsic chirp of HHG becomes of vital importance in experiments aiming to study attosecond dynamics. In work pushing towards making measurements using attosecond duration XUV pulses the chirp usually needs to be corrected13,16 if the pulses are to have the minimum possible duration. In contrast, if the harmonics directly emitted from a molecule are observed then the chirp of the electron wave packet (if the time to energy mapping is well known) can be utilized to provide exquisite temporal resolution. For each of the short trajectories the electron returns to the parent ion at a different delay time Δt, and each is associated with a different electron kinetic energy at the point of recollision (see Fig. 6 and section 1.2). This temporal spread leads to a frequency-chirped harmonic emission, with successively higher harmonics being generated at longer time delays, as directly measured by Mairesse et al.10 The long trajectories can contribute to the HHG spectrum, but in the experiments discussed here, these long trajectories were efficiently filtered out from the spectrum.11 This left us with a reliable one-to-one mapping between delay and photon frequency (Fig. 6).
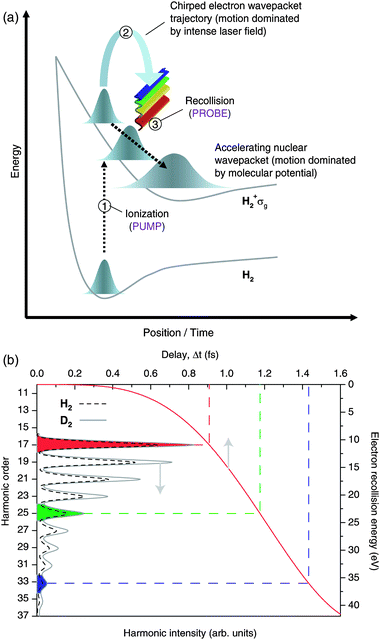 | ||
| Fig. 6 The PACER technique. (a) Ionization serves as the pump process because it launches an electron wavepacket into the continuum simultaneously with a nuclear wavepacket on the H2+ ground state potential surface (σg). The electron wavepacket then moves in response to the laser field, returning to the parent ion with an increased kinetic energy at some later time. The recollision acts as the probe of the nuclear motion that has occurred in the time delay since ionization occurred. (b) Due to the chirped nature of the returning electron wavepacket, different harmonic orders are emitted at different times after ionization, and are therefore associated with different pump–probe time delays. For example, the emission of the 17th harmonic probes the nuclear wavepacket 0.9 fs after ionisation, whereas the 33rd harmonic probes the nuclear wavepacket at a time just > 1.4.fs. | ||
The chirp of the spectrum from HHG is fundamental to the technique discussed in this section, because it allows a range of pump–probe delays to be accessed by analysis of the harmonics from a single laser pulse. Simply recording the harmonic spectrum therefore allows us to probe the internal changes that have taken place in the molecular cation over a range of pump–probe time delays, which are set by the temporal spread of the recolliding electron wavepacket. The technique has been termed PACER (probing attosecond dynamics with chirp encoded recollisions) and appears especially suited to the measurement of very fast proton rearrangement in molecules following ionization.
It is useful to make contact with a key idea of ultra-fast measurements i.e. the pump–probe experiment. In our work the ionization step can be thought of as the “pump” since, in the case of a molecule, a nuclear wavepacket is simultaneously launched at the moment of ionization, with the nuclear state making an instantaneous transition to the ground state potential of the molecular ion. The “probe” is the recollision of the electron wavepacket with the parent ion. This is in common with the earlier technique using correlated electron and nuclear wavepackets of Niikura et al.51,52 In contrast, the earlier technique used recollision induced ionization, rather than recombination followed by emission of radiation, for the probe signal, and requires a variation of laser wavelength over a significant range in order to obtain the required pump–probe delays. Our measurements inherently harness the chirp in HHG emission, so we can simultaneously measure a range of delays simply by observing the harmonic spectrum.
3.2 Investigation of chirp encoded recollision for tracking proton motion in H2 and D2
The first step of the HHG process (ionization of the molecule) simultaneously launches an electronic wavepacket into the continuum and a nuclear wavepacket onto the ground electronic potential energy surface of the molecular ion.53 At the moment of recollision of the electron wavepacket with the parent ion the probability that recombination occurs, and therefore the strength of the harmonic signal emitted, is related to the quantum mechanical overlap between the wavefunctions of the electron wavepacket and the molecular ground state. Lein predicted49 that the harmonic signal will be weaker from a molecule whose nuclei are lighter, and so move quickly, compared to that from a molecule with heavier nuclei, with therefore slower nuclear motion, since the overlap of the wavefunctions decreases as the internuclear separation increases. In addition, since successive orders of harmonics are generated at later times (if short electron trajectories are isolated as was the case in our experiments), the ratio of the harmonic signal, for instance, between D2 and H2, should increase as the harmonic order increases. The “rate” of increase of this ratio with harmonic order then yields information concerning the differing nuclear motion in the two species, and thus represents a measurement of the nuclear motion on an attosecond timescale. In our work we have studied HHG in gaseous H2 and D2 to confirm this effect, detecting a clear signature of the nuclear motion that occurs during the time interval between the ionization and recombination steps.Within the strong field approximation the strength of the harmonic signal emitted on return of the electron wave packet is given by the recombination dipole moment. For a molecule, the recombination dipole moment must be generalized to account for both the nuclear as well as electronic parts of the wavefunction. If there is significant motion of the nuclei in the short time window since ionization of the molecule occurred, then the amplitude for harmonic emission becomes:49
 | (5) |
The strength of harmonic emission is proportional to ω2|a(k)v(k)|2,24 and thus depends on the overlap of the nuclear part of the wavefunction at the times of ionization and recombination. This can be described by a nuclear correlation function
 | (6) |
The strength of the harmonic signal at different frequencies is also dependent on the momentum vs. time distribution of the returning wavepacket a(k). This has contributions from both the ionization step, and the subsequent acceleration of the wavepacket in the laser field. In PACER, the effect of the nuclear motion is isolated from any effects of the electronic parts of eqn (5) by the comparison of harmonic emission in two isotopes of the same species. This procedure circumvents the need to determine a(k), or indeed to consider details of the electronic parts of the recombination dipole that are R independent. Neither deviations of the returning electron wavepacket from a superposition of plane waves nor distortion of the molecular electronic states in the strong laser field will have a large effect upon the result as these will be essentially the same for the two molecular isotopes. The PACER technique is described in detail in ref. 11, 49 and 53. In essence, it is simply necessary to compare high harmonic generation in protonated and deuterated samples of a molecule at the same laser intensity and gas density. As an example, harmonic signals in both H2 and D2 were recorded, and any difference in the spectra could be attributed largely to the different nuclear dynamics in the two species.
Care was taken in making these measurements to ensure that both gases were delivered to the interaction region at an equal density. Interferometric measurements of the electron density were employed to confirm this equality. This required the use of a very intense laser pulse to completely ionize the molecules at the center of the focus. An auxiliary laser beam introduced as a probe from the side was incorporated in one arm of an interferometer, so that the instantaneous axial electron density could be measured by Abel inversion of the resulting interferograms. The molecular number densities measured agreed with gas flow calculations to within experimental error.
Fig. 7 shows the experimental data obtained using 8 fs pump pulses54 centered at roughly 775 nm, and focused to an intensity of 2.4 × 1014W cm−2.11 An increase in the ratio of harmonic signals is clearly observed as the harmonic order (and therefore electron return time) increases. This measurement is made over a time window 0.9–1.6 fs after the ionization event, at a temporal resolution of roughly 100 as. We compare our experimental results to calculations based on eqn (5), using Born–Oppenheimer potentials for the molecular ion. The blue line in Fig. 7a shows the ratio |cD2(τ)/cH2(τ)|2 for a randomly aligned sample. This curve has been scaled to account for the slight difference in photoionization cross sections of the two gases.55 The agreement with our experimental results is very good.
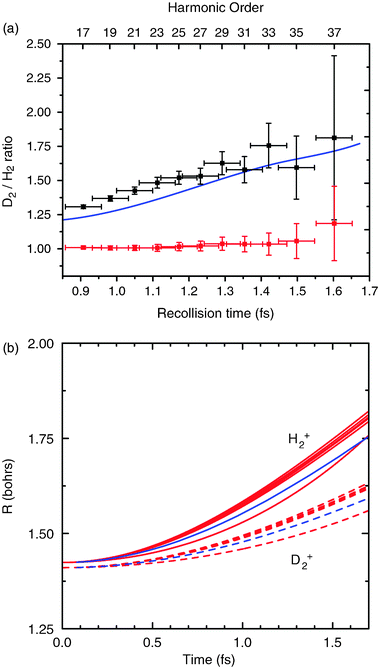 | ||
| Fig. 7 (a) Measured ratio of harmonic signals in D2 and H2 (black points). The red points show the ratio of harmonic signals in the same gas on two independent data runs, being constant at a value of 1. Blue line: calculated ratio, for a randomly aligned sample, including two-center interference. (b) Nuclear dynamics retrieved from experimental data by use of a genetic algorithm (red curves). The blue curves show the result calculated using the exact BO potentials for H2+ and D2+. | ||
The influence of the field on the dynamics of the ion has been investigated by including a static Stark shift (corresponding to the peak value of the electric field) on the BO potential.53 This is found to have an insignificant effect and was therefore neglected in subsequent calculations. Test calculations of the propagation of the nuclear wavepacket on the excited σu potential were also performed. We find that if significant excitation to the σu potential occurred, the increase in the measured ratio would be much more severe as the nuclei move apart more quickly,53 leading to very high ratios at the times accessed in our experiment. The lack of ratios greater than ∼2 in our experimental results therefore implies that we do not detect significant harmonic emission from ions populating the σu state (for any small part of the population that may be initially excited to the 2pσu potential, the small nuclear correlation function that results from the fast nuclear motion ensures that the contribution to the HHG at the times of interest is negligible). Transfer of population to the 2pσu potential is therefore neglected in the calculation shown in Fig. 7.
Calculations having both included, and neglected, two-center interference revealed that this effect is found to have a noticeable, but relatively small, contribution to the measurement.53 We estimate that using 8 fs pump pulses, the alignment of the sample is insignificant: but that for a well aligned sample, two-center interference is expected to have a larger effect. To investigate the latter effect we have performed a subsequent set of experiments where longer laser pulses were employed (30 fs) that was of sufficient intensity (3 × 1014W cm−2) to cause appreciable alignment within the molecular ensemble. Under these conditions it was found that inclusion of the two-center interference term was essential to match even qualitatively the measured ratio of harmonic emission between D2 and H2.
A genetic algorithm (GA) was used to reliably retrieve the internuclear separation vs. time information for both molecules (Fig. 7b). The GA generates the optimum ionic BO potential by calculating the wavepacket motion, and minimizing the sum of squared deviations of the correlation ratio from the measured result.11 The initial nuclear wavepackets are assumed to be identical to the vibrational ground state wavefunctions in each molecule.
The outcome of the retrieval from the data for H2 and D2 reveals the potential power of the PACER technique. The expectation value of the time dependent internuclear separation 〈R(t)〉 has been determined to a spatial resolution of ±10−11 m and temporal resolution ±100 attoseconds with an accuracy approaching these resolution values. Uncertainties in the calculated behavior in this molecule, e.g. in computing the electronic part of the dipole, are cancelled when the ratio is taken between D2 and H2 leaving only the dependence of the nuclear wavepacket motion. This is a significant advantage of the method. In addition, the method is simple to implement, since a single measurement yields all the pump–probe delays, and the correlated electronic and nuclear wavepackets are automatically synchronized, yielding the exceptional temporal resolution for relatively little experimental effort.
PACER currently has a number of limitations: for an 800 nm laser the temporal window is limited to the range ∼0.9–1.6 fs. To make the measurement, a deuterated as well as a protonated sample of the molecule is required. To get quantitative data we need to have some insight into the form of the potential energy surface (PES) so that calculations can be performed to compare with the measurements of the harmonic signal ratio or to carry out the full iterative retrieval procedure using a GA.
3.3 Extension to other molecular systems
Following the demonstration of the validity of the PACER technique for D2/H2 and the retrieval of nuclear dynamics that were in good agreement with the expected theoretical result in this molecule, we sought to apply the technique to other systems. In principle, any molecule where the dominant contribution to strong field ionization comes from an X–p bond in the molecule (where X is any other atom and p a proton) is a good candidate. However, given that the strong field ionization of many molecules remains to be worked out in detail, we decided to focus attention at first upon a system without any ambiguity as to which molecular electron was ionized. It was also required that the molecule be readily available as protonated and deuterated samples. Methane turned out to satisfy these experimental requirements well.A set of harmonic measurements in CD4 and CH4 were carried out under identical conditions, using our 8 fs pulse duration laser system. We observed behaviour consistent with our studies of D2 and H2: the harmonic yield was found to be greater in the heavier isotope, whose nuclei are expected to move more slowly, and this effect was found to be enhanced for the higher order harmonics, which probe the parent molecule at a longer time delay. The differing photoabsorption cross sections in CH4 and CD4,56 although making a small contribution to the measured ratio, cannot account for the increase in the ratio that we observe. The measured ratio of the CD4 to CH4 harmonic yield (Fig. 8a) shows a rapid rise to values >2 within 1.2 fs of the ionization. This implies a rapid rearrangement of the protons in the cation. We note that the technique is sensitive to bond angle changes as well as motion along the internuclear axis.
 | ||
| Fig. 8 Probing structural rearrangement in CH4 and CD4. (a). Ratio of harmonic signals in CD4 and CH4 (black). The error represents SE over 200 laser shots. Also shown is the control ratio of two harmonic spectra from CD4 taken separately (red). (b) Known structures of CH4 and CH4+ at equilibrium. Upon removal of an electron, it is anticipated that CH4 will rapidly evolve toward the CH4+ structure shown. | ||
It is known from theory57 and experiment58 that although the CH4 molecule has the well-known tetrahedral structure (with 109.5°-bond angles), CH4+ adopts a C2vgroup geometry, with some bond angles diminishing to <60° (Fig. 8b). It is anticipated that these structural rearrangements at the moment of ionization must be fast, as the tetrahedral structure of methane is far from the equilibrium bond angles of the ion. Our measurements provide direct evidence that the time scale for the onset of this structural rearrangement is on the order of a few femtoseconds. These results therefore confirm that this technique is not limited to probing nuclear wavepackets in diatomic molecules.
The measured ratio (Fig. 8a) is the square of the ratio of the nuclear autocorrelation functions for the two species, a quantity that can be calculated directly from the molecular potentials. Therefore, the measurement could be used to test the correctness of computed potentials. In the case of methane, the computation of the potential energy surface is complicated by the Jahn–Teller effect which, due to the degeneracy of several states of the cation, has a dominant role in this molecule.58 Therefore at this stage we cannot carry out the full program of analysis to compare with our experimental results, but hope to stimulate the interest of the theoretical quantum chemistry community to carry out these calculations.
PACER accesses nuclear dynamics far from any equilibrium state, in the first few femtoseconds after ionization, at a remarkable temporal resolution. The shape of potential energy surfaces far from equilibrium cannot be accessed by spectroscopic techniques: PACER may therefore also find application in testing the accuracy of computed potentials far from equilibrium. It is also important to note that the probe signal (harmonic emission) is sensitive to motion of the nuclei in any direction. Calculations of the dynamics of these systems are not currently available, and thus the direct measurement of these dynamics is an exciting possibility.
3.3 Extensions of chirp encoded recollision
There are many possible extensions to the work already reported that will further the usefulness of the PACER technique. For instance, the time window over which information can be gained could be extended by employing a driving laser of a longer wavelength. Work towards this goal has recently begun but it is clear from preliminary results that not only longer wavelength lasers but very short pulses are important for successful realization of this scheme. This is because the laser intensity must be high enough to ensure the ionization mechanism leads predominately to vertical transitions onto the cation PES, but that no significant alignment or fragmentation of the molecule occurs during the pulse rise time.The need for a comparison between isotopes may also be eliminated by calibration of the amplitudes of the momentum components of the electron wavepacket and the momentum dependence of the dipole. There are various possibilities to achieve this, all require a method to change the energy–time mapping in a controllable way so that the temporal dependence of the dipole (eqn (5)) can be retrieved from the measured spectrum. The most straightforward method would be to perform the same measurements over a range of laser intensities and then to perform systematic comparisons of the normalized spectra. The change in intensity varies the cut-off harmonic energy and the intrinsic chirp and so a given harmonic will be emitted at a different time if the intensity is changed (e.g. earlier if the intensity is increased). In principle, this provides a means to unravel the electron energy (momentum) dependence of the emission amplitude from any temporal dependence due to changes occurring within the ionic core. Alternatively, various techniques to use an additional external field to alter the recollision times and energy might be employed to vary the harmonic emission times systematically and so achieve a self-referenced measurement without recourse to compare different molecular species. This would allow the method to be extended to cases where a deuterated sample is not readily available (or prohibitively expensive).
By adoption of these alternative referencing methods it may also be possible to directly monitor electronic motion in bound states using the technique. It would, however, be very important to first cross check this method for appropriate molecules with the isotope comparison technique. This is because the comparison method was very effective in eliminating uncertainties in the electronic part of the dipole. If, instead, we are trying to measure the electronic part of the dipole (or at least its time dependence) we will need to carefully determine the extent to which the laser field alters the amplitude (e.g. by polarization of the ionic core in a time dependent fashion). Providing the modified technique has the property of self-referencing, i.e. the reference spectrum and the measurement for the same harmonic energy are different only as far as a change in recollision time, then reliable measurement of internal electron dynamics will be possible up to the limit where the laser field itself is disturbing those dynamics.
PACER therefore has an important role within the range of techniques for imaging very fast structural rearrangements that are currently available. We expect that the remarkable temporal resolution obtainable, together with the applicability of the method to polyatomic molecules containing protons, will place this technique as an effective tool in furthering the study of structural rearrangement in molecules.
4. Summary and conclusions
We hope that we have succeeded in offering some insight into the methods of molecular dynamic imaging using high order harmonic generation. Given the limited space it was not possible to offer an exhaustive account of either the theoretical or the experimental methods. For the former the reader is recommended to consult,1 and for the latter the reader should study the various research papers published that describe individual experiments. Instead we have tried to illuminate for readers new to the field the physical ideas at the root of the techniques and to provide an outline of the main features of the methods.The essence of structural imaging using HHG is that the intense laser field creates a recolliding continuum electron wave packet with a well defined direction and momenta that spans a broad range. Moreover, the wave packet momentum components are chirped. These features make possible both the structural retrieval described in section 2 and the extreme temporal resolution utilized in PACER and presented in section 3. It is the recolliding electron that probes the molecule through the sensitivity of the recombination amplitude to momentum and, in the case of moving nuclei, recollision time. The information is then read out through measurement of the harmonic radiation. An attraction of the methods is the experimental simplicity and the good signal to noise attainable in the measurement of HHG.
So far there have been some exciting demonstrations of the potential of these techniques,1,24,27,28,50 albeit confined to very small molecules. Doubtless the next few years will see some refinement and extension of these techniques. Some obvious areas of future development are to explore the application to significantly larger molecular systems. This will present experimental difficulties, e.g. achieving high density rotationally cooled gas phase samples and inducing efficient field-free alignment. Steps towards this were described in section 2.3. There are also some challenges to transpose the simple SFA picture to more extended molecular systems that have begun to be explored for small molecules41,48 but which will present even greater difficulties when applying these ideas to significantly larger molecules.
An obvious question is the extent to which the strong laser field distorts the structure or dynamics that we wish to measure. There is evidence from experiment11,24,45 that, for small molecules at least, the distortions due to the field are relatively small. This conclusion is backed up by recent treatments.48,53 We would not expect this to be generally true for larger molecules, where the potential difference due to the laser field across the length of the molecule will be substantial (2.7 eV Å−1 at 1014W cm−2) and so can cause significant modification to the states of the bound electrons. The assessment of the importance of the laser field distortion is a high priority for current research. Fortunately, the interest for PACER is in molecules close to the equilibrium separation, where the distortion of the molecular potential energy surfaces in a compact molecule (e.g. H2 or CH4) is rather small11 and so the effect of the field distortion of the potential surfaces on the nuclear motion is small.
Despite the many remaining challenges there is a real prospect not only that molecular imaging via HHG will become more widely applicable to the study of the structure and dynamics of small molecules, but that it may yet be useful for more complex systems. We anticipate a substantial impact upon chemical physics if that proves the case.
Acknowledgements
We are most grateful to the following people for many useful discussions: Manfred Lein, Ciprian Chirila, Luke Chipperfield, Jonathan Underwood, Misha Ivanov, Paul Corkum, David Villeneuve, Raffaele Velotta and Carlo Altucci. We are pleased to acknowledge funding from the following sources: EPSRC, RCUK and CLF (CCLRC).References
- M. Lein, J. Phys. B, 2007, 40, R135–R173 CrossRef CAS.
- M. Protopapas, C. H. Keitel and P. L. Knight, Phys. Rep., 1997, 60, 389 CrossRef.
- P. B. Corkum, Phys. Rev. Lett., 1993, 71, 1994–1997 CrossRef CAS.
- J. L. Krause, K. J. Schafer and K. C. Kulander, Phys. Rev. Lett., 1992, 68, 3535 CrossRef CAS.
- M. V. Ammosov, N. B. Delone and V. P. Krainov, Sov. Phys. JETP, 1986, 64, 1191 Search PubMed.
- X. M. Tong, Z. X. Zhao and C. D. Lin, Phys. Rev. A, 2002, 66, 033402 CrossRef.
- M. Yu. Ivanov, M. Spanner and O. Smirnova, J. Mod. Opt., 2005, 52, 165 CrossRef.
- M. Lewenstein, P. Balcou, M. Yu. Ivanov, A. L’Huillier and P. B. Corkum, Phys. Rev. A, 1994, 49, 2117 CrossRef.
- G. G. Paulus, W. Becker, W. Nicklich and H. Walther, J. Phys. B, 1994, 27, L703 CrossRef CAS.
- Y. Mairesse, A. de Bohan, L. J. Frasinski, H. Merdji, L. C. Dinu, P. Monchicourt, P. Breger, M. Kovacev, R. Taieb, B. Carre, H. G. Muller, P. Agostini and P. Salieres, Science, 2003, 302, 1540–1543 CrossRef CAS.
- S. Baker, J. S. Robinson, C. A. Haworth, H. Teng, R. A. Smith, C. C. Chirila, M. Lein, J. W. G. Tisch and J. P. Marangos, Science, 2006, 312, 424 CrossRef CAS.
- P. Antoine, A. L’Huillier and M. Lewenstein, Phys. Rev. Lett., 1996, 77, 1234–1237 CrossRef CAS.
- J. Mauritsson, P. Johnsson, E. Gustafsson, A. L’Huillier, K. J. Schafer and M. B. Gaarde, Phys. Rev. Lett., 2006, 97, 013001 CrossRef CAS.
- M. Drescher, M. Hentschel, R. Kienberger, G. Tempea, C. Spielmann, G. A. Reider, P. B. Corkum and F. Krausz, Science, 2001, 291, 1923 CrossRef CAS.
- R. Kienberger, E. Goulielmakis, M. Uiberaker, A. Baltuska, V. Yakovlev, F. Bammer, A. Scrinzi, T. Westerwalbesloh, U. Kleineberg, U. Heinzmann, M. Drescher and F. Krausz, Nature, 2004, 427, 817–821 CrossRef CAS.
- G. Sansone, E. Benedetti, F. Calegari, C. Vozzi, L. Avaldi, R. Flammini, L. Poletto, P. Villoresi, C. Altucci, R. Velotta, S. Stagira, S. De Silvestri and M. Nisoli, Science, 2006, 314, 443 CrossRef CAS.
- T. Zuo, A. D. Bandrauk and P. B. Corkum, Chem. Phys. Lett., 1996, 259, 313 CrossRef CAS.
- M. Lein, J. P. Marangos and P. L. Knight, Phys. Rev. A, 2002, 66, 051404 CrossRef.
- M. Spanner, O. Smirnova, P. B. Corkum and M. Yu. Ivanov, J. Phys. B, 2004, 37, L243 CrossRef CAS.
- S. N. Yurchenko, S. Patchkovskii, I. V. Litvinyuk, P. B. Corkum and G. L. Yudin, Phys. Rev. Lett., 2004, 93, 223003 CrossRef CAS.
- S. X. Hu and L. A. Collins, Phys. Rev. Lett., 2005, 94, 073004 CrossRef CAS.
- D. Comtois, H.-C. Bandulet, D. Pavicic, M. Meckel, A. Staudte, D. Zeidler, R. Dorner, H. Pepin, J.-C. Kieffer, D. M. Villeneuve and P. B. Corkum, Electron momentum distributions from strong field ionization of oxygen: dependence on molecular alignment, Advanced Laser Light Source Annual Report 2005–2006, INRS, Canada, p. 18, http://www.emt.inrs.ca/Francais/ALLS_Annual_Report_2005-2006.pdf Search PubMed.
- R. Velotta, N. Hay, M. B. Mason, M. Castillejo and J. P. Marangos, Phys. Rev. Lett., 2001, 87, 183901 CrossRef.
- J. Itatani, J. Levesque, D. Zeidler, H. Niikura, H. Pepin, J. C. Kieffer, P. B. Corkum and D. M. Villeneuve, Nature, 2004, 432, 867 CrossRef CAS.
- M. Lein, N. Hay, R. Velotta, J. P. Marangos and P. L. Knight, Phys. Rev. Lett., 2002, 88, 183903 CrossRef CAS.
- M. Lein, N. Hay, R. Velotta, J. P. Marangos and P. L. Knight, Phys. Rev. A, 2002, 66, 023805 CrossRef.
- T. Kanai, S. Minemoto and H. Sakai, Nature, 2005, 435, 470 CrossRef CAS.
- C. Vozzi, F. Calegari, E. Benedetti, J. P. Caumes, G. Sansone, S. Stagira, M. Nisoli, R. Torres, E. Heesel, N. Kajumba, J. P. Marangos, C. Altucci and R. Velotta, Phys. Rev. Lett., 2005, 95, 153902 CrossRef CAS.
- H. T. Yu and A. D. Bandrauk, Phys. Rev. A, 1999, 59, 539 CrossRef.
- D. G. Lappas and J. P. Marangos, J. Phys. B, 2000, 33, 4679 CrossRef CAS.
- R. de Nalda, E. Heesel, M. Lein, N. Hay, R. Velotta, E. Springate, M. Castillejo and J. P. Marangos, Phys. Rev. A, 2004, 69, 031804R.
- M. Lein, R. de Nalda, E. Heesel, N. Hay, E. Springate, R. Velotta, M. Castillejo, P. L. Knight and J. P. Marangos, J. Mod. Opt., 2005, 52, 465 CrossRef CAS.
- H. Stapelfeldt and T. Seidemann, Rev. Mod. Phys., 2003, 75, 543 CrossRef CAS.
- M. Kaku, K. Masuda and K. Miyazaki, Jpn. J. Appl. Phys., 2004, 43, L591 CrossRef CAS.
- J. Itatani, D. Zeidler, J. Levesque, M. Spanner, D. M. Villeneuve and P. B. Corkum, Phys. Rev. Lett., 2005, 94, 123902 CrossRef.
- J. Rosca-Pruna and M. J. J. Vrakking, J. Chem. Phys., 2002, 116, 6567 CrossRef.
- R. Torres, R. de Nalda and J. P. Marangos, Phys. Rev. A, 2005, 72, 023420 CrossRef.
- N. Hay, R. de Nalda, T. Halfmann, K. J. Mendham, M. B. Mason, M. Castillejo and J. P. Marangos, Phys. Rev. A, 2000, 62, 041803R.
- N. Hay, M. Castillejo, R. de Nalda, E. Springate, K. J. Mendham and J. P. Marangos, Phys. Rev. A, 2000, 61, 053810 CrossRef.
- C. Altucci, R. Velotta, E. Heesel, E. Springate, J. P. Marangos, C. Vozzi, E. Benedetti, F. Calegari, G. Sansone, S. Stagira, M. Nisoli and V. Tosa, Phys. Rev. A, 2006, 73, 043411 CrossRef.
- C. C. Chirila and M. Lein, Phys. Rev. A, 2006, 73, 023410 CrossRef.
- S. Patchovskii, Z. X. Zhao, T. Brabec and D. M. Villeneuve, Phys. Rev. Lett., 2006, 97, 123003 CrossRef.
- M. Smits, C. A. de Lange, A. Stolow and D. M. Rayner, Phys. Rev. Lett., 2004, 93, 203402 CrossRef.
- M. Smits, C. A. de Lange, A. Stolow and D. M. Rayner, Phys. Rev. Lett., 2004, 93, 213003 CrossRef.
- R. Torres, N. Kajumba, J. G. Underwood, J. S. Robinson, S. Baker, J. W. G. Tisch, R. de Nalda, W. A. Bryan, R. Velotta, C. Altucci, I. C. E. Turcu and J. P. Marangos, Phys. Rev. Lett., 2007, 98, 203007 CrossRef CAS.
- M. W. Schmidt, K. K. Baldridge, J. A. Boatz, S. T. Elbert, M. S. Gordon, J. H. Jensen, S. Koseki, N. Matsunaga, K. A. Nguyen, S. J. Su, T. L. Windus, M. Dupuis and J. A. Montgomery, J. Comput. Chem., 1993, 14, 1347 CrossRef CAS.
- R. N. Zare, Angular momentum: understanding spatial aspects in chemistry and physics, Wiley, New York, 1988 Search PubMed.
- O. Smirnova, M. Spanner and M. Yu. Ivanov, J. Mod. Opt., 2007, 54, 1019 CrossRef CAS.
- M. Lein, Phys. Rev. Lett., 2005, 94, 053004 CrossRef.
- N. L. Wagner, A. Wuest, I. P. Christov, T. Popmintchev, X. B. Zhou, M. M. Murnane and H. C. Kapteyn, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 13279 CrossRef CAS.
- H. Niikura, F. Legare, R. Hasbani, A. D. Bandrauk, M. Yu. Ivanov, D. M. Villeneuve and P. B. Corkum, Nature, 2002, 417, 917–922 CrossRef CAS.
- H. Niikura, F. Legare, R. Hasbani, M. Yu. Ivanov, D. M. Villeneuve and P. B. Corkum, Nature, 2003, 421, 826–829 CrossRef CAS.
- S. Baker, J. S. Robinson, C. A. Haworth, C. C. Chirila, M. Lein, J. W. G. Tisch and J. P. Marangos, J. Mod. Opt., 2007, 54, 1011 CrossRef CAS.
- J. S. Robinson, C. A. Haworth, H. Teng, R. A. Smith, J. P. Marangos and J. G. W. Tisch, Appl. Phys. B, 2006, 85, 525 CrossRef CAS.
- C. J. Latimer, K. F. Dunn, F. P. O’Neill, M. A. Macdonald and N. Kouchi, J. Chem. Phys., 1995, 102, 722 CrossRef CAS.
- R. E. Frey and E. R. Davidson, J. Chem. Phys., 1988, 88, 1775 CrossRef CAS.
- Z. Vager, E. P. Kanter, G. Both, P. J. Cooney, A. Faibis, W. Koenig, B. J. Zabransky and D. Zajfman, Phys. Rev. Lett., 1986, 57, 2793 CrossRef CAS.
- H. J. Worner, R. van der Veen and F. Merkt, Phys. Rev. Lett., 2006, 97, 173003 CrossRef CAS.
| This journal is © the Owner Societies 2008 |