“Imaging” combustion chemistry via multiplexed synchrotron-photoionization mass spectrometry
Craig A.
Taatjes
*a,
Nils
Hansen
a,
David L.
Osborn
a,
Katharina
Kohse-Höinghaus
b,
Terrill A.
Cool
c and
Phillip R.
Westmoreland
d
aCombustion Research Facility, Mail Stop 9055, Sandia National Laboratories, Livermore, CA 94551-0969, USA. E-mail: cataatj@sandia.gov; Fax: +1 925![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 2942276
2942276
bDepartment of Chemistry, Bielefeld University, Universitätsstraße 25, 33615, Bielefeld, Germany
cSchool of Applied and Engineering Physics, Cornell University, Ithaca, NY 14853, USA
dDepartment of Chemical Engineering, University of Massachusetts, Amherst, MA 01003-9303, USA
First published on 6th November 2007
Abstract
The combination of multiplexed mass spectrometry with photoionization by tunable-synchrotron radiation has proved to be a powerful tool to investigate elementary reaction kinetics and the chemistry of low-pressure flames. In both of these applications, multiple-mass detection and the ease of tunability of synchrotron radiation make it possible to acquire full sets of data as a function of mass, photon energy, and of the physical dimension of the system, e.g. distance from the burner or time after reaction initiation. The data are in essence an indirect image of the chemistry. The data can be quantitatively correlated and integrated along any of several dimensions to compare to traditional measurements such as time or distance profiles of individual chemical species, but it can also be directly interpreted in image form. This perspective offers an overview of flame chemistry and chemical kinetics measurements that combine tunable photoionization with multiple-mass detection, emphasizing the overall insight that can be gained from multidimensional data on these systems. The low-pressure flame apparatus is capable of providing isomer-resolved mass spectra of stable and radical species as a function of position in the flame. The overall chemical structure of the flames can be readily seen from images of the evolving mass spectrum as distance from the burner increases, with isomer-specific information given in images of the photoionization efficiency. Several flames are compared in this manner, with a focus on identification of global differences in fuel-decomposition and soot-formation pathways. Differences in the chemistry of flames of isomeric fuels can be discerned. The application of multiplexed synchrotron photoionization to elementary reaction kinetics permits identification of time-resolved isomeric composition in reacting systems. The power of this technique is illustrated by the separation of direct and dissociative ionization signals in the reaction of C2H5 with O2; by the resolution of isomeric products in reactions of the ethynyl (C2H) radical; and by preliminary observation of branching to methyl + propargyl products in the self-reaction of vinyl radicals. Finally, prospects for future research using multiplexed photoionization mass spectrometry are explored.
 Craig A. Taatjes Craig A. Taatjes | Craig A. Taatjes received a BSc in chemistry in 1985 from Calvin College and a PhD in chemical physics in 1991 from the University of Colorado, working under the supervision of Prof. Stephen R. Leone. After postdoctoral work at the Vrije Universiteit with Prof. Steven Stolte and at the FOM Institute for Atomic and Molecular Physics (AMOLF) with Prof. Aart W. Kleyn, in 1994 he joined the Combustion Research Facility of Sandia National Laboratories. His research interests include fundamental flame chemistry and the kinetics of elementary reactions that are important in combustion. |
Introduction
The detailed chemistry of combustion involves hundreds or thousands of individual chemical reactions, taking place at temperatures that range from room temperature to thousands of kelvins. Fortunately, many of these reactions are closely linked to one another, and many reactants are in partial equilibrium or quasi-steady state, substantially simplifying the picture in many cases. Furthermore, for simple technical applications, details of the chemistry are nearly immaterial; internal combustion engines were developed in the 19th century without even the fundamental knowledge of radical chain chemistry that would be developed years later. However, two areas that present some of the greatest technical challenges in modern combustion research, pollutant formation and autoignition, are intimately connected to chemistry.Dependence of pollutant emission on the nature of the fuel, for example from internal combustion engines,1–5 is well known. Pollutants are minor constituents of the overall combustion process, and the amount and toxicity of the unwanted emissions can depend on details of the combustion chemistry, as well as on physical and fluid-mechanical processes in the combustor. A particularly stubborn problem is the formation and growth of soot in hydrocarbon flames. The initiation of soot formation occurs through reactions of small, unsaturated organic radicals, especially resonantly stabilized radicals, and depends on the isomeric nature of the reaction products.6 The best-known and most important example is the recombination of propargyl (CH2![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CC˙H ↔ C˙H2–C
CC˙H ↔ C˙H2–C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CH) radicals,7 branching to form either aromatic soot precursors like phenyl and benzene or relatively innocuous linear C6H6 isomers.
CH) radicals,7 branching to form either aromatic soot precursors like phenyl and benzene or relatively innocuous linear C6H6 isomers.
Ignition chemistry has become of greater interest with the emergence of advanced engine technologies that rely on compression ignition of a premixed fuel–air charge. The ignition timing in such devices depends on the detailed fuel chemistry. For example, the oxidation reactions of alkyl radicals, or of other initial radical products of fuel decomposition, govern early heat release via“cool flame” chemistry and can substantially influence ignition times. Chain branching and propagation in these systems depend sensitively on the structure of the fuel molecule.8–11
The investigation of the chemistry of low-pressure, laminar premixed flames has long been a means of validating combustion mechanisms. The method of molecular-beam mass spectrometry (MBMS) has become a well-established tool for these studies since its first quantitative applications to flame chemistry in the 1960s and 1970s.12–18 In the past several years, MBMS machines utilizing photoionization and time-of-flight detection have introduced the possibility of detailed isomeric discrimination and multiplexed species detection.19 Most recently photoionization MBMS has been combined with tunable, bright vacuum-ultraviolet beams from synchrotron light sources, first at the Advanced Light Source (ALS) in Lawrence Berkeley National Laboratory,20,21 and later also at the National Synchrotron Radiation Laboratory in Hefei, China.22–25 These instruments provide unparalleled data throughput and isomeric resolution and have been used to identify many new isomeric species in flames, including enols in a series of hydrocarbon flames,26 C5Hn isomers in fuel-rich flames,27 triplet propargylene in a cyclopentene flame,28 and HONO in NO2-doped H2–O2 flames.29
The high throughput of the instrument at the Advanced Light Source permits data for a full characterization of a flame to be taken in the course of a single day, with mass spectra taken as a function of distance from the burner for several photon energies and as a function of photon energy at several distances from the burner. In the first years of operation of the ALS flame apparatus, more than forty different flames of two dozen fuels have been evaluated in this manner. Detailed reports of species mole-fraction profiles and comparisons to flame models are appearing in an ongoing series of publications;26,30–39 the present report highlights the information on global reaction pathways that can be gained from qualitative comparisons of the overall mass-spectrum profiles in selected flames.
More recently, a similar approach has been established to study elementary reaction kinetics and product branching with multiplexed mass spectrometry and synchrotron photoionization.40–42 In these experiments the photoionization mass-spectrometry technique developed by Gutman et al.43 is updated to include multiple-mass detection and tunable-synchrotron photoionization.
Data in both of these experiments are acquired as a function of three dimensions: mass, photoionization energy, and the natural physical dimension of the experiment (distance from the burner surface or time after photolysis). Furthermore, both experiments detect a wide range of masses simultaneously, giving a multiplex advantage over single-mass detection. Interpretation of this multidimensional data can draw inspiration from the example of ion imaging.44 The development of ion imaging in photodissociation45 and scattering46,47 dynamics has proved extraordinarily powerful not only because the simultaneous collection of data from all scattering angles is efficient, but also because the natural visualization of the processes inherent in the image data enables global features of the dynamics to be grasped almost at a glance. The purpose of the present article is to highlight the power of the multidimensional data available from multiplexed synchrotron photoionization mass-spectrometry experiments and to describe new means of visualizing combustion chemistry that these methods afford.
Experimental
The experiments described here use the Chemical Dynamics Beamline of the Advanced Light Source at Lawrence Berkeley National Laboratory. The 3-m monochromator at the Chemical Dynamics Beamline delivers about 5 × 1013photons s−1 with an energy resolution ΔE(fwhm) = 40 meV. The energy resolution is determined by measuring narrow autoionization features in Xe or O2. Higher-energy undulator harmonics are suppressed by a gas filter48 filled with Kr, Ar or He. A calibrated silicon photodiode records the photon current. Photon energies from about 7.4 eV up to the ionization threshold of the rare gas in the gas filter can be accessed under computer control.Low-pressure laminar premixed flames
The low-pressure flame apparatus, depicted in Fig. 1, is described in detail elsewhere.20 It consists of a low-pressure flame chamber, a differentially pumped flame-sampling system, and a time-of-flight mass spectrometer. A premixed laminar fuel–oxygen–Ar flame is stabilized on a sintered stainless-steel burner. Flame conditions for the flames described in this work are given in Table 1. Gases from the flame are sampled through a 200-μm orifice in a fixed quartz cone, mounted on a water-cooled flange. The burner can be moved relative to the cone in order to sample gases from different positions in the flame. The sampled gases form a molecular beam, which is skimmed and subsequently crosses the tunable-synchrotron beam in the ionization region of a pulsed-extraction time-of-flight mass spectrometer. | ||
| Fig. 1 Schematic picture of the molecular-beam sampling apparatus employing tunable-synchrotron photoionization and time-of-flight multiple-mass detection. A photograph of the combustion chamber is shown as the inset. The burner can be moved towards or away from the quartz sampling cone to probe different regions of the flame. | ||
| Fuel | Fuel/oxygen equivalence ratio, ϕ | Pressure P/Torra | Feed mole fraction of Ar | Mass flux/g m−2 s−1 |
|---|---|---|---|---|
| a 1 Torr = 133 Pa. | ||||
| Ethene | 1.90 | 30.0 | 0.440 | 20.7 |
| Ethanol | 1.96 | 35.0 | 0.317 | 32.4 |
| Dimethyl ether | 2.00 | 25.0 | 0.250 | 39.8 |
| Propyne | 1.00 | 25.0 | 0.444 | 21.4 |
| Allene | 1.00 | 25.0 | 0.444 | 21.4 |
| Methyl acetate | 1.82 | 30.0 | 0.256 | 45.6 |
| Ethyl formate | 1.82 | 30.0 | 0.256 | 45.6 |
| 1-Propanol | 1.94 | 35.0 | 0.322 | 33.0 |
| 2-Propanol | 1.94 | 35.0 | 0.322 | 33.0 |
| Cyclohexane | 2.00 | 30.0 | 0.316 | 29.4 |
| 1-Hexene | 2.00 | 30.0 | 0.316 | 29.4 |
Molecular-beam mass spectrometry has been applied to flames for decades, and the nature of the unavoidable perturbations of the flame structure have been extensively described. However, for appropriately designed sampling these perturbations can be made acceptably small, as reviewed thirty years ago by Biordi.14 The signal observed in the mass spectrometer at a given m/z is proportional to the mole fraction of the corresponding neutral species in the flame gases being sampled. The determination of mole fractions in the flame, which is the quantity of interest for comparison to models, requires only relative proportionality constants for different species at a particular position in the flame and for any given species as a function of position in the flame. A method for deriving the proportionality factor has been developed,30 which applies an elemental balance for the major species in the burned gases and uses the profile of the argon diluent, the mass fraction of which is approximately constant throughout the flame. Application of such a procedure is necessary for deriving quantitative mole-fraction profiles from molecular-beam spectrometry data.27,30,32
Mass spectra can be taken either as a function of burner position at a fixed photon energy (a “burner scan”), as shown in Fig. 2, or as a function of photon energy at a fixed burner position (an “energy scan”), as shown in Fig. 3. Binning the burner scan for each value of m/z yields the profiles of the ion signals at each mass as a function of the distance from the burner surface. After correction for isotopic contributions, fragmentation, and photoionization cross-sections, such profiles are used to generate mole-fraction profiles of individual species. The energy scan gives a relative photoionization-efficiency spectrum (i.e. ionization cross-section as a function of photon energy) for all species at that point in the flame, and is key to identifying the species responsible for the signal. Different molecules with nearly identical masses (e.g. CO and C2H4) can be distinguished based on their differing ionization energies. Isomeric species, which cannot be distinguished based on mass, exhibit photoionization-efficiency spectra that differ in onset energy and in shape. This fact forms the basis for isomeric discrimination in tunable photoionization mass spectrometry. Deconvolution of isomers of hydrocarbon radicals in flames has been accomplished by comparing measured spectra with computed Franck–Condon envelopes.27–28,32,39 Where absolute photoionization cross-sections are available,49,50 mole fractions of individual isomers can be determined.21,26,30,31,34,38
 | ||
| Fig. 2 Sections of mass spectra as a function of distance from the burner surface for a rich ethene flame, ϕ = 1.9, photon energy 10.5 eV (top); a rich ethanol flame, ϕ = 1.97, photon energy 10.5 eV (middle); and a rich dimethyl ether flame, ϕ = 2.0, photon energy 10.4 eV (bottom). The data for the individual flames are independently scaled to show best the fuel breakdown and molecular-weight growth processes; the large peaks corresponding to ionization of the fuel species are considerably saturated. | ||
 | ||
| Fig. 3 Photoionization-efficiency measurement of a stoichiometric (ϕ = 1) allene flame taken with the sampling cone held 1 mm from the burner surface. The data have been normalized by the photon current. | ||
The dependence of the photoionization efficiency on vibrational energy must be considered, as the sampling from the flame produces only mild vibrational relaxation.51 The effects of vibrational excitation are expected to be most pronounced near the ionization threshold and near the appearance thresholds for fragment ions.52 Nevertheless, direct measurements of photoionization efficiencies of vibrationally excited radicals well above the threshold show only a very weak dependence on internal energy,53 and photoionization efficiencies of known molecules sampled from the flame have exhibited a close agreement with 298 K measurements. The images in Fig. 2 are very instructive, because even from a visual inspection it is evident that ethene and the isomeric fuels ethanol and dimethyl ether exhibit strikingly different combustion chemistry under nearly identical combustion conditions.
Photoinitiated chemical kinetics
The apparatus for studying photolytically initiated reactions with tunable-synchrotron photoionization mass spectrometry54 (Fig. 4) is a adaptation of the design developed by Gutman and co-workers.43 Reactions are initiated in a slow-flow quartz reactor by uniform pulsed-laser photolysis of a suitable precursor. The contents of the reactor are continuously sampled through a small orifice in the side of the quartz tube. The molecular beam thus formed passes through an aperture into the ionization region of a miniature double-focusing (Mattauch–Herzog) mass spectrometer.55,56 Here the molecular beam crosses the tunable-synchrotron beam. The ions formed are energy-selected in an electrostatic sector and dispersed according to their mass-to-charge ratio in a 0.9 T magnet. At the exit of the magnet the ions are amplified by a microchannel plate and detected by a time- and position-sensitive delay-line detector.57,58 The inherent time response of the instrument is determined by the arrival-time broadening induced by the thermal spread in velocities in the emerging molecular beam59,60 and is on the order of 100 μs in the present apparatus. Masses in a range of approximately a factor of 6 (e.g. from m/z = 15 to m/z = 90) are detected simultaneously, and because of the continuous nature of the sector mass spectrometer, an entire time trace is collected for each photolysis pulse, giving a two-dimensional spectrum of the molecules emerging from the reactor as a function of their mass and the time relative to the photolysis. Stepping the photon energy through a desired range while accumulating these time/mass spectra builds up a three-dimensional data set, as depicted in Fig. 5.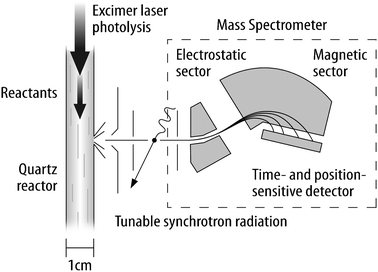 | ||
| Fig. 4 Schematic depiction (approximately to scale) of the multiplexed chemical-kinetics reactor that combines tunable-synchrotron photoionization with time-resolved multiple-mass detection. | ||
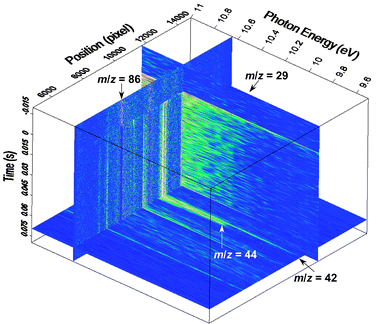 | ||
| Fig. 5 Three-dimensional dataset measured from the reaction of ethyl radicals, produced by 193 nm photolysis of 3-pentanone (diethyl ketone), with molecular oxygen. The signal has been normalized by the photon current and the pre-photolysis background has been subtracted. The position on the detector is directly, although not linearly, related to the mass-to-charge ratio of the ion. Three integrated slices through the data are shown, yielding the mass spectrum as a function of time for a given photon-energy range (left-hand vertical slice), the photoionization efficiency as a function of mass for a given reaction-time range (horizontal slice), or the photoionization efficiency as a function of reaction time for a given mass-to-charge ratio (right-hand vertical slice). The color scale is similar to that in Fig. 2 and 3. | ||
The data are normalized by the measured synchrotron photon current and can be “sliced” along different axes to investigate different aspects of the chemistry. Integrating the data over a range of photon energies gives a time-resolved mass spectrum, the counterpart of the burner scan in the flame experiments, with time after photolysis replacing distance from the burner as the natural physical dimension. These data are proportional to the time-resolved concentrations of each detected species. Such an image, seen as the left-hand vertical plane in Fig. 5, shows the prompt disappearance of the photolyte (3-pentanone, m/z = 86) at time = 0, and appearance of photoproducts and reaction products as time proceeds. Integrating the data over a range of times yields a photoionization efficiency spectrum for the constituents of the reacting mixture, the analog of the energy scan in the flame experiments. This image is the horizontal plane in Fig. 5: prominent peaks can be seen for the secondary products ketene (CH2CO, m/z = 42) and acetaldehyde (CH3CHO, m/z = 44). As in the flame experiments, this type of spectrum is important for identification of species and resolution of isomers by their photoionization efficiencies. Finally, the data can be integrated over a single mass peak to give the development of the photoionization efficiency as a function of the time after the photolysis laser. This slice is the right-hand vertical image in Fig. 5. Changes in the shape of the photoionization efficiency can reflect isomerization on the time scale of the experiment or formation of ions by two physically distinct processes, for example, direct ionization of alkyl radical reactants and dissociative ionization of alkylperoxy products in alkyl + O2 reactions.40,61
Isomer-specific flame-chemistry measurements
Many flames have been characterized with the tunable-synchrotron photoionization MBMS method, and detailed mole-fraction profiles have been published or are in preparation. The following section emphasizes some key results in the context of overall reaction mechanisms and isomer-dependent combustion chemistry.Effects of oxygenation on soot-formation pathways
Oxygenated fuel additives are effective in reducing the sooting tendencies of hydrocarbon fuels. It has been thought that the chemical basis of this effect is the interruption of the production of the carbon-rich radicals that initiate the first aromatic ring. The burner scans in Fig. 2 compare rich flames of ethene and two isomeric oxygenated fuels, ethanol and dimethyl ether. The burner scan of ethene at the top of Fig. 2 shows clearly the progress of the type of molecular-weight growth that leads to soot formation, although this flame is below the sooting limit. The fuel itself has a molecular mass of 28. Moving away from the burner, higher-molecular-weight hydrocarbons begin to be formed. Species with three carbons, in the mass range 38–44, appear very close to the burner. Four-carbon species, with m/z = 50 to m/z = 58, appear slightly farther from the burner; five-carbon species farther still, and so on. Furthermore, within each group, the more hydrogen-deficient species appear at successively larger distances as the initially-formed species lose hydrogen. This picture captures the essence of the molecular-weight growth process.This growth and dehydrogenation takes place in competition with oxidation processes that break the species down into smaller fragments. Increasing the oxidative removal of the carbon-rich radicals that are precursors to soot is one means of decreasing soot formation. However, preventing the formation of these precursors is thought to be central to the ability of oxygenates to decrease particulate formation in combustion.62 The effects of oxygenated additives on the production of soot in diesel engines has been the subject of intense study in the last several years. Comparison of engine measurements with laboratory flames suggests that the effect of oxygenate addition on the formation of soot precursors in a diesel engine is better modeled by premixed combustion than by diffusion flames.62 Under specific conditions, addition of oxygenates to the fuel side of a diffusion flame may even increase soot formation.63,64 In premixed flames, as in diesel engines, particulates62 and soot precursors34 are reduced monotonically with increasing addition of the oxygenate.
In an oxygenated fuel, the initial formation of longer carbon chains is inhibited by the presence of C–O bonds in the starting fuel. The image of the rich ethanol flame, in the center of Fig. 2, shows the decrease in the prominence of molecular-weight growth pathways compared to the ethene flame. Although the stoichiometry (given as the equivalence ratio ϕ, which is the molar ratio of the available fuel relative to the amount of fuel that can be completely consumed by the available oxygen) of the ethanol flame (ϕ = 1.97) is somewhat richer than that of the ethene flame (ϕ = 1.9), the ratio of carbon to oxygen is smaller (C/O = 0.56 compared to C/O = 0.63 in the ethene flame). However, the change in C/O is not sufficient to describe the effect, as measurements of a series of ethanol-doped propene flames, while maintaining a constant C/O value, show clear reduction in soot precursors from ethanol addition.34 Furthermore, different bonding of oxygen within the fuel molecule changes its effects on particulate formation.62 Ethanol’s isomeric counterpart, dimethyl ether, has both carbons bound to oxygen and no carbon–carbon bonds at all; in the image of a rich (ϕ = 2.0) dimethyl ether flame shown at the bottom of Fig. 2, the molecular-weight growth pathways are almost indiscernible. Detailed measurements of dimethyl ether flames, carried out using synchrotron photoionization, have been reported and compared to detailed models35 that can be used to predict the chemistry of real devices.
Cyclohexane and 1-hexene
The increasing presence of cyclic alkanes (naphthenes) in motor-vehicle fuels, especially in areas of North America where tar sands are a prominent petroleum source, has led to more consideration of the flame chemistry of naphthenes such as cyclohexane. One consequence of higher naphthene concentrations in fuels may be increased soot formation. Dehydrogenation of cyclohexane to benzene, an important step on the path to PAH (polycyclic aromatic hydrocarbons) and soot, has been observed in rapid-compression machine studies.65 Law et al.33 demonstrated that benzene formation in the preheat zone of a stoichiometric cyclohexane flame must have a substantial contribution from dehydrogenation of the fuel. This conclusion was based largely on the isomeric analysis available from the synchrotron photoionization measurements; the C6H6 in the preheat zone was found to have a much smaller fulvene-to-benzene ratio than the isomer mixture that would be characteristic of production via the propargyl-recombination reaction7 (which dominates aromatic-ring formation in linear hydrocarbon fuels).In that context it is instructive to compare a naphthene flame to that of an isomeric acyclic fuel. Fig. 6 shows a two-dimensional image of a rich cyclohexane flame (ϕ = 2) and a 1-hexene flame of the same stoichiometry and flow rates. Both images were taken at the same photon energy and have been normalized by the photon current. The molecular ion of the fuel is clearly visible at m/z = 84 in both traces. The cyclohexane flame stands off slightly farther from the burner, so many of the peaks appear shifted to larger distances. Besides this contrast, a qualitative difference in the prominence of lower-mass fragments is immediately apparent between the two fuels. The 1-hexene flame displays more pronounced sets of peaks arising from C2–C5 molecules (i.e. at lower m/z values) than does the cyclohexane flame. The 1-hexene can dissociate relatively readily into allyl (m/z = 41) + n-propyl (m/z = 43)66,67 and fragments more extensively in the preheat zone than does cyclohexane. The resultant lower-mass fragments can form aromatic species by more conventional mechanisms, and the 1-hexene flame does show a larger fulvene/benzene ratio than does the cyclohexane flame. Additionally, greater formation of polyynes is observed in the 1-hexene flame, as the small linear unsaturated species formed from the breakup of the 1-hexene recombine and dehydrogenate. This is manifested in the greater prominence of the diacetylene (C4H2) peak at m/z = 50 and the triacetylene (C6H2) peak at m/z = 74. These linear species are not as efficient in forming soot as are ring compounds, such as the benzene formed by cyclohexane dehydrogenation. Differences in the initial hydrocarbon-radical pool between the two isomeric flames continue to affect the species distribution throughout the flame. The principal thermal dissociation pathway of cyclohexane is via1-hexene,68 so the extent of the differences between the two flames is perhaps surprising, but is clearly visible from the two images. Modeling of the stoichiometric cyclohexane flame showed that cyclohexane dissociation is in fact a minor contributor in the removal of cyclohexane.33Cyclohexane is consumed almost completely by H-abstraction, followed by beta scission to 5-hexenyl (CH2CH2CH2CH2CHCH2), which decays to C2H4 and 3-butenyl (CH2CH2CHCH2), which in turn decomposes mostly to C2H4 + C2H3 with some branching to H + butadiene. The ready discrimination of the differences between these isomeric flames demonstrates the powerful role that visual representations can assume in detecting important changes in overall combustion-chemistry patterns.
 | ||
| Fig. 6 Comparison of mass spectra as a function of distance from the burner surface for ϕ = 2.0 flames of cyclohexane (top) and 1-hexene (bottom), taken at 10.5 eV photon energy. The stoichiometries and dilutions of the two flames are identical, and the two images are both normalized to the measured photon current, making profiles of individual species in the two flames comparable. | ||
Allene and propyne
The C3H4 isomers allene (CH2CCH2) and propyne (CH3–CCH) are important species in the chemistry of molecular-weight growth and soot formation because of their close relationship to the resonance-stabilized allyl and propargyl radicals. Flames of allene and propyne are very similar, exhibiting nearly identical temperature and major-species-concentration profiles.37 Again in this isomeric pair of flames, differences in the molecular-weight growth pathways can be discerned. One key difference between the two flames is the larger concentration of allyl radicals in the allene flame. Allyl radicals are formed when hydrogen atoms that diffuse upstream towards the burner add to unburned fuel molecules. Hansen et al.37 noted that the allene flame produces larger amounts of the fulvene isomer of C6H6, apparently because of the reaction of propargyl with allyl radicals. The consequences of the higher proportion of allyl radicals early in the flame are exhibited in other species distributions as well.
Fig. 7 shows a photoionization efficiency curve of m/z = 56 taken with the sampling cone 1 mm from the burner for stoichiometric (ϕ = 1) allene and propyne flames. The flames have identical stoichiometries and dilutions, their temperature profiles are essentially indistinguishable, and the photoionization-efficiency curves are normalized to the measured photon current, so the relative signal strengths should closely reflect the relative concentrations of the corresponding neutral species in the two flames. The photoionization-efficiency spectrum of the m/z = 56 species in the propyne flame is very well-matched by the known spectrum50 of cis-2-butene (trans-2-butene is assumed to be similar),69 shown as the dashed line in Fig. 7. The spectrum of the m/z = 56 species in the allene flame, however, includes a substantial contribution from the 1-butene isomer, which is a potential product of the reaction of allyl with methyl radicals. The spectrum expected from a mixture of 35% 1-butene and 65% cis-2-butene (both of which have known absolute photoionization efficiencies)50 is shown as the solid line. The observed signal deviates from this simulation at about 10.1 eV; this difference may be due to acrolein, which has an ionization energy of 10.11 eV,70 and which is a product of the rapid allyl + O reaction71 or possibly the allyl + O2 reaction at elevated temperature.72 Again, the nature of the initially formed radicals continues to influence the hydrocarbon-species distribution well downstream.
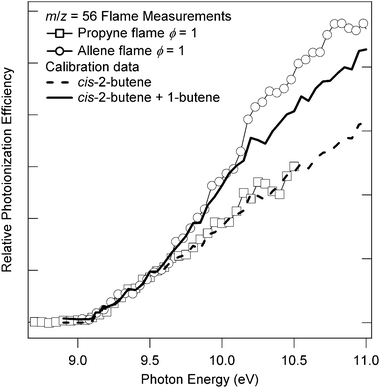 | ||
| Fig. 7 Comparison of normalized photoionization efficiencies for m/z = 56 in allene and propyne flames of identical stoichiometry and dilution. | ||
Isomeric fuel-decomposition pathways of biofuels
The use of biofuels in combustion systems appears poised to become more prominent as the limits of petroleum production and considerations of climate change are increasingly significant. The combustion chemistry of possible biofuels has not been as thoroughly investigated as that of traditional hydrocarbon fuels, however, and development and validation of kinetic models are necessary to predict the influence of biofuels on the operation of existing and emerging engine technologies. Tunable-synchrotron photoionization is well-suited to investigating fuel- and isomer-dependent chemical pathways in these systems.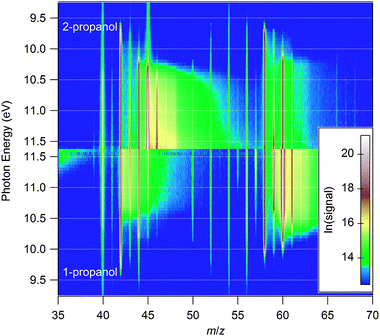 | ||
| Fig. 8 Logarithmic comparison of normalized photoionization-efficiency measurements for rich (ϕ = 1.94) flames of two propanol isomers, taken with the sampling cone just below the luminous zone of the flame. | ||
The flame chemistry of the isomeric fuels methyl acetate and ethyl formate were measured by Oßwald et al.38 using the tunable-synchrotron photoionization method. Burner-scan images taken at 11.0 eV photon energy for isomeric flames of identical equivalence ratio (ϕ = 1.82) and dilution have been normalized by the photon current. The logarithm of the ratio of the signals in the ethyl formate flame to those in the methyl acetate flame is plotted in Fig. 9. The differences in the concentration profiles are immediately apparent, with blue areas showing species favored in the methyl acetate flame and red areas showing species favored in the ethyl formate flame. The methyl acetate flame sits higher from the burner than the ethyl formate flame, which results in the blue area near 4 mm in Fig. 9, where the hydrocarbon-radical concentrations in the ethyl formate flame have dropped off but substantial radical density remains in the methyl acetate flame. Specifically favored species are clear: formaldehyde (m/z = 30), methanol (m/z = 32) and ketene (m/z = 42) are more prominent in the methyl acetate flame, whereas ethene (m/z = 28), acetaldehyde (m/z = 44), and C4 species are favored in the ethyl formate flame. These preferences can be rationalized by considering the initial decomposition pathways of the isomers, as shown in Scheme 1 below.
 | ||
| Scheme 1 Fuel decomposition pathways for two isomeric esters. | ||
 | ||
| Fig. 9 Logarithmic image of the ratio of normalized mass spectra for isomeric ester flames of identical stoichiometry and dilution, taken at 11.0 eV photon energy. The color scale is chosen so that species having higher concentration in the flame of the methyl acetate isomer appear in blue and those having higher concentration in the ethyl formate flame appear in red. The prominent blue feature at m/z = 43 arises from dissociative ionization of methyl acetate. | ||
Hydrogen-atom abstraction is likely the principal reactive-removal mechanism for the fuel. The decomposition of the radical species remaining after H abstraction will tend to produce either ketene + methoxy or acetyl + formaldehyde in the case of methyl acetate, and will form ethoxy + CO, ethene + HC(O)O or acetaldehyde + HCO for ethyl formate.38 The higher C4 concentrations in the ethyl formate flame may result from the larger ethylene formation, both as a result of the hydrogen-abstraction reactions from the fuel and because of a possible direct elimination of ethene similar to that described previously for ethyl propanoate.82 The nature of the ester apparently affects the ignition and molecular-weight growth chemistry, at least for the simple surrogate molecules considered here. It remains to be seen whether these effects will persist when coupled with the chemistry of a large hydrocarbon chain. Negligible differences in emission of particulates and nitrogen oxides have been seen when comparing the combustion of methyl and ethyl esters in heavy-duty diesel engines.83
Chemical kinetics and photoionization spectroscopy
The measurement of species profiles in well-characterized flames is an important tool for validating combustion-chemistry mechanisms. Underlying the development of these mechanisms is the measurement of the kinetics and product distributions of elementary chemical reactions. Application of these measurements to modeling combustion typically requires input from computational kinetics and theory, as it is essentially impossible to measure the necessary elementary reactions directly over the range of temperatures and pressures that are relevant to combustion. Spectroscopic and chemical-kinetics measurements taken using tunable-synchrotron photoionization can complement the flame-chemistry measurements in the effort to design predictive models for combustion. More importantly, the ability to unravel isomeric contributions in elementary reactions promises new insight into their fundamental physical chemistry. The examples highlighted below are chosen to show the power of tunable photoionization and multiple-mass detection for reactions of fundamental interest that also have relevance to current challenges in combustion chemistry.Alkylperoxy-radical photoionization
The chemistry of autoignition is relevant to the phenomenon of engine knock, the undesired spontaneous ignition of the fuel–air charge before the firing of the spark plug. Higher compression ratios in general improve the efficiency of an internal-combustion engine, but the onset of engine knock limits the feasible compression ratio in a spark-ignition engine. Recently, the emergence of advanced engine technologies that rely on compression-ignition of a premixed fuel–air charge has generated more intense interest in the chemistry of autoignition processes.9,10,84 The important low-temperature (i.e. below about 900 K) processes in autoignition center around the addition of O2 to hydrocarbon radicals, especially alkyl radicals (denoted by R). These reactions proceed via alkylperoxy (ROO) radical intermediates. The isomerization of these alkylperoxy radicals and their changing fates as temperature and pressure change underlie cool-flame chemistry, negative temperature-coefficient behavior in hydrocarbon oxidation, low-temperature chain branching and ignition.10,41,85The detection of ROO radicals by photoionization has proved difficult, and it has been suggested that this difficulty reflected low-photoionization cross-sections.86,87 The methylperoxy cation CH3OO+ was observed by electron-impact ionization88 and by photoionization with an Ar resonance lamp (11.62 and 11.83 eV).89,90 Bernstein and coworkers91 studied photoionization at 10.5 eV of several peroxy radicals in a supersonic jet, but observed only dissociative ionization for ethylperoxy and propylperoxy radicals. Measurements of alkyl + O2 reactions employing multiplexed tunable-synchrotron photoionization mass spectrometry,40,61 coupled with quantum-chemical calculations, have shed light on the photoionization processes in ROO radicals, and offer possibilities for direct monitoring of alkylperoxy radicals by ionization methods.
The formation of alkylperoxy radicals was accomplished by the reaction of alkyl radicals with O2, where the alkyl radicals were formed by 193-nm photolysis of suitable ketones. The three-dimensional data shown in Fig. 5 represent measurements of the reaction of ethyl radicals with O2. The photolysis pulse at zero time (t = 0) forms ethyl radicals from diethyl ketone. These ethyl radicals rapidly react with oxygen to form ethylperoxy radicals (with some formation of the HO2 + ethene product channel). The signals before the photolysis pulse have been subtracted from the data in Fig. 5, so that the diethyl ketone mass appears as a nearly instantaneous depletion (visible as a blue stripe on the left-hand vertical slice in Fig. 5) starting at t = 0. Photofragments appear nearly instantaneously, and reaction products are detected soon after, as the concentration of O2 in these experiments is high enough to ensure rapid reaction. These data are taken as a function of photon energy, so any product and its appearance mechanism can be identified by its mass, its kinetic time profile, and its photoionization spectrum. In the reaction of methyl radicals with O2, a clear signal from CH3OO+ was observed, permitting the measurement of the photoionization-efficiency spectrum and the ionization energy of the methylperoxy radical.40
In the case of ethyl + O2, no signal could be observed at the C2H5OO+ mass. However, the signal at m/z = 29, corresponding to C2H5+, bears the signature of ethylperoxy photoionization. Fig. 10 shows an image of the integrated m/z = 29 signal as a function of time after photolysis and photoionization energy (i.e. the right-hand vertical slice of Fig. 5). A trace resulting from further integrating this image over a range of photon energies below 10.2 eV, shown in the lower right inset, gives a rapid decay reflecting the time behavior of the ethyl-radical concentration. This signal arises from direct ionization of the ethyl radical. A trace integrated over photon energies above 10.4 eV, shown on the upper right, contains this same direct ionization contribution, but superimposed on a signal that persists to much longer time. The kinetic behavior of this long-time signal identifies it as dissociative ionization of the ethylperoxy radical.
 | ||
| Fig. 10 Time- and photon-energy-resolved measurements of the m/z = 29 signal for photolytically initiated reaction of ethyl radical with O2, with the pre-photolysis background subtracted and normalized by photon current. The evolution of the photoionization-efficiency spectrum with time after photolysis reflects overlapping contributions of direct ionization of ethyl radicals (the concentration of which decays rapidly by reaction with O2) and dissociative ionization of the ethylperoxy radical product of the ethyl + O2 reaction. These contributions can be readily seen in the integrated traces on the right, where the lower trace, integrated over photon energies up to 10.2 eV, shows only direct ionization, and the upper trace, integrated over photon energies above 10.4 eV, shows both processes. | ||
Quantum-chemical calculations support the experimental observation that the ethylperoxy radical has an unstable cation40,91 and will undergo only dissociative ionization. The ethyl cation is stabilized by hyperconjugation of the σ(Cβ–H) orbital with the empty p(Cα) orbital, as are more highly substituted carbocations,92,93 resulting in nonclassical “bridged” structures. Increased stability of the alkyl cation implies reduced stability of the alkylperoxy cation, and the cations of ethylperoxy and the larger alkylperoxy radicals are expected to be unbound. Nevertheless, tunable-synchrotron photoionization offers a means of detecting the ROO radicals by their dissociative-ionization thresholds, at least in systems where the chemistry is sufficiently simple. Furthermore, as the instability of the ROO+ species arises from hyperconjugation in the alkyl cation fragment, species in which this stabilization is reduced or impossible may yet have stable parent cations.
Isomeric products of C2H reactions
Knowledge of the products of reactions is important in modeling chemical systems, and information about the isomeric nature of the products can help unravel the mechanism of the reaction. Tunable-synchrotron photoionization is a means to accomplish isomeric product resolution, and it has begun to be applied to systems of combustion and atmospheric importance. One of these systems is the reaction of C2H with C3H4, recently investigated by Goulay and coworkers42 using the multiplexed synchrotron-photoionization technique. Fig. 11 shows a section of the mass spectra from the reaction of C2H with the propyne and allene isomers of C3H4, integrated over long reaction times, as a function of ionizing photon energy. The data are taken from the experiments of Goulay et al.42 The reaction is initiated by 193-nm photolysis of CF3CCH; however, both C3H4 reagents also absorb 193-nm photons, as do impurities in the C3H4 samples, causing some side chemistry which differs in the two systems. This side chemistry can be seen in the mass 66 and weak mass 54 peaks; because the entire relevant mass spectrum is sampled, the source of these peaks could be traced to side or secondary reactions.42 The principal product of the C2H + C3H4 reaction is C5H4 at m/z = 64. From the comparison of the photoionization-efficiency spectra in Fig. 11, it is clear that the isomeric composition of the product differs for the two reactant isomers. From the known ionization energies70 and estimated photoionization cross-sections,94 the m/z = 64 products of the C2H + propyne reaction were determined to be 15–20% ethynylallene and 80–85% methyldiacetylene, in agreement with earlier determinations of Kaiser and coworkers.95 These are the products expected from the addition to the terminal end of the triple bond. The addition to the central carbon results in the formation of diacetylene (m/z = 50) and CH3, estimated to be between 50 and 70% of the overall products.42 The reaction with allene, in contrast, produces ethynylallene and 1,4-pentadiyne, both products of terminal addition, in approximately equal yields (∼40%) with a minor role for the methyldiacetylene product expected from central addition.42 Negligible diacetylene products were observed; the m/z = 50 signal in the bottom frame of Fig. 11 arises principally from the reaction of C2H with the CF3CCH photolyte. The ability to monitor all product masses as a function of reaction time and photon energy facilitates such detailed product studies. | ||
| Fig. 11 Section of the mass spectrum of products from the 193-nm photolysis of CF3CCH in the presence of propyne (top) and allene (bottom) as a function of ionization photon energy, background-subtracted and normalized by photon current. The differing isomeric nature of the products is clear from the different shapes of the individual mass features for the two isomers. | ||
Vinyl-radical recombination
Radical–radical reactions often have a wealth of energetically allowed pathways, for example, stabilization of an initial adduct or production of other products by disproportionation. The multiplexed “imaging” of such reactions can focus on known product pathways, for example, in analysis of the isomeric composition of C6H6 from propargyl self-reaction. However the collection of full multidimensional data is particularly powerful in discovering unexpected pathways. The vinyl + vinyl reaction has been previously measured to produce both 1,3-butadiene and ethene + acetylene; these had been the only products detected.96–98 Stief and coworkers did observe methyl radicals after initiating the vinyl + vinyl reaction by F + C2H4 in a fast-flow reactor, but attributed the methyl to other reactions.98Fig. 12 displays a section of the mass spectrum taken at 9.7 eV photon energy following 193-nm photolysis of methyl vinyl ketone. Clear signal at m/z = 39, attributable to the propargyl radical, is observed, and closer inspection reveals a clearly discernable rise time, indicating that it is not a direct photolysis product. The shape of the propargyl signal is well modeled by assuming it is a product of the self-reaction of vinyl radicals, indicating substantial branching of the reaction to propargyl + methyl. This conclusion is confirmed by measurements of divinyl ketone photolysis at 193 and 248 nm, both of which show methyl and propargyl formation consistent with production from the vinyl-radical self-reaction. Preliminary analysis based on the cross-sections of Robinson et al.99 suggests that ∼30% of the reaction may proceed to this channel; however, detailed modeling and measurements under more photolysis conditions will be required to obtain a precise branching fraction. As the photolysis100,101 and pyrolysis102 of 1,3-butadiene are known to produce methyl + propargyl as dominant products, it is reasonable that the initially formed 1,3-butadiene adduct from vinyl + vinyl will also dissociate to form considerable amounts of propargyl + methyl. Formation of propargyl + methyl in this reaction may require the reevaluation of literature measurements of the overall rate constant that determined the initial vinyl-radical concentration from measurements of the end product 1,3-butadiene.96,97 Contributions from this additional channel will reduce the branching fraction to 1,3-butadiene, implying a larger initial vinyl concentration (and a smaller rate constant) in those experiments. | ||
| Fig. 12 Section of a time-resolved mass spectrum subsequent to 193-nm photolysis of methyl vinyl ketone, taken with a photon energy of 9.7 eV. | ||
Discussion and outlook
Many outstanding problems in combustion and atmospheric chemistry are amenable to the application of multiplexed photoionization mass spectrometry. Problems that are sensitive to the isomeric nature of reaction products or intermediates are particularly suited to tunable photoionization investigations. The number of isomers increases with increasing number of carbon atoms in a hydrocarbon molecule—as in the build-up process leading to soot—and it also increases when additional chemical functions may be found at different positions of a common molecular backbone, e.g.ester, alcohol or ether groups in alternative fuels.For example, the reactions of alkyl radicals with O2 proceed via internal H-atom transfer in the initially formed alkylperoxy adduct (ROO) to produce hydroperoxyalkyl radicals (denoted QOOH).41 These ephemeral radicals are responsible for low-temperature chain branching and are critical for ignition chemistry; their reactivity and stability depends on their isomeric structure. While no QOOH radical has ever been directly detected, their formation and their isomeric distribution can be inferred from measurements of the final products of their decomposition into OH radicals and carbonyl compounds or cyclic ethers.11
One specific example that is of fundamental interest and current technological importance is the reaction of cyclohexyl radicals with O2. As cycloalkanes become more prevalent in the fuel stream, ignition chemistry of such fuels, as well as their sooting characteristics (see above), are increasingly under study.65,103–112 Recently, Knepp et al.113 published an investigation of OH and HO2 formation in the reaction of cyclohexyl radicals with O2 that coupled experimental observations with detailed ab initio and master-equation calculations. The cyclohexyl-O2 potential-energy surface is rather complex, and time-resolved measurements of isomeric products would be valuable for characterizing this and other alkyl + O2 reactions. Fig. 13 and 14 show how this may be possible with tunable-synchrotron radiation. Fig. 13 shows the time-dependent C6H10 formation from pulsed-photolytic OH-initiated oxidation of cyclohexane. Under these experimental conditions, the formation of C6H10 is dominated by the cyclohexyl + O2 reaction, and the model developed by Knepp et al.113 successfully predicts the time behavior of HO2 + C6H10 product formation. Further, because the experiments use tunable-synchrotron photoionization, the isomeric distribution of products, e.g. C6H10O, can be probed. However, as shown in Fig. 14, detailed calibration measurements of the photoionization efficiency of the relevant isomers are still necessary to deconvolute the observed product spectra. This fact highlights the need for photoionization spectroscopy and cross-section measurements to proceed in concert with flame and kinetics measurements. Ongoing flame-chemistry measurements are faced with similar hurdles; reliable absolute photoionization-efficiency measurements of radical intermediates99,114,115 are particularly difficult but very valuable.
 | ||
| Fig. 13 Time-resolved production of C6H10 from OH-initiated cyclohexane oxidation compared to a prediction based on the model of Knepp et al.113 | ||
 | ||
| Fig. 14 Photoionization efficiency of the m/z = 98 (C6H10O) product in OH-initiated oxidation of cyclohexane. Ionization energies70 of several possible isomers are also shown. | ||
The extension of the kinetics measurements to higher pressure may be important in understanding chemical reactions that are mediated by collisional stabilization and dissociation of intermediate adduct species, such as in molecular-weight growth autoignition chemistry. Measurement from higher-pressure reactors will likely entail supersonic-beam sampling, for which the time response may be somewhat improved60 (although the pumping speed required might be larger). Mass sampling of premixed flames at higher pressure suffers from shrinking of the primary reaction zone, but molecular-beam mass sampling of free radicals from atmospheric pressure flames was an early application of the MBMS technique.13 Microprobe sampling of atmospheric-pressure non-premixed flames using fixed-frequency single-photon ionization116 has proved a valuable measure of the flame structure and chemistry of such systems, including measurements of toxic oxygenate emission from flames doped with different isomeric esters.117 Implementation of tunable photoionization in such systems, especially with multiple-mass detection, may provide unprecedentedly detailed probes of atmospheric and higher-pressure combustion.
Finally, other means of accomplishing the isomeric resolution attained by multiplexed tunable-synchrotron photoionization may soon become more feasible. For example, single-photon laser photoionization has been employed for both flame118–120 and kinetics121 measurements. Whereas laser photoionization typically has superior energy resolution, laser vacuum-ultraviolet light is far less readily tunable than synchrotron radiation. Perhaps future tabletop sources based on multiple harmonic generation and frequency combs122–124 will provide easily-tunable, high-resolution vacuum-ultraviolet radiation of suitable intensity for photoionization measurements. Alternatively, mass spectrometry at fixed-frequency ionization can be combined with coincident resolution of the photoelectron energy,125–127 giving a tool to monitor isomers and electronic excitation in products and intermediates.
Conclusions
The application of multiplexed tunable-synchrotron photoionization to combustion measurements yields a wealth of detailed information about the chemical transformations in elementary reactions and in flame systems. The multidimensional nature of the data provides an “image” of the chemistry that can be particularly powerful in illuminating global chemical pathways or highlighting previously-unknown reactive pathways. The multiple dimensions, along which reduced-dimensionality data can be sliced from the complete data sets, provide critical checks on proposed interpretations of the data, leading to a higher degree of certainty in the final interpretation. Further development of tunable photoionization and simultaneous multiple-mass detection technologies, in conjunction with new photoionization-spectroscopy measurements, will continue to deliver new insights into combustion.Acknowledgements
We acknowledge the outstanding work of the students and postdoctoral associates who produced the results discussed in this perspective: Tina Kasper (Sandia National Laboratories and Universität Bielefeld); Patrick Oßwald and Ulf Struckmeier (Universität Bielefeld); Matthew E. Law and Aude Morel (University of Massachusetts, Amherst); Fabien Goulay (University of California Berkeley and Lawrence Berkeley National Laboratory); Giovanni Meloni, Talitha M. Selby, and Peng Zou (Sandia National Laboratories); Juan Wang (Cornell University). The skilled technical assistance of Paul Fugazzi and Howard Johnsen (Sandia National Laboratories) was vital to realizing these experiments. We thank Musahid Ahmed and Kevin Wilson (Lawrence Berkeley National Laboratory) and Stephen R. Leone (University of California, Berkeley) for their continuing advice, wisdom, and support. This work is supported by the Division of Chemical Sciences, Geosciences, and Biosciences, the Office of Basic Energy Sciences, the U. S. Department of Energy, in part through grants DE-FG02-91ER14192 (P.R.W.) and DE-FG02-01ER15180 (T.A.C.). T.A.C. also gratefully acknowledges support from Chemical Sciences Division of the U.S. Army Research Office. The Advanced Light Source is supported by the Director, Office of Science, Office of Basic Energy Sciences, Materials Sciences Division, of the U.S. Department of Energy under Contract No. DE-AC02-05CH11231 at Lawrence Berkeley National Laboratory. Sandia is a multi-program laboratory operated by Sandia Corporation, a Lockheed Martin Company, for the National Nuclear Security Administration under contract DE-AC04-94-AL85000. We thank the Ford Motor Company for supplying the propyne used in these experiments.References
- E. W. Kaiser, W. O. Siegl, D. F. Cotton and R. W. Anderson, Environ. Sci. Technol., 1992, 26, 1581–1586 CAS.
- E. W. Kaiser, W. O. Siegl, Y. I. Henig, R. W. Anderson and F. H. Trinker, Environ. Sci. Technol., 1991, 25, 2005–2012 CrossRef CAS.
- T. J. Wallington, E. W. Kaiser and J. T. Farrell, Chem. Soc. Rev., 2006, 35, 335–347 RSC.
- D. Kayes and S. Hochgreb, Environ. Sci. Technol., 1999, 33, 3968–3977 CrossRef CAS.
- E. Zervas, X. Montagne and J. Lahaye, Fuel, 2004, 83, 2301–2311 CrossRef CAS.
- J. A. Miller and C. F. Melius, Combust. Flame, 1992, 91, 21–39 CrossRef CAS.
- J. A. Miller and S. J. Klippenstein, J. Phys. Chem. A, 2003, 107, 7783–7799 CrossRef CAS.
- H. Yamada, M. Yoshii and A. Tezaki, Proc. Combust. Inst., 2005, 30, 2773–2780 CrossRef.
- P. L. Kelly-Zion, J. E. Dec and F. Dryer, Proc. Combust. Inst., 2000, 28, 1187–1194 CrossRef CAS.
- C. K. Westbrook, Proc. Combust. Inst., 2000, 28, 1563–1577 CrossRef CAS.
- R. W. Walker and C. Morley, in Low-Temperature Combustion and Autoignition, ed. M. J. Pilling, Elsevier, Amsterdam, 1997, pp. 1–124 Search PubMed.
- K. H. Homann, M. Mochizuki and H. G. Wagner, Z. Phys. Chem., Neue Folge, 1963, 37, 299.
- T. A. Milne and F. T. Greene, J. Chem. Phys., 1966, 44, 2444–2449 CrossRef CAS.
- J. C. Biordi, Prog. Energy Combust. Sci., 1977, 3, 151–173 CrossRef CAS.
- J. C. Biordi, C. P. Lazzara and J. F. Papp, Combust. Flame, 1974, 23, 73–82 CrossRef.
- J. D. Bittner, A Molecular Beam Mass Spectrometer Study of Fuel-Rich and Sooting Benzene–Oxygen Flames, Sc.D Dissertation, Massachusetts Institute of Technology, Cambridge, MA, 1981 Search PubMed.
- P. R. Westmoreland, Experimental and Theoretical Analysis of Oxidation and Growth Chemistry in a Fuel-Rich Acetylene Flame, Ph.D. Dissertation, Massachusetts Institute of Technology, Cambridge, MA, 1986 Search PubMed.
- J. Peeters and G. Mahnen, Symp. (Int.) Combust., 1973, 14, 133–146 Search PubMed.
- J. H. Werner and T. A. Cool, Chem. Phys. Lett., 1998, 290, 81–87 CrossRef CAS.
- T. A. Cool, A. McIlroy, F. Qi, P. R. Westmoreland, L. Poisson, D. S. Peterka and M. Ahmed, Rev. Sci. Instrum., 2005, 76, 094102 CrossRef.
- T. A. Cool, K. Nakajima, T. A. Mostefaoui, F. Qi, A. McIlroy, P. R. Westmoreland, M. E. Law, L. Poisson, D. S. Peterka and M. Ahmed, J. Chem. Phys., 2003, 119, 8356–8365 CrossRef CAS.
- C. Huang, L. Wei, B. Yang, J. Wang, Y. Li, L. Sheng, Y. Zhang and F. Qi, Energy Fuels, 2006, 20, 1505–1513 CrossRef CAS.
- B. Yang, C. Huang, L. Wei, J. Wang, L. Sheng, Y. Zhang, F. Qi, W. Zheng and W.-K. Li, Chem. Phys. Lett., 2006, 423, 321–326 CrossRef CAS.
- T. Zhang, X. N. Tang, K.-C. Lau, C. Y. Ng, C. Nicolas, D. S. Peterka, M. Ahmed, M. L. Morton, B. Ruscic, R. Yang, L. X. Wei, C. Q. Huang, B. Yang, J. Wang, L. S. Sheng, Y. W. Zhang and F. Qi, J. Chem. Phys., 2006, 124, 074302 CrossRef CAS.
- C. Huang, B. Yang, R. Yang, J. Wang, L. Wei, X. Shan, L. Sheng, Y. Zhang and F. Qi, Rev. Sci. Instrum., 2005, 76, 126108 CrossRef.
- C. A. Taatjes, N. Hansen, A. McIlroy, J. A. Miller, J. P. Senosiain, S. J. Klippenstein, F. Qi, L. Sheng, Y. Zhang, T. A. Cool, J. Wang, P. R. Westmoreland, M. E. Law, T. Kasper and K. Kohse-Höinghaus, Science, 2005, 308, 1887–1889 CrossRef CAS.
- N. Hansen, S. J. Klippenstein, J. A. Miller, J. Wang, T. A. Cool, M. E. Law, P. R. Westmoreland, T. Kasper and K. Kohse-Höinghaus, J. Phys. Chem. A, 2006, 110, 4376–4388 CrossRef CAS.
- C. A. Taatjes, S. J. Klippenstein, N. Hansen, J. A. Miller, T. A. Cool, J. Wang, M. E. Law and P. R. Westmoreland, Phys. Chem. Chem. Phys., 2005, 7, 806–813 RSC.
- C. A. Taatjes, D. L. Osborn, T. A. Cool and K. Nakajima, Chem. Phys. Lett., 2004, 394, 19–24 CrossRef CAS.
- T. A. Cool, K. Nakajima, C. A. Taatjes, A. McIlroy, P. R. Westmoreland, M. E. Law and A. Morel, Proc. Combust. Inst., 2005, 30, 1681–1688 CrossRef.
- C. A. Taatjes, N. Hansen, J. A. Miller, T. A. Cool, J. Wang, P. R. Westmoreland, M. E. Law, T. Kasper and K. Kohse-Höinghaus, J. Phys. Chem. A, 2006, 110, 3254–3260 CrossRef CAS.
- N. Hansen, S. J. Klippenstein, C. A. Taatjes, J. A. Miller, J. Wang, T. A. Cool, B. Yang, R. Yang, L. Wei, C. Huang, J. Wang, F. Qi, M. E. Law and P. R. Westmoreland, J. Phys. Chem. A, 2006, 110, 3670–3678 CrossRef CAS.
- M. E. Law, P. R. Westmoreland, T. A. Cool, J. Wang, N. Hansen, C. A. Taatjes and T. Kasper, Proc. Combust. Inst., 2007, 31, 565–573 CrossRef.
- K. Kohse-Höinghaus, P. Oßwald, U. Struckmeier, T. Kasper, N. Hansen, C. A. Taatjes, J. Wang, T. A. Cool, S. Gon and P. R. Westmoreland, Proc. Combust. Inst., 2007, 31, 1119–1127 CrossRef.
- T. A. Cool, J. Wang, N. Hansen, P. R. Westmoreland, F. L. Dryer, Z. Zhao, A. Kazakov, T. Kasper and K. Kohse-Höinghaus, Proc. Combust. Inst., 2007, 31, 285–293 CrossRef.
- N. Hansen, S. J. Klippenstein, P. R. Westmoreland, T. Kasper, K. Kohse-Höinghaus, J. Wang and T. A. Cool, Phys. Chem. Chem. Phys., 2007, 10.1039/b711578d.
- N. Hansen, J. A. Miller, C. A. Taatjes, J. Wang, T. A. Cool, M. E. Law, P. R. Westmoreland, T. Kasper and K. Kohse-Höinghaus, Proc. Combust. Inst., 2007, 31, 1157–1164 CrossRef.
- P. Oßwald, U. Struckmeier, T. Kasper, K. Kohse-Höinghaus, J. Wang, T. A. Cool, N. Hansen and P. R. Westmoreland, J. Phys. Chem. A, 2007, 111, 4093–4101 CrossRef CAS.
- N. Hansen, T. Kasper, S. J. Klippenstein, P. R. Westmoreland, M. E. Law, C. A. Taatjes, K. Kohse-Höinghaus, J. Wang and T. A. Cool, J. Phys. Chem. A, 2007, 111, 4081–4092 CrossRef CAS.
- G. Meloni, P. Zou, S. J. Klippenstein, M. Ahmed, S. R. Leone, C. A. Taatjes and D. L. Osborn, J. Am. Chem. Soc., 2006, 128, 13559–13567 CrossRef CAS.
- C. A. Taatjes, J. Phys. Chem. A, 2006, 110, 4299–4312 CrossRef CAS.
- F. Goulay, D. L. Osborn, C. A. Taatjes, P. Zou, G. Meloni and S. R. Leone, Phys. Chem. Chem. Phys., 2007, 9, 4291–4300 RSC.
- I. R. Slagle, F. Yamada and D. Gutman, J. Am. Chem. Soc., 1981, 103, 149–153 CrossRef CAS.
- A. G. Suits, M. Kawasaki and W. Lawrance, Phys. Chem. Chem. Phys., 2006, 8, 2913–2914 RSC.
- D. W. Chandler and P. L. Houston, J. Chem. Phys., 1987, 87, 1445–1447 CrossRef CAS.
- A. G. Suits, L. S. Bontuyan, P. L. Houston and B. J. Whitaker, J. Chem. Phys., 1992, 96, 8618–8620 CrossRef CAS.
- T. N. Kitsopoulos, M. A. Buntine, D. P. Baldwin, R. N. Zare and D. W. Chandler, Science, 1993, 260, 1605–1610 CrossRef CAS.
- A. G. Suits, P. Heimann, X. Yang, M. Evans, C.-W. Hsu, K.-t. Lu, Y. T. Lee and A. H. Kung, Rev. Sci. Instrum., 1995, 66, 4841–4844 CrossRef CAS.
- T. A. Cool, J. Wang, K. Nakajima, C. A. Taatjes and A. McIlroy, Int. J. Mass Spectrom., 2005, 247, 18–27 CrossRef CAS.
- J. Wang, B. Yang, T. A. Cool, N. Hansen and T. Kasper, Near-threshold Absolute Photoionization Cross Sections of some Reaction Intermediates in Combustion, Int. J. Mass Spectrom., 2007, in press Search PubMed.
- M. Kamphus, N. Liu, B. Atakan, F. Qi and A. McIlroy, Proc. Combust. Inst., 2003, 29, 2627–2633.
- H. Fan and S. T. Pratt, J. Chem. Phys., 2005, 123, 204301 CrossRef.
- H. Fan and S. T. Pratt, J. Chem. Phys., 2006, 124, 114312 CrossRef CAS.
- D. L. Osborn, P. Zou, H. Johnsen, C. C. Hayden, C. A. Taatjes, D. S. Peterka, M. Ahmed and S. R. Leone, The Multiplexed Chemical Kinetic Photoionization Mass Spectrometer: A New Approach for Isomer Resolved Chemical Kinetics, 2007, in preparation.
- M. P. Sinha and A. D. Tomassian, Rev. Sci. Instrum., 1991, 62, 2618–2620 CrossRef CAS.
- M. P. Sinha and J. Houseman, Proc. SPIE-Int. Soc. Opt. Eng., 2003, 5048, 119–127.
- M. Lampton, O. H. W. Siegmund and R. Raffanti, IEEE Trans. Nucl. Sci., 1990, 37, 1548–1549 CrossRef.
- M. Lampton, O. Siegmund and R. Raffanti, Rev. Sci. Instrum., 1987, 58, 2298–2305 CrossRef CAS.
- S. B. Moore and R. W. Carr, Jr, Int. J. Mass Spectrom. Ion Phys., 1977, 24, 161–171 CrossRef CAS.
- C. A. Taatjes, Int. J. Chem. Kinet., 2007, 39, 565–570 CrossRef CAS.
- G. Meloni, T. M. Selby, F. Goulay, S. R. Leone, D. L. Osborn and C. A. Taatjes, J. Am. Chem. Soc., 2007 DOI:10.1021/ja075130n.
- C. K. Westbrook, W. J. Pitz and H. J. Curran, J. Phys. Chem. A, 2006, 110, 6912–6922 CrossRef CAS.
- C. S. McEnally and L. D. Pfefferle, Proc. Combust. Inst., 2007, 31, 603–610 CrossRef.
- C. S. McEnally, L. D. Pfefferle, B. Atakan and K. Kohse-Höinghaus, Prog. Energy Combust. Sci., 2006, 32, 247–294 CrossRef CAS.
- O. Lemaire, M. Ribaucour, M. Carlier and R. Minetti, Combust. Flame, 2001, 127, 1971–1980 CrossRef CAS.
- K. D. King, Int. J. Chem. Kinet., 1979, 11, 1071–1080 CrossRef CAS.
- C. S. McEnally and L. D. Pfefferle, Combust. Flame, 2005, 143, 246–263 CrossRef CAS.
- T. C. Brown, K. D. King and T. T. Nguyen, J. Phys. Chem., 1986, 90, 419–424 CrossRef CAS.
- H. Koizumi, K. Shinsaka, T. Yoshimi, K. Hironaka, S. Arai, M. Ukai, M. Morita, H. Nakazawa, A. Kimura, Y. Hatano, Y. Ito, Y. Zhang, A. Yagishita, K. Ito and K. Tanaka, Radiat. Phys. Chem., 1988, 32, 111–115 CrossRef CAS.
- S. G. Lias, in NIST Chemistry WebBook, NIST Standard Reference Database Number 69, ed. P. J. Linstrom and W. G. Mallard, National Institute of Standards and Technology, Gaithersburg, MD, 20899, 2005, http://webbook.nist.gov Search PubMed.
- I. R. Slagle, J. R. Bernhardt, D. Gutman, M. A. Hanning-Lee and M. J. Pilling, J. Phys. Chem., 1990, 94, 3652–3656 CrossRef CAS.
- J. Lee and J. W. Bozzelli, Proc. Combust. Inst., 2005, 30, 1015–1022 CrossRef.
- T. Kasper, Molekularstrahlmassenspektrometrie zur Analytik in Flammen oxygenierter Brennstoffe, PhD dissertation, Bielefeld University, 2006 Search PubMed.
- B. Yang, P. Oßwald, Y. Li, J. Wang, L. Wei, Z. Tian, F. Qi and K. Kohse-Höinghaus, Combust. Flame, 2007, 148, 198–209 CrossRef CAS.
- K. M. A. Refaey and W. A. Chupka, J. Chem. Phys., 1968, 48, 5205–5219 CrossRef CAS.
- G. Y. Matti, O. I. Osman, J. E. Upham, R. J. Suffolk and H. W. Kroto, J. Electron Spectrosc. Relat. Phenom., 1989, 49, 195–201 CrossRef CAS.
- B. Ruscic and J. Berkowitz, J. Chem. Phys., 1994, 101, 10936–10946 CrossRef CAS.
- J. P. Senosiain, J. A. Miller and S. J. Klippenstein, J. Phys. Chem. A, 2006, 110, 6960–6970 CrossRef CAS.
- H. Hippler and B. Viskolcz, Phys. Chem. Chem. Phys., 2000, 2, 3591–3596 RSC.
- T. Yamada, J. W. Bozzelli and T. Lay, J. Phys. Chem. A, 1999, 103, 7646–7655 CrossRef CAS.
- W. K. Metcalfe, S. Dooley, H. J. Curran, J. M. Simmie, A. M. El-Nahas and M. V. Navarro, J. Phys. Chem. A, 2007, 111, 4001–4014 CrossRef CAS.
- A. M. El-Nahas, M. V. Navarro, J. M. Simmie, J. W. Bozzelli, H. J. Curran, S. Dooley and W. Metcalfe, J. Phys. Chem. A, 2007, 111, 3727–3739 CrossRef CAS.
- R. L. McCormick, M. S. Graboski, T. L. Alleman and A. M. Herring, Environ. Sci. Technol., 2001, 35, 1742–1747 CrossRef CAS.
- F. Zhao, T. W. Asmus, D. N. Assanis, J. E. Dec, J. A. Eng and P. M. Najt, Homogeneous Charge Compression Ignition (HCCI) Engines: Key Research and Development Issues, Society of Automotive Engineers, Pittsburgh, PA, 2003 Search PubMed.
- W. J. Pitz and C. K. Westbrook, Combust. Flame, 1986, 63, 113–133 CrossRef CAS.
- C. E. McDade, T. M. Lenhardt and K. D. Bayes, J. Photochem., 1982, 20, 1–7 CrossRef.
- D. Wu and K. D. Bayes, Int. J. Chem. Kinet., 1986, 18, 547 CAS.
- I. C. Plumb, K. R. Ryan, J. R. Steven and M. F. R. Mulcahy, J. Phys. Chem., 1981, 85, 3136–3138 CrossRef CAS.
- N. Washida and K. D. Bayes, Int. J. Chem. Kinet., 1976, 8, 777–794 CrossRef CAS.
- I. R. Slagle and D. Gutman, J. Am. Chem. Soc., 1985, 107, 5342–5347 CrossRef CAS.
- H. B. Fu, Y. J. Hu and E. R. Bernstein, J. Chem. Phys., 2006, 125, 014310 CrossRef CAS.
- N. Muller and R. S. Mulliken, J. Am. Chem. Soc., 1958, 80, 3489–3497 CrossRef CAS.
- L. Radom, J. A. Pople and P. v. R. Schleyer, J. Am. Chem. Soc., 1972, 94, 5935–5945 CrossRef CAS.
- M. Bobeldijk, W. J. v. d. Zande and P. G. Kistemaker, Chem. Phys., 1994, 179, 125–130 CrossRef CAS.
- R. I. Kaiser, C. C. Chiong, O. Asvany, Y. T. Lee, F. Stahl, P. v. R. Schleyer and H. F. Schaefer III, J. Chem. Phys., 2001, 114, 3488–3496 CrossRef CAS.
- A. Fahr and A. H. Laufer, J. Phys. Chem., 1990, 94, 726–729 CrossRef CAS.
- A. Fahr, A. Laufer, R. Klein and W. Braun, J. Phys. Chem., 1990, 95, 3218–3224.
- R. P. Thorn, W. A. Payne, L. J. Stief and D. C. Tardy, J. Phys. Chem., 1996, 100, 13594–13602 CrossRef CAS.
- J. C. Robinson, N. E. Sveum and D. M. Neumark, J. Chem. Phys., 2003, 119, 5311–5314 CrossRef CAS.
- J. C. Robinson, S. A. Harris, W. Sun, N. E. Sveum and D. M. Neumark, J. Am. Chem. Soc., 2002, 124, 10211–10224 CrossRef CAS.
- H.-Y. Lee, V. V. Kislov, S.-H. Lin, A. M. Mebel and D. M. Neumark, Chem.–Eur. J., 2003, 9, 726–740 CrossRef CAS.
- S. D. Chambreau, J. Lemieux, L. Wang and J. Zhang, J. Phys. Chem. A, 2005, 109, 2190–2196 CrossRef CAS.
- S. M. Handford-Styring and R. W. Walker, Phys. Chem. Chem. Phys., 2001, 3, 2043–2052 RSC.
- S. K. Gulati and R. W. Walker, J. Chem. Soc., Faraday Trans. 2, 1989, 85, 1799–1812 RSC.
- C. Cavallotti, R. Rota, T. Faravelli and E. Ranzi, Proc. Combust. Inst., 2007, 31, 201–209 CrossRef.
- B. Sirjean, F. Buda, H. Hakka, P. A. Glaude, R. Fournet, V. Warth, F. Battin-Leclerc and M. Ruiz-Lopez, Proc. Combust. Inst., 2007, 31, 277–284 CrossRef.
- W. J. Pitz, C. V. Naik, T. N. Mhaoldúin, C. K. Westbrook, H. J. Curran, J. P. Orme and J. M. Simmie, Proc. Combust. Inst., 2007, 31, 267–275 CrossRef.
- J. P. Orme, H. J. Curran and J. M. Simmie, J. Phys. Chem. A, 2006, 110, 114–131 CrossRef CAS.
- F. Buda, B. Heyberger, R. Fournet, P. A. Glaude, V. Warth and F. Battin-Leclerc, Energy Fuels, 2006, 20, 1450–1459 CrossRef CAS.
- E. J. Silke, W. J. Pitz, C. K. Westbrook and M. Ribaucour, J. Phys. Chem. A, 2007, 111, 3761–3775 CrossRef CAS.
- D. Voisin, A. Marchal, M. Reuillon, J. C. Boettner and M. Cathonnet, Combust. Sci. Technol., 1998, 138, 137–158 CAS.
- A. El Bakali, M. Braun-Unkhoff, P. Dagaut, P. Frank, M. Cathonnet, A. M. Dean, P. R. Westmoreland, A. Fridman, A. Lifshitz, F. Dryer and W. H. Green, Proc. Combust. Inst., 2000, 28, 1631–1638 Search PubMed.
- A. M. Knepp, G. Meloni, L. E. Jusinski, C. A. Taatjes, C. Cavallotti and S. J. Klippenstein, Phys. Chem. Chem. Phys., 2007, 9, 4315–4331 RSC.
- N. E. Sveum, S. J. Goncher and D. M. Neumark, Phys. Chem. Chem. Phys., 2005, 8, 592–598 Search PubMed.
- J. C. Robinson, N. E. Sveum and D. M. Neumark, Chem. Phys. Lett., 2004, 383, 601–605 CrossRef CAS.
- C. S. McEnally, L. D. Pfefferle, R. K. Mohammed, M. D. Smooke and M. B. Colket, Anal. Chem., 1999, 71, 364–372 CrossRef CAS.
- W. R. Schwartz, C. S. McEnally and L. D. Pfefferle, J. Phys. Chem. A, 2006, 110, 6643–6648 CrossRef CAS.
- J. H. Werner and T. A. Cool, Combust. Flame, 2000, 120, 125–142 CrossRef CAS.
- J. S. Bernstein, A. Fein, J. B. Choi, T. A. Cool, R. C. Sausa, S. L. Howard, R. J. Locke and A. W. Miziolek, Combust. Flame, 1993, 92, 85–105 CrossRef CAS.
- A. McIlroy, T. D. Hain, H. A. Michelsen and T. A. Cool, Proc. Combust. Inst., 2000, 28, 1647–1653 CrossRef CAS.
- M. A. Blitz, A. Goddard, T. Ingham and M. J. Pilling, Rev. Sci. Instrum., 2007, 78, 034103 CrossRef.
- R. J. Jones, K. D. Moll, M. J. Thorpe and J. Ye, Phys. Rev. Lett., 2005, 94, 193201 CrossRef.
- C. Gohle, T. Udem, M. Herrmann, J. Rauschenberger, R. Holzwarth, H. A. Schuessler, F. Krausz and T. W. Hänsch, Nature, 2005, 436, 234–237 CrossRef CAS.
- R. T. Zinkstok, S. Witte, W. Ubachs, W. Hogervorst and K. S. E. Eikema, Phys. Rev. A, 2006, 73, 061801 CrossRef.
- D. E. Shallcross, S. Vaughan, D. R. Trease, C. E. Canosa-Mas, M. V. Ghosh, J. M. Dyke and R. P. Wayne, Atmos. Environ., 2006, 40, 6899–6904 CrossRef CAS.
- C. J. Percival, D. E. Shallcross, C. E. Canosa-Mas and J. M. Dyke, J. Photochem. Photobiol., A, 2005, 176, 250–259 CrossRef CAS.
- J. M. Dyke, M. V. Ghosh, D. J. Kinnison, G. Levita, A. Morris and D. E. Shallcross, Phys. Chem. Chem. Phys., 2005, 7, 866–873 RSC.
| This journal is © the Owner Societies 2008 |