Photo-induced proton-transfer cycle of 2-naphthol in faujasite zeolitic nanocavities
Received
22nd August 2007
, Accepted 8th October 2007
First published on 24th October 2007
Abstract
The excited-state deprotonation and ground-state reprotonation of a 2-naphthol molecule encapsulated in the zeolitic nanocavity of NaX have been studied by measuring static and time-resolved spectra of fluorescence and reflectance. The excited molecule undergoes enol dissociation within 300 ps to form an isolated ion pair, which undergoes geminate recombination in 1200 ps or separation to produce the anionic species of 2-naphtholate on the time scale of 2500 ps. Ground-state reprotonation, controlled by the diffusion rate of a proton, is then followed in 0.8 ms with an activation energy of 13 kJ mol−1.
1. Introduction
Intense interest has been focused on zeolites since they can be widely used from petroleum cracking to fine chemical synthesis.1–4 The ordered cages and channels of zeolites are potential hosts to organic and inorganic complexes, and act as nano-sized reactors in the synthesis of nanoclusters and nanowires5,6 because the properties of encapsulated guest species can be altered by varying the dielectric and charge properties of the nanoporous hosts.7–9 In this regard, arene molecules have been encapsulated into zeolitic nanocavities for several decades10–12 since the uniform pores of molecular dimensions can be compared with solvent cages. The catalytic properties of the zeolites are usually related to acid/base properties in their well-organized channels and nanocavities.13,14 Many catalytic reactions in zeolitic nanoreactors are known to begin with proton transfers at Brønsted acid/base sites.15 The acid and base sites of zeolitic nanocages are able to release and accept protons, respectively, by interacting with adsorbed guest molecules. Although proton transfers in zeolites have been reported,15,16 their dynamics and mechanism are barely understood yet. The strength, the concentration, and the nature of the active sites in acid/base catalyzed reactions have been extensively studied.13,17 Acid/base properties of zeolites are known to be dependent on the Si/Al ratio as well as on preparation and treatment methods, dehydration temperature, and zeolite structure. The acidity of a zeolite increases as the ratio of Si/Al increases, as protons replace metal cations, or as exchanged metal cations have a higher charge density.13,17
Proton transfer has been attracting considerable attention because it plays a key role in a wide variety of chemical and biological phenomena.18–22 Recent interest is focused on intrinsic proton transfer along the hydrogen-bonded bridge of 7-hydroxyquinoline or 7-azaindole complexed cyclically with a protic solvent in a nonpolar solvent or in the gas phase.18–21 The proton-transfer dynamics is determined by the size, structure and motion of a solvent cluster as well as by the nature of a prototropic group,23–30 whereas the rate of proton migration is controlled by solvent structural dynamics.31–33 The dynamics and the mechanism of excited-state deprotonation (ESD) have often been investigated with photoacids.34–39Proton-transfer studies employing various naphthol derivatives have indicated that the apparent sizes of the accepting water clusters also depend on the acidity of the proton donors.36 The solvatochromism and the triplet-state dynamics of 1-naphthol and 2-naphthol have also been studied.40–422-Naphthol having pKa of 9.5 and pKa* of 2.8 can be a good candidate for the investigation of zeolitic catalysis because it has a high fluorescence quantum yield, a unique and simple chemical structure, and strong photoacidity.43,44 Its great photoacidity is largely attributed to electron migration from its hydroxyl group to its naphthalene ring in polar media.34 Whereas its ESD rate is comparable to the rates of its fluorescence and radiationless decay in water,36–38 it is too slow to be observed in small solvent clusters.37
In this paper, we report on the dynamics of the ESD and the ground-state reprotonation (GSR) of 2-naphthol taking place in the nanoreactors of faujasite zeolites (Scheme 1). We have employed zeolitic nanocavities as complements to solvent cages to understand microenvironmental effects on ESD. As the acid/base properties of zeolites play an important role in zeolitic catalysis, interactions of zeolitic acid and base sites with the phenolic group of 2-naphthol exhibit interesting phenomena such as prototropic equilibria, photon-induced proton transfers, and cage effects. We have found that the enol dissociation of 2-naphthol to form an isolated ion pair is followed slowly by the separation of the ion pair to complete ESD, whereas the rate of GSR is controlled by the diffusion rate of a proton.
 |
| | Scheme 1 | |
2. Experimental
Materials
2-Naphthol, purchased from Merck, was used after vacuum sublimation. The NaX and NaY zeolites of high purity were synthesized following known procedures.45H+-exchanged Y (HY) was prepared by ion-exchanging NaY conventionally with NH4Cl (aq) at 350 K for 3 h two times. A Pyrex tube containing NaX or HY was jointed with a vacuum port and evacuated at 670 K for 5 h. The typical unit-cell formulae of dried NaX and HY were found to be Na86(AlO2)86(SiO2)106·264H2O and H56(AlO2)56(SiO2)136·250H2O, respectively.46 Dried NaX or HY was mixed with a known amount of 2-naphthol in a quartz tube under N2 atmosphere at room temperature in order to have a desired loading level of 0.5 molecule per supercage. The tube was evacuated, sealed, maintained at 570 K for 3 h, and cooled slowly to room temperature to obtain a uniform distribution. The intercalation of guest molecules into zeolitic supercage was confirmed with 129Xe NMR spectroscopy.7,8X-ray diffraction was also used to confirm that the zeolitic framework did not change during ion exchange, dehydration, and intercalation.
Diffuse reflectance and emission
All the static and time-resolved spectroscopic measurements were carried out at room temperature. A homemade spectrometer was used to measure diffuse-reflectance. The wavelength of a tungsten/deuterium lamp was selected using a 0.25 m monochromator (Kratos, GM252) and a sample was mounted at the tangent of an internally MgO-coated integrating sphere. The diffuse reflectance of the sample with the rejection of specular reflection was detected at a right angle from the illuminating beam using a photomultiplier tube (PMT, Hamamatsu, R374). For the measurements of emission spectra, the wavelength of a 75 W Xe lamp (Acton Research, XS 432) was selected using a 0.15 m monochromator (Acton Research, Spectrapro 150) to excite sample while the emission wavelength was selected with a 0.30 m monochromator (Acton Research, Spectrapro 300) attached with a PMT (Acton Research, PD438). The wavelength-dependent sensitivity of the emission spectrometer was not corrected.
Transient reflectance
The time-resolved spectra of transient reflectance profiles were obtained by monitoring the beam intensity of a 75 W Xe lamp (Acton Research, XS 432) after being reflected from a sample, which was excited with 320 nm pulses from a dye laser (Quantel, TDL90) pumped by a Q-switched Nd:YAG laser (Quantel, YG980), by using an intensified CCD (Princeton Instruments, ICCD-576-G) attached to a 0.5 m spectrometer (Acton Research, SpectroPro-500). For the measurements of transient-reflectance kinetic profiles, the probe beam was wavelength-selected by using a 0.25 m monochromator (Kratos, GM252), a 0.2 m double monochromator (Kratos, GM200), and a combination of optical filters, detected by using a PMT (Hamamatsu, R376), and recorded by using a 200 MHz oscilloscope (Tektronix, TDS350). The laser and the CCD or the oscilloscope were triggered with variable delays using a pulse/delay generator (Stanford Research Systems, DG535).
Time-resolved fluorescence
Fluorescence kinetic profiles were measured by using a 10 ps streak camera (Hamamatsu, C2830) attached with a CCD detector (Princeton Instruments, RTE-128-H) and an actively/passively mode-locked Nd:YAG laser (Quantel, YG701). Samples were excited with pulses of 315 nm generated from a Raman shifter, filled with CH4 gas of 15 atm and pumped by the fourth-harmonic pulses of 266 nm and 25 ps from the laser. Fluorescence and transient-reflectance kinetic constants were extracted by fitting measured kinetic profiles to computer-simulated kinetic curves convoluted with instrument temporal response functions.
3. Results and discussion
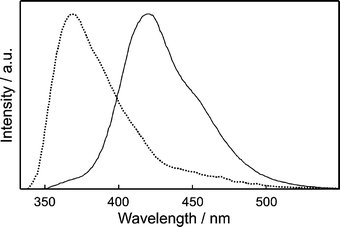
Fig. 1 shows that the diffuse reflectance spectra of 2-naphthol incorporated in HY and NaX nanocavities are quite different from each other. While the spectrum in HY shows only the normal absorption band of 2-naphthol (ROH) with the maximum at 322 nm, that in NaX shows an additional shoulder band at 363 nm as well, arising from the anionic species of 2-naphtholate (RO−). 2-Naphthol exists as the dominant species in the acidic zeolite of HY, H+-exchanged Y having a Si/Al ratio of 2.43. However, the anionic species is also considerably present with 2-naphthol in NaX because the zeolite having a Si/Al ratio of 1.23 has many basic sites to form ground-state stable 2-naphtholate. Considering that both prototropic species are significantly present at the ground states, we suggest that adsorbed molecules in NaX nanocavities have two very different microsurroundings in the strength of basicity.47
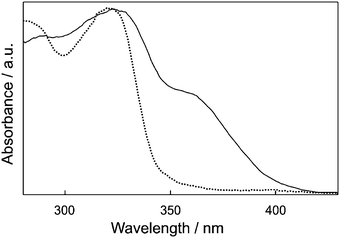 |
| | Fig. 1 Diffuse-reflectance spectra of 2-naphthol encapsulated in the nanocavities of HY (dotted) and NaX (solid), measured using bare HY and bare NaX, respectively, as the references. | |
3.3 Fluorescence kinetic profiles
On one hand, the fluorescence of 2-naphthol-adsorbed NaX at 360 nm rises instantly and decays biexponentially on the time scales of 300 and 2500 ps (Fig. 3 and Table 1). On the other hand, the fluorescence at 500 nm rises biexponentially on the time scales of 150 and 2500 ps. These facts designate that upon absorption of photons, 2-naphthol molecules encapsulated in NaX nanocavities go through the dissociation of the enol group within 300 ps to form an isolated ion pair and the subsequent separation of the ion pair in 2500 ps to complete ESD. The rise time of the fast component at 500 nm was observed to be shorter than the decay time of the fast component at 360 nm. We attribute the shortening of rise time at 500 nm to the geminate recombination of the charge-separated ion pair of RO−⋯H+. Using the time constants of 300 and 150 ps with the separation time (2500 ps) of the ion pair, we have estimated the geminate recombination time of the RO−⋯H+ to be 1200 ps, which is rather long due to the basicity of NaX. The kinetic constants observed in a short time window of 2 ns have been found to fit well to the kinetic profiles measured in a long time window of 12 ns. The excited state of 2-naphtholate decays on the time scale of 4.7 ns (Table 1), which is significantly shorter than 9 ns in water37 owing to enhanced intersystem crossing in zeolitic nanocavities.
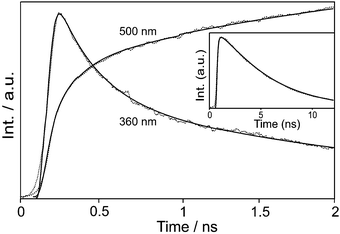 |
| | Fig. 3 Fluorescence kinetic profiles of 2-naphthol adsorbed in NaX nanocavities, excited at 315 nm and monitored at indicated wavelengths. Inset: Profile monitored at 500 nm in a long time window to show decay. Solid curves were computer-simulated using kinetic constants shown in Table 1. | |
Table 1 Fluorescence kinetic constants of 2-naphthol intercalated in NaX nanocavities, deconvoluted from the fluorescence kinetic profiles of Fig. 3
|
λ
mon
a/nm |
Rise time/ps |
Decay time/ps |
|
Monitored wavelength.
Initial fluorescence percentage of each component.
|
| 360 |
Instant |
300 (60%)b+ 2500 (40%) |
| 500 |
150 (53%)+2500 (47%) |
4700 |
Effectively no excited-state deprotonation is reported37,38 to occur for 2-naphthol in highly acidic aqueous solutions at pH < 1. However, in near-neutral aqueous solutions, deprotonation taking place with a rate constant of 1.9 × 108 s−1 is followed by reprotonation reversibly at the excited state.35 Furthermore, the deprotonation of 2-naphthol in other caging media such as AOT, protein pockets, and β-cyclodextrin has been reported to be even slower than that in water.48 The ESD of 2-naphthol molecules adsorbed in the supercages of NaX is extremely different from those in water and other caging media because Brønsted acid/base sites are heterogeneously present in the nanocages.49 The fast enol-dissociation time of 300 ps suggests that 2-naphthol molecules adsorbed in NaX nanopores are hydrogen-bonded to oxygen atoms having strongly basic properties. Upon excitation, the molecules undergo enol dissociation within 300 ps to form an isolated ion pair which separates subsequently on the time scale of 2500 ps to produce the anionic species of 2-naphtholate. The zeolitic framework of NaX dried at 670 K consists of many basic sites so that H-bonding required for the deprotonation of the phenolic group can be made by the slight rearrangement of the zeolitic framework or the adsorbed molecule. The introduction of a strong-base group induces the excited-state acidity of 2-naphthol to be very strong, allowing most molecules in zeolitic nanoreactors to undergo ESD facilely within their excited-state lifetimes.
The time-resolved spectra (Fig. 4) and kinetic profiles (Fig. 5) of transient reflectance show at least three observable transients for 2-naphthol encapsulated in NaX nanocages. The bleach band at 330 nm, owing to the depopulation of 2-naphthol, recovers with two components of 0.8 (94%) and 4 (6%) ms while the transient absorption band at 410 nm rises in 0.1 ms and decays on the time scales of 0.8 (99%) and 4 (1%) ms (Table 2). Finally, the transient absorption having a maximum at 530 nm decays biphasically with the time constants of 0.1 (80%) and 4 (20%) ms. We thus assigned the fast and the slow components of transient-absorption decay at 530 nm to the triplet-state absorption decay of 2-naphtholate and 2-naphthol, respectively. Our observed decay time of 0.1 ms is much longer than that of 0.01 ms for the lowest triplet state of the same anionic species in water.42 We attribute the slow relaxation of the triplet state mainly to the collision-free environment of the anionic species in the dried nanocavity of NaX. The triplet state of 2-naphthol decays even much more slowly than that of the anionic species because of its large energy gap to the ground state50 and its increased ππ* character.51 Note that the triplet-state transient absorption of 2-naphthol monitored at 4 ms is shifted to the blue by 25 nm from that of 2-naphtholate with a maximum at 523 nm, monitored at 0.03 ms (Fig. 4). Transient absorption at 410 nm is ascribed to the absorption of the anionic species phototransformed from 2-naphthol. Thus, we assert that the reprotonation of the anionic species takes place on the time scale of 0.8 ms. The reprotonation time in NaX, which is known to be slightly basic, is slightly shorter than that of 1 ms in neutral water.41
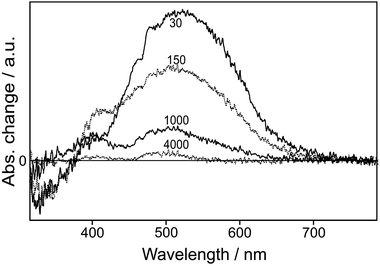 |
| | Fig. 4 Time-resolved transient-reflectance spectra of 2-naphthol encapsulated in NaX nanopores, measured at time delays given in microseconds after excitation at 320 nm. | |
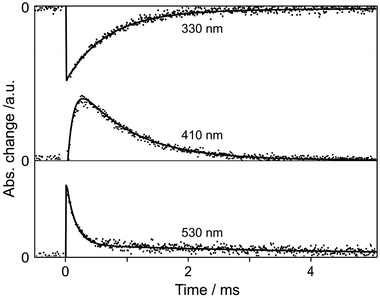 |
| | Fig. 5 Transient-reflectance kinetic profiles of 2-naphthol encapsulated in NaX supercages, monitored at shown wavelengths after excitation at 320 nm. The solid curves were computer-simulated using the kinetic constants of Table 2. | |
Table 2 Transient-absorption kinetic constants, deconvoluted from the transient-reflectance kinetic profiles of Fig. 5, of 2-naphthol in NaX supercages
|
λ
mon/nm |
Rise time/ms |
Decay time/ms |
|
Wavelength giving transient bleach instead of transient reflectance.
Initial absorption percentage of each component.
The Arrhenius plot of the inverse of this time, presented in Fig. 6, gives 13 kJ mol−1 and 2.4 × 105 s−1 for the activation energy and the pre-exponential factor, respectively.
|
| 330a |
Fast |
0.8 (94%)b+ 4 (6%) |
| 410 |
0.1 |
0.8 (99%)c+ 4 (1%) |
| 530 |
Fast |
0.1 (80%)+ 4 (20%) |
3.5 Arrhenius plot
Fig. 5 and Table 2 have shown that the reprotonation rate of the anionic species is faster in collectively basic NaX than in neutral water. This hints that protons diffuse from acid sites to reprotonate the anionic species. In order to check this idea, we have monitored the temperature dependence of the reprotonation-rate constants. The small pre-exponential factor of 2.4 × 105 s−1(Fig. 6 and Table 2) also supports the restrictively collisional environment of the anionic species in NaX. The observed activation energy of 13 kJ mol−1 is in the range of 12–15 kJ mol−1 reported for the activation energy of H+ diffusion in water,52 indeed as already speculated with the result of Fig. 5. We have also tried to plot the kinetics at 410 nm in Fig. 5 on a semilog scale in order to check diffusion.53 Although the fast component of 0.8 ms is interfered by the slow component of 4 ms, it really shows the temporal behavior of diffusion by the Smoluchowski theory.54 In zeolite NaX, voids of small β-cages and large supercages are found.13,17 Since diffusion within zeolites is thought to proceed by thermally activated jumps from one site to an adjacent site, the geometry of the zeolite pores is also considered to play an important role. However, the size of a proton is very small relative to that of a pore or 2-naphtholate. Therefore, the rate of protonation is determined by the diffusion rate of protons, as it was found that the activation energy estimated from Fig. 6 is in the range of the activation energy of H+ diffusion.52
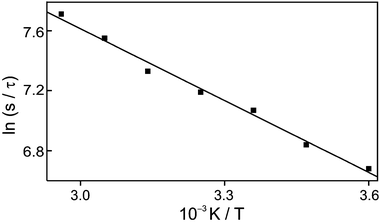 |
| | Fig. 6 Arrhenius plot for the decay rate constant of transient reflectance, monitored at 410 nm, of 2-naphthol encaged in NaX nanocavities. | |
3.6 Summary scheme
Fig. 7 summarizes the mechanism that a photon drives a 2-naphthol molecules encaged in the supercages of zeolite NaX to undergo a proton transfer cycle involving excited and ground states. Deprotonation at S1 is completed by charge separation following enol dissociation whereas reprotonation at S0 is controlled by the diffusion rate of a proton. Upon absorption of a photon a 2-naphthol molecule goes through enol dissociation within 300 ps to form RO−⋯H+. The ion pair undergoes geminate recombination in 1200 ps or charge separation on the time scale of 2500 ps. The T1 states of 2-naphthol and 2-naphtholate, populated via intersystem crossing within the lifetime of their respective S1 states, relax nonradiatively via intersystem crossing to their respective S0 states in 4 and 0.1 ms, respectively. The dominant fraction of photobleached ground-state 2-naphthol is replenished by the reprotonation of 2-naphtholate at S0 on the time scale of 0.8 ms with the diffusional activation energy of 13 kJ mol−1.
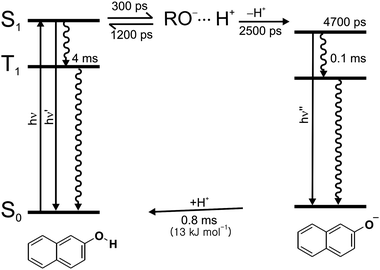 |
| | Fig. 7 Schematic representation of the excited-state deprotonation dynamics of 2-naphthol and the ground-state reprotonation dynamics of 2-naphtholate in NaX nanocavities. | |
4. Conclusions
We have presented a detailed microscopic picture of the photo-induced proton-transfer cycle of 2-naphthol adsorbed in faujasite zeolitic nanocavities. We have characterized the adsorption states of 2-naphthol on heterogeneous zeolite surfaces and discriminated reaction intermediates as well as reaction pathways and rates by measuring diverse time-resolved and static spectra. The dynamics of ESD is governed by the initial microenvironment of molecules whereas that of relaxation at the lowest triplet state and GSR is determined by the overall properties of the hosting zeolite. Excited-state 2-naphthol encapsulated in a faujasite zeolitic nanocavity undergoes enol dissociation within 300 ps. The dissociated ion pair recombines geminately in 1200 ps or subsequent ion separation in 2500 ps to complete ESD. The lowest triplet states of 2-naphthol and 2-naphtholate relax in 4 and 0.1 ms, respectively. The dominant fraction of photobleached 2-naphthol is replenished by the reprotonation of 2-naphtholate at the ground state on the time scale of 0.8 ms with the diffusional activation energy of 13 kJ mol−1.
Acknowledgements
This work was financially supported by a Korean Research Foundation Grant (KRF-2004-015-C00230). Y.S.L. also acknowledges the scholarship of the BK 21 Program.
References
- M. Choi, H. S. Cho, R. Srivastava, C. Venkatesan, D.-H. Choi and R. Ryoo, Nat. Mater., 2006, 5, 718 CrossRef CAS.
- H. Frei, Science, 2006, 313, 309 CrossRef CAS.
- J. S. Lee, Y.-J. Lee, E. L. Tae, Y. S. Park and K. B. Yoon, Science, 2003, 301, 818 CrossRef CAS.
- Z. Lai, G. Bonilla, I. Diaz, J. G. Nery, K. Sujaoti, M. A. Amat, E. Kokkoli, O. Terasaki, R. W. Thompson, M. Tsapatsis and D. G. Vlachos, Science, 2003, 300, 456 CrossRef CAS.
- W.-S. Chae, J.-H. Yoon, H. Yu, D.-J. Jang and Y.-R. Kim, J. Phys. Chem. B, 2004, 108, 11509 CrossRef CAS.
- A. Goldbach and M.-L. Saboungi, Acc. Chem. Res., 2005, 38, 705 CrossRef CAS.
- O.-H. Kwon, H. Yoo, K. Park, B. Tu, R. Ryoo and D.-J. Jang, J. Phys. Chem. B, 2001, 105, 4195 CrossRef CAS.
- O.-H. Kwon, H. Yu and D.-J. Jang, J. Phys. Chem. B, 2004, 108, 3970 CrossRef CAS.
- J.-S. Lee, S. H. Joo and R. Ryoo, J. Am. Chem. Soc., 2002, 124, 1156 CrossRef CAS.
- D. Lesthaeghe, B. De Sterck, V. Van Speybroeck, G. B. Marin and M. Waroquier, Angew. Chem., Int. Ed., 2007, 46, 1311 CrossRef CAS.
- I. Daems, A. Méthivier, P. Leflaive, A. H. Fuchs, G. V. Baron and J. F. M. Denayer, J. Am. Chem. Soc., 2005, 127, 11600 CrossRef CAS.
- S. Mintova, V. D. Waele, U. Schmidhammer, E. Riedle and T. Bein, Angew. Chem., Int. Ed., 2003, 42, 1611 CrossRef CAS.
- R. A. van Santen and G. J. Kramer, Chem. Rev., 1995, 95, 637 CrossRef CAS.
- H. M. Kao, H. M. Liu, J. C. Jiang, S. H. Lin and C. P. Grey, J. Phys. Chem. B, 2000, 104, 4923 CrossRef CAS.
- T. N. Truong, J. Phys. Chem. B, 1997, 101, 2750 CrossRef CAS.
- J. T. Fermann and S. M. Auerbach, J. Phys. Chem. A, 2001, 105, 2879 CrossRef CAS.
- A. Corma, Chem. Rev., 1995, 95, 559 CrossRef CAS.
- O.-H. Kwon, Y.-S. Lee, B. K. Yoo and D.-J. Jang, Angew. Chem., Int. Ed., 2006, 45, 415 CrossRef CAS.
- O.-H. Kwon, Y.-S. Lee, H. J. Park, Y. Kim and D.-J. Jang, Angew. Chem., Int. Ed., 2004, 43, 5792 CrossRef CAS.
-
(a) C. Tanner, C. Manca and S. Leutwyler, Science, 2003, 302, 1736 CrossRef CAS;
(b) C. Tanner, M. Thut, A. Steinlin, C. Manca and S. Leutwyler, J. Phys. Chem. A, 2006, 110, 1758 CrossRef CAS.
-
(a) K. Sakota, Y. Komoto, M. Nakagaki, W. Ishikawa and H. Sekiya, Chem. Phys. Lett., 2007, 435, 1 CrossRef CAS;
(b) K. Sakota, N. Inoue, Y. Komoto and H. Sekiya, J. Phys. Chem. A, 2007, 111, 4596 CrossRef CAS.
- A. J. Kirby, Acc. Chem. Res., 1997, 30, 290 CrossRef CAS.
- L. M. Tolbert and K. M. Solntsev, Acc. Chem. Res., 2002, 35, 19 CrossRef CAS.
- K. M. Solntsev, L. M. Tolbert, B. Cohen, D. Huppert, Y. Hayashi and Y. Feldman, J. Am. Chem. Soc., 2002, 124, 9046 CrossRef.
- N. Agmon, J. Phys. Chem. A, 2005, 109, 13 CrossRef CAS.
- K. M. Solntsev, C. E. Clower, L. M. Tolbert and D. Huppert, J. Am. Chem. Soc., 2005, 127, 8534 CrossRef CAS.
- H. Yu, O.-H. Kwon and D.-J. Jang, J. Phys. Chem. A, 2004, 108, 5932 CrossRef CAS.
- O.-H. Kwon, H. Doo, Y.-S. Lee and D.-J. Jang, ChemPhysChem, 2003, 4, 1079 CrossRef CAS.
- O.-H. Kwon, T.-G. Kim, Y.-S. Lee and D.-J. Jang, J. Phys. Chem. B, 2006, 110, 11997 CrossRef CAS.
-
(a) O.-H. Kwon and D.-J. Jang, J. Phys. Chem. B, 2005, 109, 8049 CrossRef CAS;
(b) O.-H. Kwon and D.-J. Jang, J. Phys. Chem. B, 2005, 109, 20479 CrossRef CAS.
- D. Marx, M. E. Tuckerman, J. Hutter and M. Parrinello, Nature, 1999, 397, 601 CrossRef CAS.
- M. E. Tuckerman, D. Marx, M. L. Klein and M. Parrinello, Science, 1997, 275, 817 CrossRef CAS.
- T. G. Kim, Y. Kim and D.-J. Jang, J. Phys. Chem. A, 2001, 105, 4328 CrossRef CAS.
- N. Agmon, W. Rettig and C. Groth, J. Am. Chem. Soc., 2002, 124, 1089 CrossRef CAS.
- B. Cohen, P. Leiderman and D. Huppert, J. Phys. Chem. A, 2002, 106, 11115 CrossRef CAS.
- L. M. Tolbert and J. E. Haubrich, J. Am. Chem. Soc., 1994, 116, 10593 CrossRef CAS.
- W. R. Laws and L. Brand, J. Phys. Chem., 1979, 83, 795 CrossRef CAS.
- S. P. Webb, S. W. Yeh, L. A. Philips, M. A. Tolbert and J. H. Clark, J. Am. Chem. Soc., 1984, 106, 7286 CrossRef CAS.
- T. G. Fillingim, N. Luo, J. Lee and G. W. Robinson, J. Phys. Chem., 1990, 94, 6368 CrossRef CAS.
- K. M. Solntsev, D. Huppert and N. Agmon, J. Phys. Chem. A, 1998, 102, 9599 CrossRef CAS.
- M. Gutman, E. Nachliel, E. Gershon, R. Giniger and E. Pines, J. Am. Chem. Soc., 1983, 105, 2210 CrossRef CAS.
- U. Lachishi, M. Ottolenghi and G. Stein, Chem. Phys. Lett., 1977, 48, 402 CrossRef CAS.
- G. A. Brucker and D. F. Kelley, J. Chem. Phys., 1989, 90, 5243 CrossRef CAS.
- C. Clower, K. M. Solntsev, J. Kowalik, L. M. Tolbert and D. Huppert, J. Phys. Chem. A, 2002, 106, 3114 CrossRef CAS.
-
D. W. Breck, Zeolite Molecular Sieve: Structure, Chemistry, and Use, John Wiley and Sons, New York, 1974, ch. 4 Search PubMed.
- A. Di Lella, N. Desbiens, A. Boutin, I. Demachy, P. Ungerer, J.-P. Bellat and A. H. Fuchs, Phys. Chem. Chem. Phys., 2006, 8, 5396 RSC.
- D. Murphy, P. Massiani, R. Frank and D. Barthomeuf, J. Phys. Chem., 1996, 100, 6731 CrossRef CAS.
-
(a) H.-R. Park, B. Mayer, P. Wolschann and G. Köhler, J. Phys. Chem., 1994, 98, 6158 CrossRef CAS;
(b) M. H. Kleinman, J. H. Flory, D. A. Tomalia and N. J. Turro, J. Phys. Chem. B, 2000, 104, 11472 CrossRef CAS;
(c) M. Gutman, Methods Enzymol., 1986, 127, 522 CAS;
(d) E. Bardez, E. Monnier and B. Valeur, J. Phys. Chem., 1985, 89, 5031 CrossRef CAS.
- S. R. Blaszkowski and R. A. van Santen, J. Phys. Chem., 1995, 99, 11728 CrossRef CAS.
- H. Fidder, M. Rini and E. T. J. J. Nibbering, J. Am. Chem. Soc., 2004, 126, 3789 CrossRef CAS.
-
N. J. Turro, Modern Molecular Photochemistry, University Science Book, Mill Valley, CA, 1991 Search PubMed.
-
K. J. Laidler, Chemical Kinetics, Harper & Row Publishers, New York, 3rd edn, 1987, ch. 6 Search PubMed.
-
(a) O. F. Mohammed, D. Pines, E. T. J. Nibbering and E. Pines, Angew. Chem., Int. Ed., 2007, 46, 1458 CrossRef CAS;
(b) D. Pines and E. Pines, J. Chem. Phys., 2001, 115, 951 CrossRef CAS;
(c) E. Pines, B.-Z. Magnes and T. Barak, J. Phys. Chem. A, 2001, 105, 9674 CrossRef CAS.
- I. V. Gopich, K. M. Solntsev and N. Agmon, J. Chem. Phys., 1999, 110, 2164 CrossRef CAS.
Footnotes |
| † Present address: Korea Research Institute of Standards and Science, Daejeon 305-600, Korea. |
| ‡ Present address: A. A. Noyes Laboratory of Chemical Physics, California Institute of Technology, Pasadena, California, 91125, USA. |
|
| This journal is © the Owner Societies 2008 |
Click here to see how this site uses Cookies. View our privacy policy here.