Reactive uptake of acetic acid on calcite and nitric acid reacted calcite aerosol in an environmental reaction chamber
Amy Preszler
Prince
c,
Paul D.
Kleiber
b,
Vicki H.
Grassian
*ac and
Mark A.
Young
*c
aDepartments of Chemical and Biochemical Engineering, The University of Iowa, Iowa City, IA 52242, USA
bDepartments of Physics and Astronomy, The University of Iowa, Iowa City, IA 52242, USA
cDepartments of Chemistry, The University of Iowa, Iowa City, IA 52242, USA
First published on 21st November 2007
Abstract
The heterogeneous chemistry of gas-phase acetic acid with CaCO3(calcite) aerosol was studied under varying conditions of relative humidity (RH) in an environmental reaction chamber. Infrared spectroscopy showed the loss of gas-phase reactant and the appearance of a gaseous product species, CO2. The acetic acid is observed to adsorb onto the calcite aerosol through both a fast and a slow uptake channel. While the fast channel is relatively independent of RH, the slow channel exhibits enhanced uptake and reaction as the RH is increased. In additional experiments, the calcite aerosol was exposed to both nitric and acetic acids in the presence of water vapor. The rapid conversion of the particulate carbonate to nitrate and subsequent deliquescence significantly enhances the uptake and reaction of acetic acid. These results suggest a possible mechanism for observed correlations between particulate nitrate and organic acids in the atmosphere. Calcium rich mineral dust may be an important sink for simple organic acids.
1. Introduction
The chemical composition of organic species in the atmosphere is extremely complex and, in addition to methane, includes aliphatic chains, alcohols, aromatics, phenols and organic acids, amongst many others. A significant portion of the non-methane hydrocarbon species, approximately 25%, is comprised of organic acids.1 The two most abundant organic acids are formic acid, HCOOH, and acetic acid, CH3COOH (HAc). These acids are often discussed together in the literature as field measurements have shown that their mixing ratios are correlated, although their relative concentrations exhibit marked diurnal and seasonal variations.1–5 While the field data suggest common sources and sinks for both species, a clear understanding of the relevant reaction pathways for these simple organic acids is currently lacking.1,4,6Known anthropogenic sources of atmospheric organic acids include direct emission from motor vehicles7,8 and biomass burning.8,9 Primary biogenic emissions from plants5,10 and soil microbes11–13 are known to be sufficient to influence global fluxes. Secondary production from the photochemical oxidation of atmospheric hydrocarbons, of both biogenic and anthropogenic origin, is also a major source of organic acids. Photochemical reaction mechanisms leading to the formation of formic and acetic acids have been discussed in the literature.1,4,6,14–18
Acetic acid has been detected in the atmosphere as a gas-phase species, in rain and fog water, and in association with particulate matter. Gas-phase measurements of acetic acid concentrations are highly variable and depend on geographic location, season, and time of day1,5,6,19 A major removal mechanism for atmospheric acetic acid is via wet deposition. Acetic acid is readily soluble in water, exhibiting a large Henry’s law constant,4 8800 M atm−1, and is observed in precipitation samples worldwide.1,6,8,19–25 While a great deal of focus has been given to inorganic acids in rainwater, acetic and formic acids can contribute approximately 25% of the free acidity in the United States and greater than 60% in remote regions.24,26
An alternative loss pathway involves dry deposition of acetic acid to particulates and surfaces. In a 1989 study, Grosjean estimated that 91% of the total budget for acetic acid deposition in the Southern California air basin was through dry deposition.27 However, it was also pointed out that the estimates for removal fluxes are greater than those for production by a factor of two to four and it is unclear as to whether the discrepancy can be ascribed to overestimation of the deposition rates or if the corresponding rates of production were being underestimated. This study,27 and others,8,28,29 indicate that the dry deposition pathways for acetic acid are not well understood.
There has been some work addressing acetic and formic acid adsorption on transition metals as these systems have proven to be ideal for fundamental surface studies.30–35 Investigations of organic vapor adsorption on mineral surfaces at varying RH values have concluded that for most organics the interaction is dominated by adsorption to the surface of the water film present under ambient conditions but that uptake decreases with increasing RH.36–39 However, these results may not be relevant for small polar organic compounds such as acetic acid. Several studies involving organic acids have focused on adsorption from the aqueous phase to a solid mineral surface, representative of processes occurring in soils and sediments.40–43 Experiments on the dissolution kinetics of calcite in acidic solutions have also been performed44,45 and a recent study by Sabbioni et al. analyzed small organic anions in association with damage to monuments and buildings.29
A more recent and relevant investigation from our laboratory by Carlos-Cuellar et al. measured the uptake coefficients for representative organic species, including acetic acid, on model mineral compounds under dry conditions.46 Comparison of the measured uptake coefficients for acetic acid to the known rates for homogeneous photolytic loss pathways shows that heterogeneous uptake can be competitive with photolysis.46 As discussed by Carlos-Cuellar et al., it is suggested that an increase in relative humidity will increase the acetic acid uptake coefficient.46 Al-Hosney et al. have conducted similar studies of formic acid reactivity with carbonate.47 However, more laboratory studies addressing the heterogeneous reactions of acetic acid with environmental surfaces, such as mineral aerosols, under relevant RH and temperature conditions are clearly still needed as significant questions remain concerning the atmospheric cycling of acetic acid.
Organic acids in the atmosphere have been correlated with mineral aerosol and nitrate in field studies. During the Atlanta SuperSite Project, Lee and co-workers analyzed 380![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 single aerosol particles in the 0.35–2.5 μm size range using laser mass-spectrometry.28 Approximately 40% of the particles analyzed contained fragments associated with organic acids, such as formic and acetic acid. Nitrate and oxidized organic species were found to be more often present in the larger particles measured.48 The most common particulates in the size range studied contained a mixture of organics and sulfate. However, mineral aerosols comprised of aluminosilicates, as well as other metal oxides, were also present. The majority of the mineral aerosols (95%) also had contributions from organic acids, in addition to other water-soluble species such as sulfate (95%) and nitrate (80%).28 Another study by Russell et al. using single particle X-ray spectroscopy has observed correlations between large calcium containing particles and fragments indicative of organic acids.49
000 single aerosol particles in the 0.35–2.5 μm size range using laser mass-spectrometry.28 Approximately 40% of the particles analyzed contained fragments associated with organic acids, such as formic and acetic acid. Nitrate and oxidized organic species were found to be more often present in the larger particles measured.48 The most common particulates in the size range studied contained a mixture of organics and sulfate. However, mineral aerosols comprised of aluminosilicates, as well as other metal oxides, were also present. The majority of the mineral aerosols (95%) also had contributions from organic acids, in addition to other water-soluble species such as sulfate (95%) and nitrate (80%).28 Another study by Russell et al. using single particle X-ray spectroscopy has observed correlations between large calcium containing particles and fragments indicative of organic acids.49
In the current study, we report on our exploration of the reactivity of the organic acid, HAc, towards calcium carbonate (calcite) aerosol, a model mineral dust component. Experiments were performed in an environmental reaction chamber that allowed the effects of varying RH to be assessed while the dust sample was aerosolized, maintaining isolated particle conditions. Reactivity was determined through the use of IR absorption spectroscopy. In addition to studying the effects of RH on the uptake and reaction of HAc, the interaction of acetic acid with processed mineral dust aerosol was investigated using mixed samples of HAc and HNO3 in the presence of water vapor.
2. Experimental
The experimental apparatus and techniques used in the current study have been discussed in detail in previous publications.50–52 Briefly, the environmental reaction chamber has an internal volume of 151 L and almost all of the exposed internal surfaces have either been coated with Teflon or are fabricated from glass or Teflon, rendering the chamber relatively inert. The chamber can be evacuated and filled with reagent gases from an attached sample vacuum line. Buffer gas is provided by air from a commercial purge gas generator which is initially very dry, <1% RH, but can be humidified to the desired RH using a water bubbler. The chamber is usually filled with the gases of interest at the selected RH and brought to near atmospheric pressure with the purge gas. The mixture is allowed to sit in the chamber for a period of hours to insure passivation and to characterize wall reactions.A known mass of mineral dust is placed in a sample cartridge which is connected by stainless steel tubing to a nozzle with a partial impactor plate positioned at the nozzle exit. The dust is generally held under vacuum for a period of several hours to remove moisture in the sample. When the experiment is to be initiated, a slide valve separating the sample from the main chamber is opened and the sample cartridge is rapidly pressurized by a short pulse of inert gas, forcing the powder through the nozzle and into the chamber. The impactor ensures efficient deagglomeration of the powder and facilitates aerosolization. The pulsed introduction of powder rapidly mixes the chamber contents in less than a minute and marks the initiation of the reaction.
The progress of the chemistry is monitored by absorption spectroscopy using a modified Fourier Transform infrared spectrometer (FTIR) where an external IR beam is directed through a chamber side arm sealed with Ge windows and onto an external mid-band HgCdTe (MCT) detector. The instrument was operated at 8 cm−1 resolution and spectra were averaged for 256 scans, yielding a temporal resolution of approximately 52 s. In addition to following the kinetics and identifying products, IR spectroscopy was used for a Beer’s Law analysis to quantitatively determine the partial pressure of water and other reactant/product species (e.g. HAc, HNO3, CO2) in the chamber. The chamber temperature and total pressure were also recorded during the experiment.
Acetic acid (EMD Chemicals, glacial, 99.9985%) and water (Fischer Chemical, Optima) samples for the bubbler were placed in glass bulbs and then passed though several freeze–pump–thaw cycles before use. Gaseous nitric acid samples were produced in the laboratory by mixing H2SO4(96.0%) and HNO3(79.5%) in a 3∶1 ratio. The resulting solution was then passed through several freeze–pump–thaw cycles. Typical HAc pressures used in these studies ranged from 138–140 mTorr while lower pressures, <10 mTorr, were employed in some experiments. When nitric acid was included as a reagent, its initial partial pressure was approximately 118 ± 2 mTorr, although the pressure just prior to dust introduction had a larger variation due to RH dependent wall loss. The CaCO3 powder was obtained from a commercial source (OMYA, UF). We measured the BET specific surface area as 10.1 ± 0.3 m2 g−1 using a BET apparatus (Quantachrome, Nova 1200 Multipoint). The crystal phase of the calcium carbonate samples was confirmed to be calcite by X-ray powder diffraction. Calcium acetate powder (Alfa Aesar, Puratronic) was also used in one experiment. Typical powder loadings were in the range of 0.7–0.8 mg.
3. Results
The reactive uptake of HAc onto calcite aerosol was studied in the environmental reaction chamber using initial pressures of 138–140 mTorr and varying the RH, as detailed above. Typical results from IR spectroscopic probes of the reaction progress are contained in Fig. 1. For reference, a spectrum of acetic acid only is shown in Fig. 1a. Some absorption features due to HAc are indicated in the figure and others are summarized in Table 1. Bands due to acetic acid dimers were assigned on the basis of pressure dependence studies and literature references.53–56 The spectra of Fig. 1b and c show the results of exposing calcite aerosol to HAc after a reaction time of 1 h under dry conditions, <1% RH, (b), and with moisture present, 53% RH, (c). These spectra have the prominent carbonate band at 1445 cm−1 labeled. Other characteristic particulate carbonate absorptions are listed in Table 1. Both spectra have had gas phase water absorptions subtracted. | ||
| Fig. 1 FTIR spectral data for the acetic acid–CaCO3 system showing a reference spectrum for acetic acid in the reaction chamber, (a), and spectra of acetic acid–CaCO3 mixtures after a reaction time of 1 h under conditions of <1% RH, (b), and 53% RH (c). The initial acetic acid pressure, ≈135 mTorr, is the same in all cases. Gas phase water absorptions have been subtracted from all spectra and the data in (b) and (c) has been offset for clarity. The positions of various bands are indicated. | ||
| Species | Absorptiona/cm−1 | Mode description |
|---|---|---|
| a Frequencies determined from the current work. | ||
| CH3COOH (g, monomer) | 3582 | s (OH) |
| 1790 | s (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) O) |
|
| 1399 | b (CH3) | |
| 1179 | b (COH) | |
| 990 | b (CH3) | |
| HNO3(g) | 3550 | ss (OH) |
| 2995 | as (NO2)+ ss (NO2) | |
| 1708 | as (NO2) | |
| 1316 | ss (NO2) | |
| 887 | ipb (NO2) | |
| CO2(g) | 2349 | as |
| 668 | b | |
| CaCO3(particulate) | 2510 | |
| 1795 | ss + ipb | |
| 1445 (1600–1200) | as | |
| 878 | opb | |
| 713 | ipb | |
| Ca(NO3)2(particulate) | 1436, 1358 | as |
| 1045 | ss | |
| 820 | opb | |
| Ca(CH3COO)2(particulate) | 1551 | as (COO) |
| 1455 | ss (COO) | |
| H2O (a) | 3430 (3660–2900) | ss, as |
| 1645 | b |
As can be observed in the data of Fig. 1, significant HAc remains even after long exposure to calcite aerosol. However, detailed analysis reveals that some of the HAc has been lost due to reactions with the dust. In addition, a clear feature at 2349 cm−1 assigned to CO2 is present and appears to increase in intensity with longer reaction times and elevated RH values. Spectral subtraction of gas phase HAc, water, and particulate carbonate bands shows that carbon dioxide is the only product species that could be identified under these reaction conditions.
The raw IR spectral data indicate that gas-phase acetic acid is lost upon introduction of calcite aerosol into the atmospheric reaction chamber and gaseous CO2 is produced. In order to assess the time-dependent partial pressure of these species, IR spectra are collected over the course of an experiment and analyzed. To monitor HAc, the strong feature at 1790 cm−1 was isolated using a spectral subtraction procedure to remove calcite and water contributions which was necessary due to spectral overlap of the relevant features. The net absorbance (peak height) of the acetic acid band centered at 1790 cm−1 was measured for each spectrum and compared to a previously determined Beer’s Law calibration. The resulting HAc partial pressure, before and after aerosol addition, can be plotted as a function of time. The upper panel of Fig. 2 shows the results for a series of experiments at different RH values. A similar analysis procedure was used to quantify CO2 production through the integrated absorbance of the 2349 cm−1 band and the resultant measurements are summarized in the lower panel of Fig. 2. In order to account for wall reactions, separate experiments at similar acetic acid pressures and RH values but without the calcite aerosol were conducted. A fit to the wall loss data was then used to correct the HAc and CO2 pressures for times, t > 0 (i.e. after the CaCO3 dust has been introduced into the chamber).
 | ||
| Fig. 2 Partial pressure of reactant acetic acid, (A), and product CO2, (B), as a function of time under conditions of <1% RH, (a), 21% RH, (b), and 53% RH, (c). All of the data for times after introduction of the CaCO3 aerosol, t > 0, have been corrected for wall reactions. The solid lines show fits to the data in the case of the higher RH experiments, (b) and (c). | ||
The HAc pressure manifests a rapid drop upon exposure to the calcite aerosol on a time scale too fast to resolve with the current instrument. The initial acetic acid consumption is approximately the same for each RH value. Subsequent to the initial drop in the dry experiment, <1% RH, Fig. 2a, little or no additional HAc decay is observed. However, for the higher RH experiments, Fig. 2b and c, further HAc consumption can be seen. The acetic acid time course data appears to decay exponentially before approaching a linear decay at longer times. To facilitate quantification of the observed HAc pressure, the data were fit to an exponential plus linear decay function. The fitting function, which is empirical in nature, was selected to describe the approach to zero-order kinetics (vide infra) for the slower reaction pathway. The resultant fits are shown as solid lines overlying the experimental data in Fig. 2.
Similarly, production of CO2 shows a rapid increase in the first 5–10 min after dust exposure followed by a slower, longer time production process. In the case of dry conditions, <1% RH, Fig. 2a, the CO2 pressure barely increases beyond this initial value but, in the case of the higher RH experiments, Fig. 2b and c, continued production of CO2 is readily measured. Analogous to the HAc data, we have fit these experiments to an exponential plus linear growth function and the fits are indicated by the solid lines in Fig. 2.
Several experiments were conducted at lower initial acetic acid pressures, approximately 7–9 mTorr, in an attempt to further characterize the kinetics of the heterogeneous reaction in terms of the uptake coefficient, γ. The results shown in Fig. 3 indicate that we were still unable to resolve the initial fast decay. The amount of HAc adsorbed is similar to that observed for the initial drop of acetic acid at higher starting pressures in Fig. 2 and does not appear to change greatly under different RH conditions. Due to the spectral interference detailed above, vide supra, we could not accurately measure the HAc loss at longer times with these relatively low concentrations. We can, however, estimate a lower limit for the uptake coefficient, γ, from our data using,51,52,57
 | (1) |
![[c with combining macron]](https://www.rsc.org/images/entities/i_char_0063_0304.gif) is the mean speed of HAc and τ is the characteristic decay time of the reactant. Assuming τ≈ 1 min, corresponding to the scan time of our FTIR, we then estimate a lower limit for the uptake coefficient of γ≥ 5 × 10−6 for the initial loss of HAc.
is the mean speed of HAc and τ is the characteristic decay time of the reactant. Assuming τ≈ 1 min, corresponding to the scan time of our FTIR, we then estimate a lower limit for the uptake coefficient of γ≥ 5 × 10−6 for the initial loss of HAc.
 | ||
| Fig. 3 Time dependence of the acetic acid pressure for lower initial pressures under conditions of (a) <1% RH, (b) 21% RH, and (c) 53% RH, (c). | ||
Representative IR data for the mixed experiments, whereby both HAc and HNO3 were present in the chamber and reacted with calcite aerosol, are shown in Fig. 4. In the upper panel we have plotted a reference spectrum, (a), of an acetic/nitric acid mixture in the chamber. Characteristic HNO3 absorptions, in addition to the previously discussed HAc features, are observed and the band frequencies are listed in Table 1. We have previously studied the interaction of HNO3 with calcite aerosol and typical data is shown in (b) for a RH of 43%. Under these conditions, all of the initial nitric acid, approximately 120 mTorr, is consumed as no HNO3 absorptions are visible in the spectrum. Instead, broad bands due to adsorbed water and new features corresponding to particulate nitrate58,59 are observed, indicative of the rapid reaction to form deliquesced calcium nitrate product. The appearance of an intense CO2 absorption is further evidence of the reaction. Relevant absorptions due to nitrate and adsorbed water are listed in Table 1.
 | ||
| Fig. 4 FTIR data from the HAc/HNO3/CaCO3 experiments. In the upper panel, (A), we show a reference spectrum for an acetic/nitric acid mixture, (a). Also shown are spectra characterizing the exposure of calcite aerosol to HNO3 only at 43% RH, (b), and a mixture of acetic and nitric acid at 41% RH, (c). In the lower panel, (B), expanded spectral data for calcite reacting with HNO3 only at 43% RH, (b), and a mixture of HNO3 and acetic acid at 41% RH, (c), are shown. A reference spectrum of calcium acetate aerosol is also included, (a). Gas phase water absorptions have been subtracted and spectrum (c) in the lower panel, (B), has been further processed to subtract acetic acid adsorptions. The positions of various bands are indicated. | ||
Spectrum (c) in the upper panel displays the results of exposing calcite aerosol to a HAc/HNO3 mixture at a RH of 41%. The spectrum is generally similar to that recorded for the nitric acid only experiment, (b), but with the appearance of additional absorptions recognized as arising from HAc. To further characterize the reaction, an expanded and enhanced view of the IR data is shown in the lower panel of Fig. 4 for the HNO3 only, (b), and mixture, (c), experiments. In addition to subtraction of the gas phase water bands, spectrum (c) has been further processed to remove residual acetic acid features. While increasing the spectral noise, the subtraction process shows that both spectra have most features in common except for the appearance of a new band at ≈1551 cm−1 in the case of the HAc/HNO3 mixture. To aid in identification of this feature, we recorded a reference spectrum of calcium acetate, Ca(CH3COO)2, aerosol, as depicted in (a). Based on this comparison and literature references for acetate IR absorptions,58,60 we assigned the new feature to the formation of acetate product in the mixture experiments. More frequencies are listed in Table 1.
We analyzed the acetic acid and CO2 product concentrations as a function of time in the mixed reagent gas experiments using the procedure outlined for the HAc only data. The results are displayed in Fig. 5 for the HAc and CO2 partial pressures in the upper and lower panels, respectively. The total loss of reactant acetic acid over the course of an experiment is clearly much larger when HNO3 is present, as can be seen from comparing the data of curves (b) and (c) with the results from an HAc only experiment, (a). Furthermore, the extent of HAc consumption increases when the RH is increased from 21%, (b), to 41%, (c). The fast initial drop in HAc pressure is still observable at 21% RH but is suppressed at the higher RH. Similar to the HAc only data, the decay curves appear to be linear at longer times where the 41% RH data show an exponential approach to linearity. The longer time HAc loss data was fit to either a linear decay, in the case of the 21% RH data, or an exponential plus linear decay function, for the 41% RH curve, and the resultant fits are shown as the solid lines in Fig. 5.
 | ||
| Fig. 5 Partial pressure of reactant acetic acid, (A), and product CO2, (B), as a function of time for the HAc/HNO3/CaCO3 experiments under conditions of 21% RH, (b), and 53% RH, (c). In addition, shown for reference, are data from Fig. 2 for an acetic acid only experiment at 53% RH, (a), and, in the lower panel, (B), results from a nitric acid only experiment at 44% RH, (d). All of the data for times after introduction of the CaCO3 aerosol, t > 0, have been corrected for wall reactions. The solid lines show fits to the data. | ||
The CO2 time course data show an immediate jump in pressure upon introduction of the calcite aerosol, similar to the results observed for the HNO3 only experiment, represented by curve (d). However, unlike the experiment with only nitric acid present where complete consumption of HNO3 immediately produces a fixed, stoichiometric amount of carbon dioxide, the mixture experiments show continued production of gaseous CO2 product as the HAc is consumed. The total amount of carbon dioxide produced is larger for the higher RH experiment. In addition, the amount of CO2 present at longer reaction times is larger than the sum of the contributions from the nitric acid only, curve (d), and the acetic acid only results, curve (a), recorded with similar starting reactant pressures. In other words, the yield of product carbon dioxide is enhanced when nitric acid is present and the enhancement increases with larger RH values. As with the HAc loss data, the 21% RH CO2 production curve was fit to a straight line and the 41% RH data was fit to an exponential plus linear growth function. The curve fits are shown by the solid lines.
4. Discussion
HAc–CaCO3 reaction
The results presented illustrate that acetic acid is irreversibly taken up by calcite aerosol and yields a gas phase product, CO2. The overall reaction can be described in analogy to that of HNO3 as, | (2) |
A convenient method for characterizing uptake and reaction for both the fast and slow channels in the HAc/CaCO3 system is to quantify the surface coverage in terms of the amount of HAc lost and the “reaction” coverage calculated from the CO2 product yield and assuming the 1∶1 stoichiometry between carbon dioxide and reacted Ca sites as suggested by eqn (2). We further normalize the coverage in terms of the number of monolayers by using a site density for CaCO3 of ns= 6.5 × 1014 sites cm−2,51,61 corresponding to one monolayer. The coverage can then be calculated as,
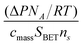 | (3) |
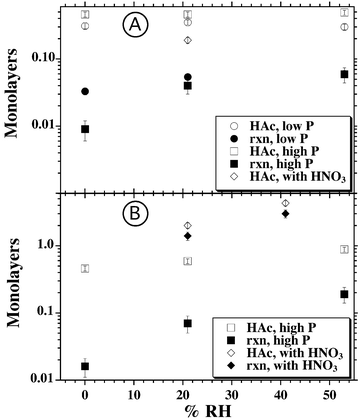 | ||
| Fig. 6 Semi log plots of the RH dependence for the acetic acid uptake and the number of reacted layers, calculated from CO2 production. The upper panel, (A), shows the results for the fast component of the HAc uptake and reaction. The lower panel, (B), shows similar data for the longer time behavior for both the acetic acid only and the HAc/HNO3 mixture experiments. | ||
As can be discerned in the upper panel of Fig. 6, the amount of HAc adsorbed onto calcitevia the fast reaction pathway is only slightly dependent on initial acetic acid pressure and is relatively independent of RH. While calcite is not hygroscopic, RH values of 50% should correspond to a few monolayers of water coverage on the surface. Apparently, under these conditions, there is minimal interference between coadsorbing water and acetic acid. Since the uptake coefficient for HAc onto dry calcite is likely to be orders of magnitude lower than that for H2O,46 it may be that water and acetic acid do not initially adsorb to the same sites and that water does not completely cover the available surface sites. Alternatively, the acetic acid may efficiently displace adsorbed water.
The lower limit to the uptake coefficient we have estimated for HAc onto dry CaCO3, γ≥5 × 10−6, is consistent with previous measurements of the uptake of acetic acid onto various model dust surfaces using a Knudsen cell technique.46 Although calcite was not investigated in this study, other dust surfaces yielded uptake coefficients of (2–0.2)× 10−3 for HAc under dry conditions. Similarly, a value of γ=(3 ± 1)× 10−3 for formic acid uptake by calcite has been observed.47 The acetic acid uptake coefficient is probably larger at the higher relative humidity conditions that are more typical of the troposphere. For instance, the uptake of nitric acid onto bulk CaCO3 powder samples was found to increase with relative humidity from a value of ≈10−4 measured under dry conditions.62 The CaCO3 surface is also likely to be more reactive towards acetic acid, particularly in the presence of water, and the resultant uptake larger than for the relatively unreactive metal oxide surfaces used in our previous Knudsen cell work. In companion experiments, IR spectroscopy has also been used to identify surface acetate on metal oxides exposed to acetic acid.46 Again, this would be in agreement with the overall reaction suggested by eqn (2). Previous measurements have shown an increased amount of surface formate from calcite exposure to formic acid when the RH is raised.47
The number of reacted layers in the upper panel of Fig. 6, determined from the amount of CO2 evolved in the fast reaction, is also independent of the initial acetic acid pressure although it appears that increased RH does increase the production of CO2. It is likely that the fast reaction pathway is facilitated by surface adsorbed water. The reaction coverage increases by about a factor of 1.5 from 21% to 53% RH, a change in RH that almost doubles the measured water coverage.62 Under the driest conditions, barely any CO2 above background levels is observed so whatever residual surface adsorbed water remains on the calcite is insufficient for appreciable reaction. The measured stoichiometry is also far from that suggested by eqn (2) even when surface water is present. We calculate a ratio of acetic acid reacted to CO2 produced of 8 ± 2 for the 53% RH experiment while eqn (2) would predict a stoichiometric ratio of 2∶1. Clearly, much of the acetic acid is adsorbed onto the surface as a reaction intermediate that we cannot detect via our IR probe method. It may be that the reaction pathway is initiated by ligand exchange on the hydroxyl-terminated Ca surface sites,47,63–67
 | (4) |
Interestingly, in the mixed HAc/HNO3 studies, evidence for the fast uptake of acetic acid can still be observed in the 21% RH experiment. The extent of the uptake is less than in the HAc only experiment, as demonstrated by the relevant data points in the upper panel of Fig. 6. At 41% RH in the mixed gas experiment, this channel is totally suppressed and there is no indication of a fast, initial, HAc uptake. Despite the extensive processing of the carbonate aerosol by HNO3 due to calcium nitrate formation and deliquescence, these results suggest that there are still available surface sites onto which the HAc can rapidly adsorb. It may be that the HNO3 reaction does not lead to a uniform coating of the calcite particle surface but, rather, preferentially reacts at certain sites. The lack of synergistic effects between SO2 and HNO3 uptake was previously suggested to be possible evidence for unreacted particulate surface in mixed SO2/HNO3 experiments.57
In all instances, the calculated coverages for the fast reaction channel are less than unity; about 0.5 in the case of HAc uptake and much lower, 0.06, for the number of reacted layers at 53% RH. Thus, this reaction pathway appears to be surface limited, even at RH values typical of the ambient troposphere. The saturation coverages are on the order of (3.1 ± 0.3)× 1014 molecules cm−2 for acetic acid uptake. This value can be compared with the saturation coverage for HAc on less reactive hematite and alumina particles of (6 ± 4)× 1013 molecules cm−2 measured in a Knudsen cell study, albeit at a much lower acetic acid pressure, 6 μTorr.46
The time course data in Fig. 2 indicates that the slower pathway leading to HAc uptake and CO2 evolution is, in contrast to the fast channel, very sensitive to the RH. Similarly, the RH dependence can be observed through the calculated coverages, as summarized by the data in the lower panel of Fig. 6. The HAc uptake increases by approximately a factor of two in going from dry conditions to 53% RH. The reactive coverage increases even more, by greater than an order of magnitude, for the same increase in experimental RH. Correspondingly, the ratio of HAc reacted to CO2 produced is 5 ± 1 at 53% RH. If only the reaction due to the slower channel is considered, correcting the data for the uptake and reaction from the fast process, the ratio decreases to 3.0 ± 0.2, approaching the value of 2∶1 expected from the overall reaction of eqn (2).
The observed increase in reaction extent with increase in RH is similar to our previous observations with the SO2/CaCO3 system.57 In those studies, the reactive uptake of SO2 onto calcite, quantified by measurement of CO2 product from the reaction,
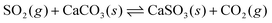 | (5) |
 | (6) |
The fast component, corresponding to t≤ 10 min, for CO2 production from the acetic acid reaction appears to depend on the amount of surface adsorbed water on the calcite. Similarly, carbon dioxide evolution from the reaction of SO2 with calcite exhibits a fast, initial rise that has been ascribed to a process facilitated by surface water. In both systems, a subsequent, slower reaction is observed that eventually becomes linear, suggestive of zero-order kinetics due to saturation of the surface. The measured reaction rate, υ, can be expressed in terms of a surface reaction rate constant, k, as
 | (7) |
| RH/% | ||||
|---|---|---|---|---|
| <1 | 21 | 53 | ||
| HAc only | HAc loss (×109 cm−2 s−1) | 1.7 ± 0.2 | 3.3 ± 0.4 | |
| CO2 production (×108 cm−2 s−1) | 4.9 ± 0.5 | 4.1 ± 0.9 | 56 ± 5 | |
| 21 | 41 | |||
|---|---|---|---|---|
| HAc/HNO3 mix | HAc loss (×1010 cm−2 s−1) | 9.8 ± 0.9 | 11 ± 1 | |
| CO2 production (×1010 cm−2 s−1) | 6.5 ± 0.6 | 6.8 ± 0.6 |
The calculated reaction rates exhibit greater sensitivity to the RH than is apparent from the coverage data. In particular, the rate of CO2 production increases by an order of magnitude when the RH is approximately doubled from 21 to 53%. Overall, these rates appear to be lower than the corresponding areal rates calculated from our SO2/CaCO3 experiments. Extrapolating the SO2 results to 53% RH suggests a rate of 2.1 × 1010 cm−2 s−1 compared to the CO2 production rate from the HAc reaction of (5.6 ± 0.5)× 109 cm−2 s−1 at the same RH, about four times smaller. The measured areal rates for the HAc reaction are only a fraction of the bulk phase value for calcite dissolution at neutral pH, as was the case of our previous SO2 experiments.57
HAc–HNO3–CaCO3
The results for the mixed reagent gas experiments, with both HAc and HNO3 present in the reaction chamber, clearly reveal evidence of an enhancement in the HAc reaction with calcite. The reaction with HNO3 is very rapid, viaeqn (6), converting the calcite particle to nitrate which deliquesces under the RH conditions of these experiments, promoting further reaction by exposing new surface sites from the particle bulk and increasing the amount of surface water, which further facilitates reaction by establishing a quasi-liquid phase. The IR spectra of Fig. 4 show evidence of the aerosol processing in the form of new spectral features due to particulate nitrate and surface adsorbed water. The fast HAc channel can still compete with the HNO3 reaction at 21% RH, as we have discussed (vide supra), but the HNO3 reaction is sufficiently extensive at 41% RH that this pathway is suppressed. We have previously determined that, with similar initial HNO3 pressures, the number of adsorbed water layers increases from about 10–12 to about 16–19 as the RH is raised from 20 to 40%.51 The dominant loss mechanism for HAc is the slower reaction pathway, as seen by the data in Fig. 5. Thus, the HAc reaction in the mixed gas experiments can be considered to be between gaseous acetic acid and a highly processed calcite particle with a partial, or total, coating of concentrated nitrate solution.The resultant reaction leads to more uptake of HAc and production of CO2 than is the case with HAc alone. The data reflect the enhanced reactivity as Fig. 5 shows more HAc uptake at long reaction times with the processed aerosol (≈5× larger relative to the 53% RH acetic acid data) and the amount of CO2 evolved is greater than the sum of carbon dioxide produced from the HNO3 and HAc only experiment (curves (a) and (d) in the lower panel of Fig. 5) under similar initial conditions. The increased reaction extent is similarly exhibited by the data in the lower panel of Fig. 6 which indicates that the HAc and reaction coverages are larger than the corresponding measurements in the absence of HNO3. The reaction coverages for the mixed gas experiments were determined by subtracting the amount of CO2 produced from the HNO3 reaction, knowing the initial nitric acid pressure and assuming all of the HNO3 is consumed according to eqn (6). Now, the number of reacted layers resulting from acetic acid chemistry is greater than a monolayer, about 3–4 at 41% RH, indicating the reaction can extend beyond the surface into the bulk of the particle. Finally, the presence of particulate acetate absorption features in the IR spectrum of Fig. 4 shows that the HAc reaction with nitric acid processed calcite aerosol is enhanced.
The derived areal reaction rates for the mixed reagent gas experiments are contained in Table 2. The rates are larger than the HAc only results for both HAc loss and CO2 production. The rates are still far smaller than bulk dissolution rates measured for calcite. Interestingly, the areal rates are approximately the same for the 21 and 41% RH measurements, implying that the amount of adsorbed water (10–20 layers) is sufficient to at least establish a consistent quasi-liquid like environment on the particle surface for the HAc reaction at either RH. The ratio of the HAc loss rate to the CO2 production rate is 1.6 ± 0.1 for the 41% RH experiment. The implied stoichiometric ratio is similar to that calculated from quantitative measurements of the HAc and CO2 pressures but is intermediate between the expected stoichiometric ratio from eqns (2) and (4).
Our results illustrate that in the presence of HNO3 and sufficient RH, at least greater than the DRH of calcium nitrate, the uptake and reaction of HAc is enhanced, clear evidence of a cooperative, synergistic effect between the two acidic gases. In contrast, our study of the SO2 reaction with calcite aerosol and HNO3 manifested no such effect; the production of CO2 from sulfur dioxide was not enhanced and the SO2 reaction extent was similar to that with unprocessed carbonate particles. It may be informative to consider the reason for such a contrast. The aqueous phase solubility of HAc and SO2, both acidic gases, will depend on the relevant effective Henry’s law constant, K*H, which, in turn, will be pH dependent. Lower pH conditions will decrease K*H for either gas due to their acidic nature, suppressing uptake by the aqueous phase. However, uptake of HAc can also have a contribution from CaAc+ since calcium acetate is highly soluble, unlike calcium sulfite, where44,70
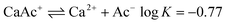 | (8) |
 | (9) |
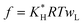 | (10) |
 | ||
| Fig. 7 The calculated effective Henry’s law constant, KH*, upper panel, (A), and aqueous phase mole fraction, Xaq, lower panel, (B), for acetic acid, with and without the formation of CaAc+, and SO2. | ||
Atmospheric implications
There are several conclusions from the current work that may bear on the role of heterogeneous processes in atmospheric cycles of acetic acid. It is also likely that these findings are equally applicable to formic acid, another relatively abundant organic acid that is highly correlated with acetic acid in field measurements.1,6 The loss of atmospheric acetic acid due to reactive uptake on available calcite aerosol surfaces, and other mineral dust particles, may be competitive with homogeneous, loss pathways, especially in dusty urban and desertified environments. Based on the current work, and the previous Knudsen cell studies for associated metal oxide components of mineral dust, the acetic acid uptake coefficient for the fast reaction pathway is sufficiently large that dust may be a significant sink compared to photolytic pathways.46 The uptake coefficient can be expected to be larger under higher RH conditions typical of the ambient troposphere. However, the fast reaction channel saturates at sub-monolayer coverages and results in relatively little processing of the calcite aerosol, corresponding to only about one-fifth of a monolayer at 53% RH. The acetic acid saturation coverages we have determined would be reached in approximately 0.25–3 h for an acetic acid concentrations of 10 ppb and assuming an uptake coefficient of γ≈ 10−5–10−4.Perhaps of greater significance is the slower reaction pathway we have identified which also saturates the surface but leads to continued acetic acid consumption and CO2 production. While RH values in the troposphere will usually be below the DRH of the calcium acetate product, the slower reaction is sensitive to RH and will be enhanced under typical tropospheric conditions. Continued processing of the aerosol would extend beyond the initially exposed surface area into the bulk of the particle, suggesting that dust aerosol mass may be a more critical consideration that surface area in atmospheric chemistry models that seek to incorporate heterogeneous dust interactions. In addition, it should be noted that the heterogeneous process converts acetic acid to CO2, an important greenhouse gas, although it is unlikely to constitute a major source.
The reaction between acetic acid and calcite aerosol that has been processed in the atmosphere to increase particulate hygroscopicity, for instance via HNO3 chemistry, will result in enhanced HAc uptake and reaction. The buffering capacity of the carbonate and the formation of soluble acetate product will provide a thermodynamic driving force for acetic acid partitioning to the particle phase. Subsequent reaction can be extensive and extend beyond the surface into the bulk of the product. However, the calcite dissolution rates appear to be orders of magnitude less than the bulk rate, at least under our experimental conditions. Finally, there is a clear synergistic effect between HNO3 and acetic acid when reacting with calcite aerosol. The increased affinity of calcium carbonate for acetic acid may partially rationalize the correlation between particulate nitrate and organic acids observed in field measurements.28,48
5. Conclusion
The reaction of acetic acid with calcite aerosol under varying conditions of relative humidity has been examined. The uptake of acetic acid by calcite and the production of gas phase CO2 is observed to occur under both dry and wet conditions and exhibits distinct fast and slow reaction pathways. The fast pathway is characterized by an uptake coefficient of γ≥ 5 × 10−6 and saturates the surface at sub-monolayer coverages. The fast uptake is relatively independent of initial pressure and RH. The corresponding production of CO2 is facilitated by surface adsorbed water which increases with RH. The slower reaction pathway also eventually saturates the surface but exhibits continued uptake and CO2 evolution. The reaction yield increases with RH but extensive processing of the calcite aerosol is limited by the very high DRH of the calcium acetate product. Experiments exposing calcite aerosol to mixtures of HAc and HNO3 reveal a strong synergistic effect leading to enhanced consumption of acetic acid and concomitant processing of the particle bulk. These results suggest that heterogeneous chemistry involving mineral dust may be a significant sink for atmospheric organic acids and may help explain observed correlations between particulate nitrate and organics.Acknowledgements
This material is based upon work supported by the National Science Foundation under Grant No. CHE-0503854. Any opinions, findings, and conclusions or recommendations expressed in this material are those of the authors and do not necessarily reflect the views of the National Science Foundation.References
- P. Khare, N. Kumar, K. M. Kumari and S. S. Srivastava, Rev. Geophys., 1999, 37, 227–248 CrossRef CAS.
- R. J. Kieber, B. Peake, J. D. Willey and G. B. Avery, Atmos. Environ., 2002, 36, 3557–3563 CrossRef CAS.
- T. Reiner, O. Mohler and F. Arnold, J. Geophys. Res., [Atmos.], 1999, 104, 13943–13952 CrossRef CAS.
- J. H. Seinfeld and S. N. Pandis, Atmospheric Chemistry and Physics: From Air Pollution to Climate Change, John Wiley & Sons, New York, 1998 Search PubMed.
- R. W. Talbot, B. W. Mosher, B. G. Heikes, D. J. Jacob, J. W. Munger, B. C. Daube, W. C. Keene, J. R. Maben and R. S. Artz, J. Geophys. Res., [Atmos.], 1995, 100, 9335–9343 CrossRef CAS.
- A. Chebbi and P. Carlier, Atmos. Environ., 1996, 30, 4233–4249 CrossRef CAS.
- K. Kawamura, L. L. Ng and I. R. Kaplan, Environ. Sci. Technol., 1985, 19, 1082–1086 CAS.
- R. W. B. Talbot, K. M. Harriss, R. C. Cofer III and W. R., J. Geophys. Res., [Atmos.], 1988, 93, 1638–1652 CrossRef CAS.
- J. P. Lacaux, J. Loemba-Ndembi, B. Lefeivre, B. Cros and R. Delmas, Atmos. Environ., 1992, 26A, 541–551 CrossRef CAS.
- R. W. Talbot, M. O. Andreae, H. Berresheim, P. Artraxo, M. Garstang, R. C. Harriss, K. M. Beecher and S. M. Li, J. Geophys. Res., [Atmos.], 1990, 95, 16955–16969 CrossRef.
- G. Enders, R. Dlugi, R. Steinbrecher, B. Clement, R. Daiber, J. v. Eijk, S. Gab, M. Haziza, G. Helas, U. Herrmann, J. Kesselmeier, D. Kotzias, K. Kourtidis, H.-H. Kurth, R. T. McMillen, G. Roider, W. Schurmann, U. Teichmann and L. Torres, Atmos. Environ., 1992, 26, 171–189 CrossRef.
- W. C. Keene and J. N. Galloway, Tellus B, 1988, 40B, 322–334 CrossRef CAS.
- E. Sanhueza and M. O. Andreae, Geophys. Res. Lett., 1991, 18, 1707–1710 CrossRef CAS.
- S. Madronich, R. B. Chatfield, J. G. Calvert, G. K. Moortgat, B. Veyret and R. Lesclaux, Geophys. Res. Lett., 1990, 17, 2361–2364 CrossRef CAS.
- S. Madronich and J. G. Calvert, J. Geophys. Res., [Atmos.], 1990, 95, 5697–5715 CrossRef CAS.
- D. J. Jacob and S. C. Wofsy, J. Geophys. Res., [Atmos.], 1988, 93, 1477–1486 CrossRef CAS.
- H. A. Khwaja, Atmos. Environ., 1995, 29, 127–139 CrossRef CAS.
- D. Grosjean, E. Grosjean and E. L. Williams, II, Environ. Sci. Technol., 1994, 28, 186–196 CrossRef CAS.
- M. O. Andreae, R. W. Talbot, T. W. Andreae and R. C. Harriss, J. Geophys. Res., [Atmos.], 1988, 93, 1616–1624 CrossRef CAS.
- F. Maupetit and R. J. Delmas, J. Geophys. Res., [Atmos.], 1994, 99, 16491–16500 CrossRef CAS.
- K. C. Weathers, G. E. Likens, F. H. Bormann, S. H. Bicknell, B. T. Bormann, B. C. Daube, Jr, J. S. Eaton, J. N. Galloway, W. C. Keene, K. D. Kimball, W. H. McDowell, T. G. Siccama, D. Smiley and R. A. Tarrant, Environ. Sci. Technol., 1988, 22, 1018–1026 CAS.
- M. O. Andreae, R. W. Talbot, H. Berresheim and K. M. Beecher, J. Geophys. Res., [Atmos.], 1990, 95, 16987–16999 CrossRef CAS.
- J. P. Lacaux, R. Delmas, G. Kouadio, B. Cros and M. O. Andreae, J. Geophys. Res., [Atmos.], 1992, 97, 6195–6206 CAS.
- W. C. Keene and J. N. Galloway, Atmos. Environ., 1984, 18, 2491–2497 CrossRef CAS.
- R. W. Talbot, M. O. Andreae, H. Berresheim, D. J. Jacob and K. M. Beecher, J. Geophys. Res., [Atmos.], 1990, 95, 16799–16811 CrossRef CAS.
- W. C. Keene, J. N. Galloway and J. D. Holden, Jr, J. Geophys. Res., [Atmos.], 1983, 88, 5122–5130 CrossRef CAS.
- D. Grosjean, Environ. Sci. Technol., 1989, 23, 1506–1514 CrossRef CAS.
- S.-H. Lee, D. M. Murphy, D. S. Thomson and A. M. Middlebrook, J. Geophys. Res., [Atmos.], 2002, 107 DOI:10.1029/2000JD000011.
- C. Sabbioni, N. Ghedini and A. Bonazza, Atmos. Environ., 2003, 37, 1261–1269 CrossRef CAS.
- A. R. Garcia, J. L. Da Silva and L. M. Ilharco, Surf. Sci., 1998, 415, 183–193 CrossRef CAS.
- J. C. Hemminger, Int. Rev. Phys. Chem., 1999, 18, 387–417 CrossRef CAS.
- P. Baumann, H. P. Bonzel, G. Pirug and J. Werner, Chem. Phys. Lett., 1996, 260, 215–222 CrossRef CAS.
- Q. Gao and J. C. Hemminger, J. Electron Spectrosc. Relat. Phenom., 1990, 54–55, 667–676 CrossRef CAS.
- B. A. Sexton, Chem. Phys. Lett., 1979, 65, 469–471 CrossRef CAS.
- R. J. Madix, J. L. Falconer and A. M. Suszko, Surf. Sci., 1976, 54, 6–20 CrossRef CAS.
- K. U. Goss, Environ. Sci. Technol., 1993, 27, 2127–2132 CrossRef CAS.
- K.-U. Goss and R. P. Schwarzenbach, J. Colloid Interface Sci., 2002, 252, 31–41 CrossRef CAS.
- C. M. Roth, K.-U. Goss and R. P. Schwarzenbach, J. Colloid Interface Sci., 2002, 252, 21–30 CrossRef CAS.
- K. U. Goss, Environ. Sci. Technol., 1992, 26, 2287–2294 CrossRef CAS.
- J. D. Kubicki, L. M. Schroeter, M. J. Itoh, B. N. Nguyen and S. E. Apitz, Geochim. Cosmochim. Acta, 1999, 63, 2709–2725 CrossRef CAS.
- S. J. Hug and B. Sulzberger, Langmuir, 1994, 10, 3587–3597 CrossRef CAS.
- E. C. Yost, M. I. Tejedor-Tejedor and M. A. Anderson, Environ. Sci. Technol., 1990, 24, 822–828 CAS.
- M. V. Biber and W. Stumm, Environ. Sci. Technol., 1994, 28, 763–768 CAS.
- C. N. Fredd and H. S. Fogler, Chem. Eng. Sci., 1998, 53, 3863–3874 CrossRef CAS.
- D. E. Nierode and B. B. Williams, Soc. Petrol. Eng. J., 1971, 11, 406–418 Search PubMed.
- S. Carlos-Cuellar, P. Li, A. P. Christensen, B. J. Krueger, C. Burrichter and V. H. Grassian, J. Phys. Chem. A, 2003, 107, 4250–4261 CrossRef CAS.
- H. A. Al-Hosney, S. Carlos-Cuellar, J. Baltrusaitis and V. H. Grassian, Phys. Chem. Chem. Phys., 2005, 7, 3587–3595 RSC.
- S.-H. Lee, D. M. Murphy, D. S. Thomson and A. M. Middlebrook, J. Geophys. Res., [Atmos.], 2003, 108, 8417–8424 CrossRef.
- L. M. Russell, S. F. Maria and S. C. B. Myneni, Geophys. Res. Lett., 2002, 29(16), 1779 CrossRef.
- A. P. Prince, PhD thesis, University of Iowa, 2003.
- A. P. Prince, V. H. Grassian, P. Kleiber and M. A. Young, Phys. Chem. Chem. Phys., 2006, 9, 622–634 Search PubMed.
- A. P. Prince, J. Wade, V. H. Grassian, P. Kleiber and M. A. Young, Atmos. Environ., 2002, 36, 5729–5740 CrossRef.
- M. Haurie and A. Novak, J. Chim. Phys. Phys.-Chim. Biol., 1966, 63, 1584–1586 CAS.
- A. Burneau, F. Genin and F. Quiles, Phys. Chem. Chem. Phys., 2000, 2, 5020–5029 RSC.
- J. Chocholousova, J. Vacek and P. Hobza, J. Phys. Chem. A, 2003, 107, 3086–3092 CrossRef CAS.
- M. Haurie and A. Novak, J. Chim. Phys. Phys.-Chim. Biol., 1965, 62, 146–157.
- A. P. Prince, P. Kleiber, V. H. Grassian and M. A. Young, Phys. Chem. Chem. Phys., 2007, 9, 3432 RSC.
- K. Nakamoto, Infrared Spectra of Inorganic and Coordination Compounds, John Wiley and Sons, New York, 1963 Search PubMed.
- K. Nakamoto, Infrared and Raman Spectra of Inorganic and Coordination Compounds, John Wiley and Sons, New York, 1986 Search PubMed.
- F. Quiles and A. Burneau, Vib. Spectrosc., 1998, 16, 105–117 CrossRef CAS.
- G. M. Underwood, P. Li, H. Al-Abadleh and V. H. Grassian, J. Phys. Chem. A, 2001, 105, 6609–6620 CrossRef CAS.
- A. L. Goodman, G. M. Underwood and V. H. Grassian, J. Geophys. Res., [Atmos.], 2000, 105, 29053–29064 CrossRef CAS.
- H. A. Al-Hosney and V. H. Grassian, J. Am. Chem. Soc., 2004, 126, 8068–8069 CrossRef CAS.
- S. I. Kuriyavar, R. Vetrivel, S. G. Hegde, A. V. Ramaswamy, D. Chakrabarty and S. Mahapatra, J. Mater. Chem., 2000, 10, 1835–1840 RSC.
- S. L. Stipp and J. M. F. Hochella, Geochim. Cosmochim. Acta, 1991, 55, 1723–1736 CrossRef CAS.
- S. L. Stipp, Geochim. Cosmochim. Acta, 1999, 63, 3121–3131 CrossRef CAS.
- H. A. Al-Abadleh, H. A. Al-Hosney and V. H. Grassian, J. Mol. Catal. A: Chem., 2005, 228, 47–54 CrossRef.
- I. N. Tang and K. H. Fung, J. Chem. Phys., 1997, 106, 1653–1660 CrossRef CAS.
- J. Schonherr and M. Luber, Plant Soil, 2001, 236, 117–122 Search PubMed.
- C. A. Colman-Porter and C. B. Monk, J. Chem. Soc., 1952, 4363–4368 RSC.
| This journal is © the Owner Societies 2008 |