Electronic spectra of radicals in a supersonic slit-jet discharge by degenerate and two-color four-wave mixing
Fabio J. Mazzottia, Elena Achkasovaa, Richa Chauhana, Marek Tulejb, Peter P. Radib and John P. Maier*a
aDepartment of Chemistry, University of Basel, Klingelbergstrasse 80, CH-4056, Basel, Switzerland. E-mail: j.p.maier@unibas.ch
bPaul Scherrer Institute, CH-5232, Villigen PSI, Switzerland
First published on 25th October 2007
Abstract
Four-wave mixing techniques have been used for the measurement of electronic transitions of cold transient species generated in a supersonic slit-jet discharge expansion. The origin band of the d3Πg–a3Πu system of C2 and Ã2Π3/2–![[X with combining tilde]](https://www.rsc.org/images/entities/char_0058_0303.gif) 2Π3/2 electronic transition of HC4S were recorded. A signal-to-noise ratio of 104 in the spectra was achieved, resulting in detection limits of 1010 cm−3 for these two molecules. Application of selective two-color resonant four-wave mixing is used for the spectral assignment utilizing the double-resonance nature of the method. The combination of these techniques with a slit source proves to be a sensitive approach for the detection of transient molecules in a molecular beam discharge.
2Π3/2 electronic transition of HC4S were recorded. A signal-to-noise ratio of 104 in the spectra was achieved, resulting in detection limits of 1010 cm−3 for these two molecules. Application of selective two-color resonant four-wave mixing is used for the spectral assignment utilizing the double-resonance nature of the method. The combination of these techniques with a slit source proves to be a sensitive approach for the detection of transient molecules in a molecular beam discharge.
Introduction
Electronic spectra of unsaturated carbon-chain radicals and ions produced in a supersonic discharge have been extensively studied during the last two decades by laser techniques, with important implications for astrophysics.1 These sources offer the advantage of producing a full class of carbon-bearing radicals, starting with a simple precursor, namely acetylene. Furthermore, the internal temperature of the molecules produced in a discharge followed by a supersonic jet expansion into vacuum is low enough to enable precise analysis of their spectra because the ground-state population is limited to the lowest J levels.Owing to the low density of the species present in the plasma, sensitive optical techniques are required for such measurements. Linear techniques such as absorption-based cavity ringdown spectroscopy (CRDS),2 laser-induced fluorescence (LIF)3,4 and resonant-enhanced multi-photon ionization (REMPI)5 and its double-resonance variant6 were successfully applied to the study of several of these systems. Although it is relatively easy to implement, CRDS is based on long line-of-sight absorption and has no selectivity, especially when spectral features overlap. Furthermore, CRDS is inappropriate for detecting transitions which are broadened by the short lifetime of upper states.7 LIF is inherently background-free and has extensively been applied to plasmas in luminous and harsh environments; however, it is only suited for systems which exhibit reasonably large fluorescence quantum yields and cannot be used to study systems that have unstable (dissociative or predissociative) excited electronic states. REMPI is limited to neutral molecules but its mass-selectivity provides unambiguous assignment for different species. In the case of complex, overlapping spectral features, linear techniques fail to unambiguously assign rotational lines within the same spectral system.
The use of a technique which would not share these disadvantages would be a valuable spectroscopic tool to study transients. Degenerate four-wave mixing (DFWM) and its variant two-color resonant implementation (TC-RFWM) appear to be good candidates for this task. The technique offers many advantages. First of all, it is background-free and produces a coherent signal which can be discriminated from scattered radiation.8 It has also proven to be highly sensitive, with obtained signal-to-noise (S/N) ratios as high as 104 with the species concentrations usually present in a discharge.9 Furthermore, it benefits from a large dynamic range. It does not suffer from fluorescence quenching10,11 and offers the unsurpassed possibility to perform resonant spectroscopy which, given simple selection rules, leads to spectra that are unambiguously assigned, even in the case of overlapping lines within the same system. This has lead to the understanding of molecular structure and dynamics in the case of high vibrational excitation.12,13 The TC-RFWM spectrum of highly excited states of NH3 was recorded in a room temperature gas cell14 with ten-fold improved sensitivity compared to previous measurements made with DFWM and rotational constants of ground-state vibrational levels of HCO were accurately determined,15 proving that TC-RFWM provided an increase in spectral sensitivity and selectivity compared to the degenerate case. It is, however, a technique difficult to implement which suffers practically from complex saturation effects such as line broadening with laser power. In fact, because the intensity at which a line saturates is inversely proportional to the strength of the observed transitions, the net result can vary for different spectral regions and lines if care is not taken in controlling the laser power. Moreover, it sometimes requires a powerful laser source capable of delivering millijoules per pulse and it is very sensitive to beam alignment as well as beam profile.
In a pioneer experiment, the potential of DFWM for monitoring the OH radical in combustion environments was reported.16 Following this work, four-wave mixing (FWM) measurements have been carried out in the gas phase as a diagnostic in combustion8,17–19 and in reacting plasmas.20,21 However, up until now, the FWM technique was used for measurements in molecular free-jet expansions only for a limited class of relatively small molecules and radicals, namely formaldehyde,22,23 SO2,24,25 SiC2,26 C2H2,27 CS2,28 C312,29 generated by laser vaporization, predissociation spectroscopy of NO230,31 and, more recently, C3 by degenerate and two-color variant in a supersonic discharge.9
In the present work, we report the detection of C2 and of HC4S in a supersonic slit-jet discharge with degenerate and two-color resonant FWM. The two-color scheme provides unambiguous assignment of the rotational spectra for HC4S. To date, measurements done with the FWM technique on C2 have been carried out only in high-temperature (3000 K) oxy-acetylene flames.32–34 Sulfur-terminated carbon chains are of interest in astronomy due to the relatively large cosmic abundance of this non-metallic element.3 Recently, the electronic spectrum of the Ã2Π3/2–2Π3/2 system of HC4S produced in a supersonic discharge has been studied by LIF4 and dispersed fluorescence.3 In the latter, the authors report detection of several vibrational states in the electronic ground state, while in the former a rotationally resolved spectrum is achieved with a 0.02 cm−1 laser linewidth and spectroscopic constants are derived for several of these vibronic transitions.
Description of four-wave mixing
The theory of DFWM involves the third-order susceptibility tensor.35 In spite of the fact that the higher-order polarizations responsible for nonlinear optical phenomena are small, the signal intensities in the four-wave mixing process can be large due to its strong dependence on laser power, concentration and interaction length. Moreover, if one or more of the interacting frequencies are close to one- or multiphoton transitions, nonlinear susceptibilities can be greatly enhanced. FWM may be thought of as a resonant variant of coherent anti-Stokes Raman spectroscopy (CARS).21 A qualitative explanation of the phenomenon is that two beams (referred to as pump beams) interfere in the medium inducing macroscopic variations in the ground- and excited-state density and/or polarization, forming a Bragg grating. When the third beam (probe) is resonant with a second transition (it is the same in the degenerate case) having in common upper- in stimulated-emission pumping (SEP) spectroscopy or lower-state (UP transition), it gets reflected off the grating and its direction is given by the Bragg phase-matching condition.36Fig. 1 shows energy-level diagrams for the three aforementioned spectroscopic techniques. The technique works well even with short-lived upper states since the grating formed by the lower-state density modulations and causing probe-beam reflection remains long-lived. The coherent laser-like signal beam allows a high collection efficiency and stray light is efficiently suppressed by remote probing. Under the unsaturated regime, the TC-RFWM intensity is given by:37 | (1) |
 | ||
| Fig. 1 Schemes of the three different spectroscopic techniques; non-resonant coherent anti-Stokes Raman spectroscopy (CARS), degenerate four-wave mixing (DFWM) and two-color resonant four-wave mixing (TC-RFWM). Although similar to CARS, FWM is resonant in nature and as such allows for stronger non-linear effects. Unlike laser-induced fluorescence, the coherent nature of the signal allows rejection of scattered light and plasma emission by detection at large distances (4 meters in our case). | ||
Formula 1 was used for making two-color spectral simulations: pump transitions were weighted by a Gaussian lineshape centered at the pump wavelength (FWHM was chosen as measured in the experimental spectrum). The corresponding SEP and UP transitions were then weighted by the factor NiB1B3, convoluted with a Gaussian and added in the spectrum which was squared so that intensity of isolated lines is in accordance with Formula 1. Ni is computed by assuming a Boltzmann distribution of the rotational states and the G factor, which levels off for J > 3, was not taken into account.
A forward “box” (as beams are directed through the diagonals of a rectangular parallelepiped) cross-beams CARS geometry (known as BOXCARS,38see Fig. 2) configuration was chosen, for which virtually all generated photons can be detected, in contrast to the Doppler-free phase-conjugate arrangement,39 for example, resulting in an increased sensitivity. The polarization of the four interacting beams was vertical in the laboratory frame.
 | ||
| Fig. 2 Degenerate and two-color resonant four-wave mixing was carried out in the forward BOXCARS configuration. I1 and I3 are the pump beams (of the same frequency), I2 is the probe beam (of the same frequency as the pump in degenerate case) and I4 is generated along the “dark” direction, of the same frequency as the probe. The beams are longitudinal to the slit enabling a longer path-length and an enhanced signal. The Bragg grating which is less smeared out by the radical velocity due to the expansion is created by interference of I1 and I3 and is parallel to the vertical flow of the radical jet. | ||
Experimental
Ion source
Radicals were generated by pulsing a 0.7–1% acetylene (with 0.3% CS2 for HC4S measurements) mixture in argon through the 30 × 0.3 mm (or 30 × 1.0 mm) slit-jet nozzle. A voltage of −600 to −1000 V was applied to the electrodes. The timing between the gas pulse, discharge and laser was optimized according to the species studied. Backing pressure of 6–10 atmospheres was used with a chamber pressure of about 40 mTorr. The nozzle itself was mounted on a precision x–y translation stage which enabled variation of observation height (2 to 12 mm) as well as of lateral distance. Experiments carried out with a circular orifice proved that the slit-jet provides two orders of magnitude of improvement in signal intensity, as was already pointed out when a backwards-box DFWM configuration in conjunction with a CS2 free-jet slit expansion was used.28 This enhancement in sensitivity is not due to an increased concentration of the species but rather to a larger interaction volume. It is worth noting that FWM signal intensity is proportional to the square of the path-length of the beams through the jet so that a 10-fold-longer interaction zone produces a signal that is 100 times more intense.Four-wave mixing setup
A dye laser pumped by the third harmonic of a Nd:YAG laser operated at 20 Hz produced pulses of 8 ns duration. Coumarin 307 was used allowing continuous coverage of the 485–550 nm spectral range with a 0.10 cm−1 linewidth. An intracavity etalon enabled reduction of the laser linewidth by a factor of 5. The laser energy was of the order of 1 mJ per pulse and variable neutral-density filters were used to reduce the laser-beam intensity because saturation of the FWM signal results in broadening of the absorption lines.40 Calibration was done with a wavemeter. The beam was spatially filtered in order to obtain a homogeneous profile. Broadband dielectric mirrors and beamsplitters divided the light into three collinear components and a lens of 1000 mm focal length overlapped the beams in the center of the vacuum chamber, near the tip of the nozzle. The crossing angle was 1.7° while the beam diameter was about 2 mm before the focusing lens, defining an overlapping region with the plasma produced by the slit source of 30 mm in length. The collection optics consisted of a series of spatial filters set on a 4-meter path which efficiently rejected the unwanted stray and scattered light. Alignment was performed by positioning a thin quartz cell containing a dye (absorber) in the interaction region. The output from the detector was sent to a fast 400 MHz digital oscilloscope. The area of the PMT signal was calculated and recorded. Typically, spectra were obtained by averaging the FWM signal on 20–40 laser shots. For the two-color experiment, a dye laser with a 0.15 cm−1 linewidth pumped by a separate Nd:YAG laser was used as the probe. Timing between lasers was electronically controlled by a pulse generator. A schematic diagram of the TC-RFWM setup is shown in Fig. 3. | ||
| Fig. 3 Schematic of the experimental setup used for two-color resonant four-wave mixing. The two Nd:YAG lasers are triggered electronically by a pulse generator. R represents broadband dielectric reflectors, P is a prism, BS is a 50% broadband beamsplitter, SP represents spatial filters, L is a 1000 mm lens and M is a mask. The photodiode (PD) triggers the oscilloscope which sends data to a computer. Control of the timing between lasers, valve opening and the high-voltage (HV) pulse applied on the source electrodes is done with a computer analogical card. The signal IS travels along a 4-meter path consisting of spatial filters, resulting in efficient elimination of scattered light and emission from plasma. | ||
Cavity ringdown spectroscopy setup
A standard CRDS setup for pulsed radiation was used.2 The cavity was formed by two highly reflective plano-concave mirrors with 1-meter radii of curvature (99.98% reflectivity at 500 nm) separated by a distance of 52 cm. The input laser beam was matched to the TEM00 mode. The radiation leaking out from the rear mirror was collected by a photodiode with amplifier, in front of which a broadband filter rejected the emission from the discharge. The ringdown curves were fitted by a home-made high-speed program. As CRDS is not a zero-background technique, the discharge was intermittently turned off every other laser pulse so that correction of the background was possible. Typical ringdown times measured were about 18–20 μs with an empty cavity and about 15 μs when the discharge was turned on with no absorption at the laser wavelength.Results and discussion
Detection of C2 by degenerate four-wave mixing
The recordings of the d3Πg–a3Πu electronic system of C2 by DFWM in high-temperature flames have been reported,33,34 and make the molecule well suited for initial experiments. A spectrum of the C2 Swan-system origin band is shown in Fig. 4a. The numerous transitions between the triplet states produce a high density of lines, with a clear progression of triplets in the high Js of the R branch due to the spin multiplicity.33. The spectral contour fitting made with PGOPHER software41 by using spectroscopic constants available in the literature42 leads to an estimate of the rotational temperature of about 140 K. The DFWM signal detected was intense, resulting in S/N of the order of 104 for the strongest transitions. Fig. 4b also depicts a spectrum recorded by DFWM at a lower plasma temperature (approximately 40 K); to achieve this, the discharge voltage was lowered to −400 V and the probed region was around 12 mm downstream. Other vibronic transitions were readily detected within the spectral region of the dye. As an example, the 1–0 band is shown in Fig. 4c, obtained with a temperature of 100 K. This exemplifies the potential of DFWM for probing the radical plasma temperature in luminous and hostile environments. | ||
| Fig. 4 (a) 0–0 d3Πg–a3Πu system of C2 measured by degenerate four-wave mixing. The simulation indicates a rotational temperature of 140 K. Notice the high signal-to-noise ratio (104 for the strongest transitions). (b) 0–0 band recorded at low temperature, 40 K. (c) 1–0 band with an estimated temperature of 100 K. | ||
Detection of C2 by two-color four-wave mixing
The wavelength of the pump laser was tuned to a transition of C2 while the probe laser was scanned, resulting in spectra (Fig. 5) which can be unambiguously analyzed. When tuned to the rR11(3) transition: 2 SEP and 2 UP corresponding transitions were observed, one of each in the P and Q branches. The experimental linewidth of the lines is nearly twice the linewidth of the probe laser, indicating that the experiments were done in the saturated regime. Under these conditions, the reproducibility in intensity of the spectral lines was improved as well as S/N since the FWM signal is not as highly dependent on shot-to-shot laser variations as in the unsaturated regime.43 Also shown are spectra obtained when the transition located at 515.020 nm is excited, which corresponds to the weak line rQ21(6). With higher laser power, weak components originating from nearby lying transitions, i.e. the 60-fold-stronger rR33(6) transition and 7-fold-weaker sR21(2) are visible in the spectrum. This is interpreted as the laser-pump wavelength (FWHM of 0.004 nm) lying in the wings of the cubed FWM Lorentzian lineshape of the rR33(6) strong line, which is shifted 0.007 nm to the blue end of the spectrum (and of the sR21(2) line, shifted 0.001 nm to the red). When laser power is appropriately reduced, it is possible to excite only one transition (Fig. 5, trace c) and the two-color spectrum is unambiguously assigned to rQ21(6). This illustrates the importance of laser-power control when performing spectroscopy with FWM. It should be emphasized that the main advantage of the two-color RFWM compared to standard absorption techniques in analyzing complex molecular spectra: it provides excellent spectral selectivity and offers enough sensitivity for the detection of transient species produced in a supersonic discharge.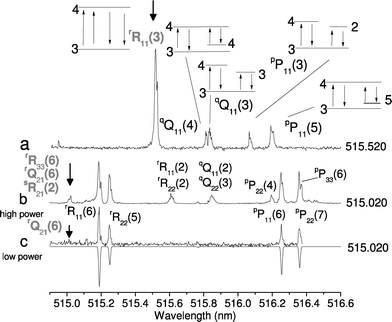 | ||
| Fig. 5 (a) Two-color four-wave mixing spectrum of C2 showing five transitions (the bold arrow indicates the pump wavelength). Two have a common lower state in the Q and P branches respectively. Two exhibit a common upper state and the degenerate transition at the R11(3) line position. J numbers of lower and upper states are shown. (b) Two-color spectrum recorded with pump wavelength at 515.020 nm (indicated by the bold arrow). According to the laser linewidth and saturation degree, excitation of several transitions may occur for the four-wave mixing technique (see text). Thus, the laser intensity should be controlled carefully. (c) The two-color spectrum recorded at the same pump frequency as (b) with lower laser intensity, along with simulation (inverted trace). Notice the lower S/N ratio because of decreased intensity. The simulation assumes Gaussian lineshapes, with FWHM of 0.15 cm−1 for probe and pump transitions. Rotational temperature was set to 140 K. | ||
Detection of HC4S with DFWM
To date, FWM was not applied to middle-size or long carbon chains in a discharge. A strong FWM signal was observed when the laser was tuned to the origin band of the Ã2Π3/2–2Π3/2 electronic transition of HC4S. If the laser power was too high (several hundreds of μJ), saturation ensued. The spectrum would then display a broad envelope, showing no rotational structure. When the laser-power intensity was reduced (to 20–100 μJ), the P branch could be resolved without an intracavity etalon, as can be seen in Fig. 6a. The spectra were recorded 12 mm from the nozzle slit, where the temperature of the radicals is lower. With an intracavity etalon, the scan obtained (Fig. 6b) shows good agreement for the intensity and line position with the simulation. For these preliminary measurements, the rms of the fit of the experimental line positions is 0.008 cm−1 which is smaller than the laser linewidth and gave spectroscopic constants which agreed with previous measurements.4 About 50 lines were used in the fit with J up to 40.5 in the P branch, and only the rotational constants in the upper and lower states were varied. In addition, a weaker spectral component was observed in the spectra, shifted 11.5 cm−1 from the origin band toward the red end of the spectrum, with a lower (10.5 cm−1) shift for DC4S. This feature is believed to be a vibronic transition from one of the lowest skeletal bending modes, which are symmetry-allowed for the dipole moment only for overtones and combinations.4 Preliminary experiments suggest that improved molecular constants including centrifugal distortion and spin–orbit coupling are obtainable by combining CRDS and four-wave mixing techniques. | ||
| Fig. 6 (a) The Ã2Π3/2–2Π3/2 origin band of HC4S and DC4S recorded by degenerate four-wave mixing. The linewidth of the laser was 0.1 cm−1, small enough to resolve the individual rotational lines in the P branch. In the HC4S spectrum, a vibronic component is present (see text). (b) Degenerate four-wave mixing spectrum of HC4S recorded with an intracavity etalon. The inverted trace is the square of the simulation with a rotational temperature of 30 K and a Gaussian linewidth of 0.038 cm−1, slightly larger than the estimated laser linewidth of 0.020 cm−1. The rms of the fit in the line positions is 0.008 cm−1 (see text). The relative intensity of the experimental spectrum was best matched to the simulation by contour fitting. | ||
Detection of HC4S by two-color four-wave mixing
When the pump laser was set to the spectrally unresolved band head, many corresponding transitions could be observed in the P branch as shown in Fig. 7, where a simulated two-color spectrum is also plotted. The intensity pattern of the isolated lines in the P branch is well reproduced by the simulation. However, the band head which consists of many overlapping lines and for which the total enhancement of the intensity depends on FWM process is broader than the simulation and a special treatment would be more appropriate. This spectrum illustrates the ability of TC-RFWM to disentangle overlapping spectral features in a congested band head. For this, it is sufficient to tune the pump laser on any of the observed P transitions and only two corresponding R transitions will be present in the TC spectrum if the correct saturation conditions are met.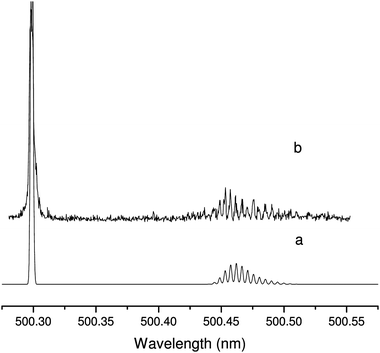 | ||
| Fig. 7 Two-color four-wave mixing spectrum of the Ã2Π3/2–2Π3/2 origin band of HC4S (trace b) when the pump laser is tuned to the band head (500.304 nm) along with a simulation (trace a). The simulation linewidth was chosen to reproduce the 0.06 cm−1 FWHM of the experimental lines in the P branch. The rotational temperature was set to 22 K and the intensity was normalized on the strongest transition of the P branch. The observed band head is much broader than the simulation because of overlapping lines (see text). | ||
Sensitivity of four-wave mixing
CRDS measurements were used to obtain the value of the concentrations of C2 and HC4S in the discharge. Owing to its capability of recording absolute absorption,44 CRDS is a self-calibrated technique. By using the oscillator strength previously measured (f = 0.029),45 a concentration of 1012 molecules per cm3 for C2 (a3Πu state) in the plasma is estimated at a temperature of 140 K. With S/N of about 104 for DFWM, the technique has a detection limit of 1010 molecules per cm3. This gives a sensitivity below 109 molecules per cm3 per rotational state, which is about the lowest detection limit achieved with this technique reported in the literature.46 In the case of HC4S, using values of transition moments obtained by ab initio computations,47 a density of about 1011 molecules per cm3 is inferred. The S/N ratio of 103 yields a detection limit below 1010 molecules per cm3. These results confirm the potential of the FWM technique for measuring unsaturated carbon chains in a planar-expansion discharge.Conclusion
This paper presents electronic spectra of C2 and HC4S produced in a slit-jet discharge by the four-wave mixing technique. DFWM and TC-RFWM were used in the forward BOXCARS geometry. The technique produces a coherent beam which can be discriminated from laser scattering and plasma emission, resulting in high S/N spectra and a DFWM sensitivity among the highest achieved.26 It is illustrated that the two-color approach, applied to the d3Πg–a3Πu system of C2 and the Ã2Π3/2–2Π3/2 electronic transition of HC4S, provides a spectroscopic tool for unambiguous assignment while offering the possibility of disentangling overlapping components. The potential of using four-wave mixing in slit-jet discharges for studying radicals at low temperature is asserted.
Acknowledgements
This work has been supported by the Swiss National Science Foundation (Project 200020-115864/1) and the Swiss Department of Energy (BFE, Contract 100708).References
- J. P. Maier, J. Phys. Chem. A, 1998, 102, 3462 CrossRef CAS.
- H. Linnartz, T. Motylewski and J. P. Maier, J. Chem. Phys., 1998, 109, 3819 CrossRef CAS.
- N. J. Reilly, G. C. Cupitt, S. H. Kable and T. W. Schmidt, J. Chem. Phys., 2006, 124, 194310 CrossRef CAS.
- M. Nakajima, Y. Sumiyoshi and Y. Endo, Chem. Phys. Lett., 2002, 351, 359 CrossRef CAS.
- G. Bazalgette Courreges-Lacoste, J. P. Sprengers, J. Bulthuis, S. Stolte, T. Motylewski and H. Linnartz, Chem. Phys. Lett., 2001, 335, 290.
- T. W. Schmidt, A. E. Boguslavskiy, T. Pino, H. Ding and J. P. Maier, Int. J. Mass Spectrom., 2003, 228, 647 CrossRef CAS.
- E. Achkasova, M. Araki, A. Denisov and J. P. Maier, J. Mol. Spectrosc., 2006, 237, 70 CrossRef CAS.
- R. L. Farrow and D. J. Rakestraw, Science, 1992, 257, 1894 CAS.
- M. Tulej, M. Meisinger, G. Knopp, A. M. Walser, T. Gerber and P. P. Radi, J. Raman Spectrosc., 2007, 38, 1022 CrossRef CAS.
- T. J. Butenhoff and E. A. Rohlfing, J. Chem. Phys., 1993, 98, 5460 CrossRef CAS.
- J. R. Dunlop and E. A. Rohlfing, J. Chem. Phys., 1994, 100, 856 CrossRef CAS.
- S. Williams, J. D. Tobiason, J. R. Dunlop and E. A. Rohlfing, J. Chem. Phys., 1995, 102, 8342 CrossRef CAS.
- J. D. Tobiason, J. R. Dunlop and E. A. Rohlfing, J. Chem. Phys., 1995, 103, 1448 CrossRef CAS.
- M. N. R. Ashfold, D. W. Chandler, C. C. Hayden, R. I. McKay and A. J. R. Heck, Chem. Phys., 1995, 201, 237 CrossRef CAS.
- P. P. Radi, M. Tulej, G. Knopp, P. Beaud and T. Gerber, J. Raman Spectrosc., 2003, 34, 1037 CrossRef CAS.
- P. Ewart and S. V. O’Leary, Opt. Lett., 1986, 11, 279 CAS.
- R. L. Farrow and D. J. Rakestraw, Science, 1992, 257, 1894 CAS.
- S. Williams, R. N. Zare and L. A. Rahn, J. Chem. Phys., 1994, 101, 1093 CrossRef CAS.
- S. Williams, D. S. Green, S. Sethuraman and R. N. Zare, J. Am. Chem. Soc., 1992, 114, 9122 CrossRef CAS.
- D. S. Green, T. G. Owano, S. Williams, D. G. Goodwin, R. N. Zare and C. H. Kruger, Science, 1993, 259, 1726 CAS.
- T. G. Owano, C. H. Kruger, D. S. Green, S. Williams and R. N. Zare, Diamond Relat. Mater., 1993, 2, 661 CAS.
- M. Tulej, M. Meisinger, G. Knopp, A. M. Walser, P. Beaud, T. Gerber and P. P. Radi, J. Raman Spectrosc., 2006, 37, 376 CrossRef CAS.
- M. Tulej, M. Meisinger, G. Knopp, A. M. Walser, P. Beaud, T. Gerber and P. P. Radi, J. Raman Spectrosc., 2006, 37, 680 CrossRef CAS.
- A. Okazaki, T. Ebata and N. Mikami, Chem. Phys. Lett., 1995, 241, 275 CrossRef CAS.
- D. Y. Chen, R. W. Fan, X. J. Yang, Q. K. Zheng and Q. Z. Qin, Chin. Phys. Lett., 2004, 21, 295 CrossRef CAS.
- T. J. Butenhoff and E. A. Rohlfing, J. Chem. Phys., 1992, 97, 1595 CrossRef CAS.
- Y. Tang and S. A. Reid, Chem. Phys. Lett., 1996, 248, 476 CrossRef CAS.
- T. Muller and P. H. Vaccaro, Chem. Phys. Lett., 1997, 266, 575 CrossRef.
- T. J. Butenhoff and E. A. Rohlfing, J. Chem. Phys., 1992, 97, 1595 CrossRef CAS.
- Y. Tang and S. A. Reid, Chem. Phys. Lett., 1998, 292, 691 CrossRef CAS.
- Y. Tang, J. P. Schmidt and S. A. Reid, J. Chem. Phys., 1999, 110, 5734 CrossRef CAS.
- K. Nyholm, M. Kaivola and C. G. Aminoff, Opt. Commun., 1994, 107, 406 CrossRef CAS.
- C. F. Kaminski, I. G. Hughes and P. Ewart, J. Chem. Phys., 1997, 106, 5324 CrossRef CAS.
- G. M. Lloyd and P. Ewart, J. Chem. Phys., 1999, 110, 385 CrossRef CAS.
- F. Di Teodoro and E. F. McCormack, J. Chem. Phys., 1999, 110, 8369 CrossRef CAS.
- T. J. Butenhoff and E. A. Rohlfing, J. Chem. Phys., 1993, 98, 5460 CrossRef CAS.
- S. Williams, E. A. Rohlfing, L. A. Rahn and R. N. Zare, J. Chem. Phys., 1997, 106, 3090 CrossRef CAS.
- A. C. Eckbreth, Appl. Phys., 1978, 32, 421 CAS.
- R. L. Abrams, J. F. Lam, R. C. Lind, D. G. Steel and P. F. Lao, Phase conjugation and high-resolution spectroscopy by resonant degenerate four-wave mixing, in Optical Phase Conjugation, ed. R. A. Fisher, Academic, New York, 1983, pp. 211-284 Search PubMed.
- R. T. Bratfalean, G. M. Lloyd and P. Ewart, J. Opt. Soc. Am. B, 1999, 16, 952 Search PubMed.
- C. M. Western, PGOPHER, a program for simulating rotational structure, University of Bristol, 2007, http://pgopher.chm.bris.ac.uk Search PubMed.
- A. Tanabashi, T. Hirao, T. Amano and P. F. Bernath, Astrophys. J., Suppl. Ser., 2007, 169, 472 CrossRef CAS.
- R. W. Field, E. Hirota, J. P. Maier and S. Tsuchiya, Nonlinear Spectroscopy for Molecular Structure Determination, Blackwell Science, Oxford, 1998 Search PubMed.
- P. Zalicki and R. N. Zare, J. Chem. Phys., 1995, 102, 2708 CrossRef CAS.
- G. Stark and S. P. Davis, Z. Phys. A, 1985, 321, 75 CAS.
- L. Lehr and P. Hering, IEEE J. Quantum Electron., 1997, 33, 1465 CrossRef CAS.
- J. R. Flores, J. Phys. Chem. B, 2003, 107, 9711 CrossRef CAS.
| This journal is © the Owner Societies 2008 |