The dynamics and orientation of a lipophilic drug within model membranes determined by 13C solid-state NMR
Received 21st August 2007, Accepted 5th October 2007
First published on 31st October 2007
Abstract
Methods for determining how a drug interacts with cellular membranes at the molecular level can give valuable insight into the mode of action of the drug and its absorption, distribution and metabolism profile. A procedure is described here to determine the orientation and location of the lipophilic drug trifluoperazine (TFP) intercalated into dimyristoylphosphatidylcholine (DMPC) bilayers, by using a novel combination of high-resolution solid-state nuclear magnetic resonance (SSNMR) methods to observe signals from 13C within the drug at natural abundance. SSNMR measurements of 1H–13C dipolar couplings for TFP and selective broadening of 13C NMR peaks by paramagnetic Mn2+ together suggest a model for the location, orientation and dynamics of the drug within lipid bilayers that offers an explanation for the lysoprotective effect of the drug at low concentrations. The experiments described are straightforward to implement and can be used for the routine analysis of drug–membrane interactions to provide useful information for drug design and structure refinement.
Introduction
The interactions between drug compounds and cellular membranes play a critical role in drug absorption, distribution, metabolism and excretion (ADME).1,2 There are also numerous instances of drugs that exert their pharmacological response, or give rise to undesirable side-effects, as a direct result of interacting with membrane phospholipids. For example, the pharmacology of the anticonvulsant valproic acid3 and the anticancer agent adriamycin4 are both believed to involve direct interactions with lipid bilayers. Other drugs, such as benzodiazepenes, accumulate at membrane surfaces,5 which may counter pharmacological efficacy of the compound by lowering its effective concentration and reducing its bioavailability. Moreover, compounds such as the anti-parasitic drug praziquantel and phenyldantoin alter the physicochemical properties of cellular membranes and model phospholipid vesicles, having significant consequences for membrane fluidity.6 Consequently there is interest within preclinical drug discovery to develop methods to characterise drug–membrane interactions beyond the simple quantification of lipid solubility.7NMR spectroscopy is a useful tool for probing the location of small lipophilic molecules within the phospholipid bilayers of multilamellar vesicles (MLVs) or large unilamellar vesicles (LUVs) that are used routinely as model membrane systems. For instance, NOESY methods have been combined with magic-angle spinning (MAS) to provide site-specific measurements of intermolecular cross relaxation rates between the aromatic protons of drugs and phospholipid protons that place constraints on the position of drug within the bilayer.8–11 In principle the signals from 13C nuclei present in virtually all drug compounds at natural abundance can also be exploited to gain insight into the location and orientation of drugs within phospholipid membranes. Although the sensitivity and natural abundance of 13C is much lower than for 1H, there are several features of 13C spectroscopy that are attractive for studies of drug–membrane interactions. The 13C chemical shift dispersion for the membrane-associated species is usually large compared to the line widths when solid-state NMR (SSNMR) line narrowing methods are applied and, consequently, the peaks for the partitioned species in the 13C spectrum are often less overlapped than in the corresponding 1H spectrum. SSNMR methods can be applied in a variety of ways to provide site-specific information about drugs in membrane environments. For example Villalain and co-workers used 13C MAS NMR and paramagnetic gadolinium ions to measure the insertion depth of triclosan in lipid bilayers by measuring changes in the 13C T1 relaxation profile of both the lipids and drugs.12
Here a novel combination of 13C SSNMR experiments has been used to provide information about the location and dynamics of drugs partitioned into lipid membranes. The procedure is used to examine the membrane location and orientation of the anti-psychotic drug trifluoperazine (TFP), a calmodulin antagonist that is well-known to interact with cell membranes.13 The chemical structure of TFP consists of a hydrophobic tricyclic ring and a more hydrophilic piperidine tail (Fig. 1). At low concentrations TFP protects erythrocytes from hypotonic lysis but at higher concentrations the drug can form micelles that haemolyse erythrocytes and solubilize phospholipid bilayers.13 It is shown how the 13C detection of 13C–1H dipolar couplings combined with paramagnetic relaxation measurements give valuable constraints on the orientation and depth of insertion of TFP into dimyristoylphosphatidylcholine (DMPC) bilayers and also reveal how the dynamics of the drug may contribute to its effects on biological membranes.
Experimental
Materials
Trifluoperazine dihydrochloride and DMPC were purchased from Sigma-Aldrich, Ltd (UK) and [acyl-2H54]L-α-dimyristoylphosphatidylcholine (DMPC-d54) was purchased from Avanti Polar Lipids.Multilamellar vesicle samples were prepared by dissolving 50 mg DMPC in chloroform–methanol and drying the sample down to a lipid film under argon and then under high vacuum overnight. A solution of TFP (5 mM in 20 mM phosphate, 1 mM EDTA, pH 7.4 was added to the dry lipid (50 mg) and the lipid mixture was vortexed and subjected to 5 cycles of freeze–thawing to distribute the drug evenly within the vesicles. In some experiments the paramagnetic relaxation agent MnCl2 was added to the vesicles and the freeze–thawing procedure was followed as above.It was necessary to begin with a high initial concentration of TFP in solution so that suspension of the lipid in the drug solution would achieve a high enough TFP/DMPC molar ratio (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :50) to detect an adequate signal from the drug in the NMR experiments. The initial volume of the TFP solution was kept relatively low (500 μL) so as to shift the partition equilibrium of the drug toward the membrane when the lipid was added. The partition coefficient (log PPC) for TFP in phosphatidylcholine LMVs was estimated to be 3.88 by following an adaptation of the 19F NMR method described by Omran et al.14 in which the 19F T1 relaxation times for TFP were measured at different lipid concentrations to yield a binding curve.15 From this value, the fraction F of the total drug concentration within the DMPC membranes can be calculated from
:50) to detect an adequate signal from the drug in the NMR experiments. The initial volume of the TFP solution was kept relatively low (500 μL) so as to shift the partition equilibrium of the drug toward the membrane when the lipid was added. The partition coefficient (log PPC) for TFP in phosphatidylcholine LMVs was estimated to be 3.88 by following an adaptation of the 19F NMR method described by Omran et al.14 in which the 19F T1 relaxation times for TFP were measured at different lipid concentrations to yield a binding curve.15 From this value, the fraction F of the total drug concentration within the DMPC membranes can be calculated from
| |  | (1) |
where
Va and
Vm are the volumes of the aqueous and
membrane phases. Here,
Va is 500 μL and
Vm is estimated to be 50 μL (from the hydrated density of 1 g mL
−1 for DMPC), so for
PPC of 10
3.88 over 98% of the
drug resides within the
membrane at equilibrium. To reduce the volume of the aqueous phase, the excess
water was removed by
centrifugation and the viscous
membrane sediment was transferred to an
NMR sample rotor.
NMR methods
Solid-state NMR experiments were performed on a Bruker Avance 400 spectrometer operating at a magnetic field of 9.3 Tesla and corresponding to resonance frequencies of 400.52 MHz for 1H, 161.23 for 31P, 61.62 MHz for 2H and 100.13 MHz for 13C. The sample temperature was maintained at 30 °C (±0.5 °C) in all experiments. Samples were confined to the central region of a 4 mm external diameter zirconia rotor. Proton spectra were obtained without water suppression with MAS at a spinning frequency (νr) of 5 kHz (±2 Hz). Wide line 2H spectra were recorded without sample spinning using the quadrupole echo (90x–τ–90y–τ–acquisition)16 with a 90° pulse length of 4 μs, delay τ of 22 μs and a recycle delay of 1 s. Proton decoupled 13C cross-polarization magic-angle spinning (CP-MAS) experiments were performed at a MAS rate of 3.4 kHz. Hartmann–Hahn cross-polarization from 1H to 13C was achieved over a 1.6 ms contact time at a field of 65 kHz for both nuclei, and protons were decoupled during signal acquisition at a field of 85 kHz. Two-pulse phase-modulated (TPPM) proton decoupling17 was applied during the acquisition period. A recycle delay of 2 s was used in all experiments. Measurements of 13C–1H dipolar coupling strengths were performed using the constant time dipolar and chemical shift correlation experiment (CT-DIPSHIFT)18 shown in Fig. 2(a). In a series of 8 experiments, peak intensities were measured for 8 t1 intervals, where t was incremented from zero to 1/νr. The 8 spectra (Sn), each defined by 1024 points, were then replicated 16 times and concatenated (S1, S2… S8, S1, S2… S8, etc.), to give a 1024 × 128 matrix, which was Fourier transformed in the indirect dimension to produce the pseudo-two dimensional spectra, with the 13C chemical shift information in the direct dimension and C–H dipolar coupling information in the indirect dimension. |
| | Fig. 2 The CT-DIPSHIFT experiment. (a) In this experiment, after 1H–13C Hartmann–Hahn cross-polarization (CP) the 13C magnetization is then allowed to evolve over two rotational cycles (2/νr), applying an 8 μs π pulse to the 13C spins at the end of the first rotational cycle to refocus the chemical shifts at the end of the evolution period. Homonuclear decoupling of protons is applied for a defined interval t1 during the first rotor cycle of the dipolar evolution using the frequency switched Lee-Goldburg (FSLG) sequence.28 Following the period t1, continuous wave proton decoupling is applied at a field of 85 kHz until the end of the acquisition period (t2). (b) A simulated dipolar profile calculated for a C–H group oriented at an angle θCH of 10° with respect to a single axis of rapid rotation. The profile was calculated for a MAS frequency of 3.4 kHz. (c) A C–H dipolar side-band pattern obtained by reproducing the dipolar profile over several sample rotation cycles followed by Fourier transformation. | |
Simulation of two-dimensional CT-DIPSHIFT spectra
The modulation of signal amplitude A for the 13C spin of a given C–H group over the evolution period t1 of the CT-DIPSHIFT experiment is given by | |  | (2) |
where δCH is the rigid limit dipolar anisotropy (scaled to 11800 Hz by homonuclear decoupling), ΩML is the set of Euler angles describing the orientation of each molecule in the magnetic field and 〈…〉 denotes a statistical powder average over all possible molecular orientations. In the case of a molecule undergoing anisotropic rotation about a principal axis within a lipid bilayer, A(t1) becomes additionally dependent on the angle θCH, defining the orientation of the C–H bond relative to the rotational axis.The value of δCH for a given spin for a drug undergoing rapid anisotropic motions within a lipid bilayer is dynamically averaged and can be replaced by a scaled value (δAV) given by:19,20
| |  | (3) |
SCH is an order parameter describing the orientation and angular fluctuations of the
drug in the bilayer and can be expressed as
| |  | (4) |
where
Smol is a measure of the angular excursions of the principal rotational axis with respect to the bilayer normal.
Smol takes values from 0, representing the isotropic extreme and 1 indicating that no additional angular excursions of the rotational axis occur.
Generally, profiles of signal amplitudes over t1 of the CT-DIPSHIFT experiment (e.g.Fig. 2(b)) can be calculated for different values of SCH by replacing δCH in eqn (2) with δav calculated from eqn (3) and (4). Simulations show that the dipolar profiles are sensitive to SCH and thus can provide information about the orientational order of a drug in lipid bilayers. Dipolar side-band patterns can be obtained by concatenating identical dipolar profiles for several sample rotation cycles and then performing a Fourier transformation (Fig. 2(c)). The side-band pattern in the dipolar spectrum reflects the value of δAV, with the intensities of the outer side-bands decreasing with increased motional scaling of δ. In the CT-DIPSHIFT spectra, the direct dimension holds the chemical shift information and the indirect dimension shows the side-band patterns of the dipolar spectrum.
To simulate CT-DIPSHIFT spectra for the specific case of TFP within DMPC membranes, a non-linear least squares routine was first used to fit Lorentzian lines to the peaks of interest in the experimental 13C NMR spectrum. Two-dimensional spectra were then constructed from each of the calculated Lorentzian lines individually using the following procedure. Each individual line was used to generate a series of amplitude modulated lines by multiplying the initial line by signal amplitudes at different t1 times taken from the calculated dipolar profile for a given value of SCH. A total of 64 such spectra were generated from amplitudes at 64 consecutive and equally spaced evolution times covering 8 rotational cycles. These spectra represent the time domain in the indirect dimension of the two-dimensional spectra. Fourier transformation of the 64 amplitude modulated peaks produced a two-dimensional spectrum for each of the simulated peaks of interest. The final spectrum was then obtained by adding together all the two-dimensional spectra.
Results and discussion
Initial characterization
In order to achieve a suitable lipid/drug molar ratio for the NMR studies it was necessary to initially dissolve TFP in aqueous solution to a much higher concentration (5 mM) than the apparent critical micellar concentration (CMC) of the drug (20–30 μM). The CMC of TFP was deduced previously from decreases in fluorescence emission intensity for the drug observed around the 20–30 μM concentration range13 and the micelles formed have been shown to solubilize erythrocyte and model lipid membranes. Here it was important that TFP did not self-associate under the initial conditions of the experiments because solubilization of the DMPC vesicles by TFP micelles would destroy the lipid bilayer and complicate the interpretation of the NMR spectra. The aggregation state of TFP in solution was therefore monitored over concentrations ranging from 1 μM to 5 mM using fluorescence and 1H NMR spectroscopy. It was found that the intensity of TFP fluorescence did not change over this concentration range and the 1H NMR line widths for the drug in aqueous solution remained narrow up to 5 mM (data not shown). As drug self-association would be expected to be marked by an abrupt decrease in fluorescence and a concomitant broadening of the NMR lines, these findings are together consistent with TFP remaining monomeric in aqueous solution up to a concentration of 5 mM under the conditions of these experiments.After adding the 5 mM TFP solution to DMPC membranes, wide line 31P NMR was used to probe the lipid phosphate groups in order to determine whether the high concentration of drug disrupts the lipid bilayer. The 31P spectra of DMPC alone and in the presence of TFP at drug/lipid molar ratios of up to 1:50 showed line shapes characteristic of bilayer membranes before and after adding TFP. A very small central component appeared in the spectrum (shown at ∼15 ppm) after adding TFP, which is characteristic of a negligible fraction of smaller vesicles, mixed micelles or other non-bilayer structures. Nevertheless, the overall shape of the spectrum indicated that the vast majority of the lipid bilayer sample was not perturbed or solubilized by the drug (Fig. 3(a)). Similarly, wide line 2H NMR spectra of chain-perdeuterated DMPC (DMPC-d54) showed a slight overall broadening after adding TFP, but there was no evidence from the measurable (i.e. the innermost and outermost) quadrupole splittings that any significant disordering of the lipid chains occurred (Fig. 3(b)). These observations are consistent with the drug remaining in a monomeric, non-solubilizing form within the lipid bilayer.
 |
| | Fig. 3 Wide line NMR spectra of DMPC membranes before and after the addition of TFP. (a) 31P NMR spectra before and after the addition of TFP at the lipid/drug molar ratios shown. Spectra were obtained by accumulating 2048 scans. (b) 2H NMR spectra of DMPC-d54 alone and after the addition of TFP at a lipid/drug molar ratio of 50:1. Spectra were obtained by accumulating 40960 scans. | |
Next, 1H and 13C NMR spectra were obtained (with MAS) from the 1:50 TFP/DMPC membrane sample to compare the resolution of the peaks for the aromatic moiety of the drug in the two spectra. The aromatic signals do not overlap with any of the lipid resonances and so they are in principle useful reporters of the drug environment within the membrane if the peaks are sufficiently resolved from each other to be assigned to specific positions in the aromatic ring. Peaks for the aromatic protons are clearly visible in the 1H spectrum after just 2 minutes of data collection (Fig. 4(a)), but the chemical shift range is narrow and the lines are broad because of strong residual 1H–1H dipolar couplings. Consequently there is significant peak overlap and only one peak can be attributed to a specific aromatic proton (assigned from the spectrum of the drug in aqueous solution). The 13C spectrum by comparison shows a much greater dispersion of the peaks for the tricyclic ring of TFP (Fig. 4(b)) and although some of the peaks overlap, several others are resolved enough for it to be possible to assign them to specific ring carbons (C5, C6, C8, C9, C12 and C14) by comparison with the solution spectrum. Thus despite the much longer acquisition times needed to obtain good signal to noise (6–10 h), the 13C spectrum contains more site-specific information about the drug than does the 1H spectrum.
 |
| | Fig. 4 High-resolution NMR spectra of DMPC membranes containing TFP at a lipid/drug molar ratio of 50:1. Proton (a) and 13C (b) NMR spectra were obtained with MAS at a spinning rate of 3.4 kHz. The 2H spectrum was obtained after accumulating 128 scans and the 13C spectrum was obtained with CP-MAS by accumulating 40960 scans. The insets show the peaks assigned to the tricyclic ring of TFP, labelled according to the carbon atom numbering system given in Fig. 1. | |
Dynamics of TFP in lipid bilayers
The signals for TFP in the 13C spectrum in Fig. 4(b) can be attributed exclusively to the drug within DMPC bilayers and any drug remaining in the residual aqueous phase at equilibrium is not detected. This is because the spectrum was obtained with 1H–13C Hartmann–Hahn cross-polarization (HHCP), which does not detect small molecules in aqueous solution as their rapid isotropic tumbling orientationally averages the dipolar couplings that are necessary to generate observable signals.21 Molecules that are partitioned into lipid bilayers, however, assume the anisotropic dynamics of the membrane environment and as a result the dipolar couplings are not averaged to zero, thereby giving rise to detectable signals. The peak intensities for some of the protonated aromatic carbon atoms of TFP at HHCP contact times from 1 ms to 10 ms give profiles that resemble the shape of profiles for the lipid glycerol carbons, reaching maximum intensity at contact times of 4–6 ms (Fig. 5). This similarity suggests that the dynamics of TFP are close to those of the lipid molecules, and also indicates that the drug is not rapidly exchanging between the membrane and aqueous phases, which would shift the maxima to much longer contact times similar to the profiles of non-protonated (e.g., lipid carbonyl or TFP C9) carbons.22 |
| | Fig. 5 Peak intensities at Hartmann–Hahn contact times ranging from 1–10 ms measured from the 13C CP-MAS NMR spectrum of DMPC membranes containing TFP at a lipid/drug molar ratio of 50:1. Values are expressed as a percentage of maximum intensity for each peak over the contact time range. (a) Peak intensities for positions C6, C9, C12 and C14 of the TFP aromatic ring. (b) Values for the glycerol backbone (G1, G2, G3) and carbonyl (CO) carbons of DMPC. Error bars represent the level of the noise. | |
The dynamic behaviour of drugs and other lipophilic molecules within the relatively ordered environment of a lipid bilayer usually involves their rapid anisotropic reorientation about a principal axis of rotation, usually assumed to be parallel with the bilayer normal.23,24 By measuring the angle θCH representing the inclination of individual aromatic C–H bonds of TFP relative to this principal rotational axis, it is possible to draw conclusions about the orientation and order of the drug within DMPC bilayers. Orientational information was obtained using the pseudo-two-dimensional CT-DIPSHIFT experiment to measure the C–H dipolar anisotropy for specific aromatic ring positions of TFP in the DMPC membranes. The dipolar anisotropy was measured only for the aromatic carbons C6, C12 and C14 because they are easily distinguishable in the 13C spectrum and all have bonded protons to which they are coupled.
To demonstrate the sensitivity of the dipolar side-band pattern to molecular orientation a molecular reference frame is first defined for TFP, with the y-axis bisecting the S and N ring atoms and the z-axis bisecting the two outer ring centroids (Fig. 6(a)). Simulations of CT-DIPSHIFT spectra for the tricyclic ring of TFP rotating about either of these two orthogonal axes reveal characteristic differences in the side-band patterns for C6, C12 and C14 (Fig. 6(b) and (c)). Recalling eqn (3) and (4), if it is assumed that Smol equals 1 because there are no additional motional fluctuations of the rotational axis with respect to the bilayer normal, then for rotation of the phenothiazine ring about y, θCH is approximately 0° for C6 and C14 and 60° for C12. In this case |SCH| is 1.0 for C6 and C14 (i.e. δAV = δCH = 11 800 Hz) and |SCH| is 0.125 for C12 (δAV = 1475 Hz). The simulated DIPSHIFT spectrum for rotation about y gives rise to observable intensity in up to the third-order (±3) side-bands for C6 and C12, covering a frequency range of 20 kHz in the indirect dimension, whereas the central line predominates for C14 and little or no side-band intensity is observed (Fig. 6(b)). For rotation of the tricyclic ring about z, θCH equals 90° for C6 and C14 and 30° for C12. Correspondingly |SCH| is 0.5 for C6 and C14 (i.e., δAV = 5400 Hz) and |SCH| is for C12 (δAV = 7375 Hz) and thus rotation about z gives rise to observable intensity only up to the second-order (±2) side-bands for C6, C12 and C14 (Fig. 6(c)).
 |
| | Fig. 6 Simulations demonstrating the sensitivity of the CT-DIPSHIFT experiment to the orientation of TFP relative to its principal axis of anisotropic rotation within a lipid bilayer. (a) The molecular reference frame for TFP with respect to the structure of the phenothiazine ring. (b) A simulated two-dimensional CT-DIPSHIFT spectrum corresponding to the signals for C6, C12 and C14 of TFP, calculated for rapid molecular rotation about axis y and assuming that the axis of motion does not undergo additional motional fluctuations (i.e., Smol is equal to 1). (c) A simulated spectrum for rotation about z. The horizontal dashed lines highlight the positions of side-bands (±1, ±2 and ±3) relative to the central line at 0 Hz. | |
The experimental DIPSHIFT spectrum for the TFP/DMPC sample is shown in Fig. 7(a). A comparison of the spectrum with simulated spectra for different order parameters (Fig. 7(b)) reveals the best agreement be for |SCH| values of 0.8–0.9 for C12 and 0.2–0.3 for C6 and C14 (the middle two simulated spectra). It is possible to calculate values of θCH and Smol by exploiting the fixed geometry of the C–H bonds in the tricyclic ring (i.e. the C–H bonds of C6 and C14 are oriented at 120° with respect to the C–H bond of C12). Hence,
| |  | (5) |
and
| |  | (6) |
and by solving the two simultaneous equations for the boundary condition 0 ≤
Smol≤ 1 values of
θCH (14°–18°) and
Smol (0.92–0.97) were found. The data are therefore consistent with a principal axis of rotation bisecting the
xz plane and inclined at an angle of 16°± 2° relative to the
z axis (
Fig. 7(c)). Moreover, the value of
Smol is close to 1, which supports the assumption that there are no additional motional fluctuations of the rotational axis with respect to the bilayer normal. If the angle of rotation is coincident with the bilayer normal, then
TFP is oriented with the three ring system extending into the bilayer, although it is not known at this stage how the
piperidine group is positioned relative to the tricyclic moiety or whether the CF
3 group points toward or away from the
lipid polar head-
groups.
 |
| | Fig. 7 Determination of the orientation of TFP in DMPC bilayers using the two-dimensional CT-DIPSHIFT experiment. (a) The experimental CT-DIPSHIFT spectrum for TFP in DMPC bilayers. Each of the one-dimensional spectra used to construct the two-dimensional DIPSHIFT spectrum was the result of accumulating 40960 scans. (b) Simulated spectra calculated for order parameters SCH of (from left to right) 0.1–0.4 for C6/C14 and 1.0–0.7 for C12. (c) The tricyclic ring of TFP oriented with the z-axis inclined at an angle of 16° relative to the axis of rotation. | |
Orientation and insertion depth of TFP
Further 13C experiments were carried out on the TFP/DMPC membrane sample to validate the conclusions of the CT-DIPSHIFT experiment and to confirm the orientation of the phenothiazine ring and position of the piperidine group within the bilayer. Peak intensities in the spectra were compared before and after adding 100 μM Mn2+ in order to probe the approximate depth of various positions in the TFP molecule in the bilayer relative to the surface of the membrane. Paramagnetic ions such as Mn2+ and lanthanides bind to lipid head-groups and enhance nuclear T2 relaxation times for nuclear spins within the bilayer to an extent that depends on the ionic concentration and the distance of the relaxing nucleus from the membrane surface. Thus the addition of Mn2+ diminishes the peak intensities for sites close to the membrane surface, such as for the lipid head-groups and glyceryl backbone, to a greater extent than peaks for sites closer to the centre of the bilayer. This effect has been exploited to probe the depth of drugs and peptides in lipid bilayers, by observing selective changes in peak intensities, line widths or relaxation times for different parts of the molecule.12,25,26The 13C NMR spectrum of the sample before and after the addition of Mn2+ indicates that peaks for the lipid head-group, glyceryl backbone and acyl chain carbons C2 and C3 have been abolished, whereas peaks for acyl chain carbons closer to the centre of the bilayer remain visible despite being diminished in intensity (Fig. 8(a)). The selective effect reflects the distance of the lipid sites from the membrane surface. The peak intensities for TFP are also affected selectively by the addition of Mn2+. The overlapping peaks for the piperidine ring and alkyl linker group in the 40–50 ppm range are all abolished after the addition of Mn2+ (Fig. 8(b)) suggesting that these regions of the molecule lie close to the membrane surface. Some of the peaks for the tricyclic moiety are also abolished (C2, C3, C4, C5 and C6), whereas others remain visible but are much lower in intensity (C8, C9, C12, C14) relative to the spectrum before addition of Mn2+. The peak at 127 ppm also remains visible, but it is not clear whether the residual intensity arises from C1, C11 or both. Despite this one uncertainty in the peak assignments, overall these results are consistent with the CF3-substituted ring being oriented toward the bilayer interior and the ring defined by C1–C6 being oriented toward the membrane surface. The peaks for the CF3-substituted ring of TFP are diminished by approximately the same proportion as the peaks for C12, C13 and C14 of DMPC, suggesting that the substituted ring resides close to the centre of the lipid bilayer.
 |
| | Fig. 8 An experiment to determine the orientation of TFP in DMPC bilayers by observing 13C NMR peak intensities before and after adding 100 μM MnCl2. (a) The 13C spectrum of DMPC membranes containing TFP at a lipid/drug molar ratio of 50:1 showing peaks for the lipid only before (top) and after (bottom) the addition of MnCl2. Peaks correspond to lipid acyl chain segments (C2, C3, C4–C11, C12, C13 and C14), carbonyl (C1), glyceryl backbone (G1, G2, G3) and choline headgroup (H1, H2). (b) A region of the spectra in (a) highlighting the peaks from the pheothiazine moiety of TFP before (top) and after (bottom) the addition of MnCl2. The spectra were vertically expanded by 10 times (×10) or fifty times (×50) as shown. The spectra were the result of accumulating 81920 scans. | |
A model of TFP in DMPC bilayers
The information above was used to derive a model of TFP in DMPC membranes in which drug is oriented with the tricyclic ring approximately parallel with the bilayer normal and the piperidine ring facing up toward the membrane surface (Fig. 9). As the phenothiazine moiety rotates anisotropically about an angle approximately parallel with its long axis, the rotational trajectory of the entire molecule traces an inverted cone that would have the effect of creating more space between the head groups of the surrounding phospholipids. The piperidine ring and alkyl linker are also likely to add to this effect by undergoing additional segmental/rotational motions as the molecule sweeps through 360°. The lysoprotective effect of monomeric TFP at low concentrations may originate from the dynamic behaviour of the drug in the membrane. By driving the lipid head groups apart the drug may force the acyl chains to pack together deeper within the hydrophobic core of the lipid bilayer creating a more tight sealed barrier.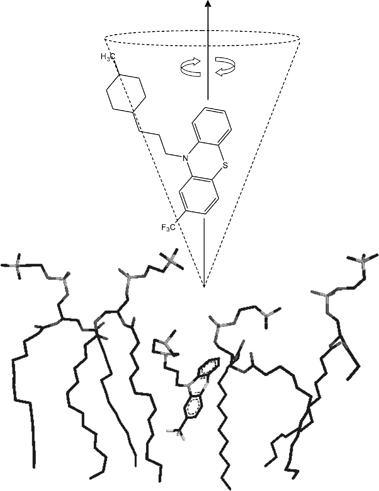 |
| | Fig. 9 A model for the orientation of TFP within a single leaflet of DMPC bilayers showing the proposed rotational trajectory of the drug. | |
Conclusions
In summary, this work has shown how a novel combination of 13C SSNMR methods can provide information about the orientation and location of small organic molecules within lipid bilayers. By taking advantage of the NMR signals from 13C at natural abundance, paramagnetic relaxation experiments using water-soluble Mn2+ have provided information about the depth of insertion and orientation of TFP within lipid bilayers. It is worth noting that the insertion depth of drugs could also be measured in principle from within the membrane by using membrane-soluble oxygen at high partial pressures to give rise to position-dependent paramagnetic effects,27 although the experimental technique is perhaps less straightforward to implement than the method described here. Critically this work shows that further information about the orientation and dynamics of TFP obtained using the CT-DIPSHIFT experiment can offer insights into the membrane-perturbing behaviour, and possibly the pharmacological effects, of the drug at low concentrations. The procedure is easily adaptable for routine analysis of a variety of lipophilic drugs to investigate how the pharmacological and toxicological properties of drugs are derived from their interactions with cell membranes.Abbreviations
Acknowledgements
The BBSRC are acknowledged for a studentship to M.P.B. and for funding in support of the 400 MHz NMR facility.References
- O. G. Mouritsen and K. Jorgensen, Pharm. Res., 1998, 15, 1507–1519 CrossRef CAS.
- J. K. Seydel, E. A. Coats, H. P. Cordes and M. Wiese, Arch. Pharm., 1994, 327, 601–610 CrossRef CAS.
- A. Kessel, B. Musafia and N. Ben-Tal, Biophys. J., 2001, 80, 2536–2545 CrossRef CAS.
- T. Soderlund, A. Jutila and P. K. J. Kinnunen, Biophys. J., 1999, 76, 896–907 CrossRef CAS.
- D. A. Garcia and M. A. Perillo, Biochim. Biophys. Acta, 1999, 1418, 221–231 CAS.
- S. V. P. Malheiros, N. C. Meirelles and E. de Paula, Biophys. Chem., 2000, 83, 89–100 CrossRef CAS.
- Y. Xu, V. E. Yushmanov and P. Tang, Biosci. Rep., 2002, 22, 175–196 CrossRef CAS.
- J. F. Ellena, R. N. Dominey, S. J. Archer, Z. C. Xu and D. S. Cafiso, Biochemistry, 1987, 26, 4584–4592 CrossRef CAS.
- L. L. Holte and K. Gawrisch, Biochemistry, 1997, 36, 4669–4674 CrossRef CAS.
- H. A. Scheidt, A. Pampel, L. Nissler, R. Gebhardt and D. Huster, Biochim. Biophys. Acta, 2004, 1663, 97–107 CAS.
- A. Siarheyeva, J. J. Lopez and C. Glaubitz, Biochemistry, 2006, 45, 6203–6211 CrossRef CAS.
- J. Guillén, A. Bernabeu, S. Shapiro and J. Villalaín, Eur. Biophys. J. Biophys. Lett., 2004, 33, 448–453 Search PubMed.
- S. V. P. Malheiros, E. de Paula and N. C. Meirelles, Biochim. Biophys. Acta, 1998, 1373, 332–340 CAS.
- A. A. Omran, K. Kitamura, S. Takegami, M. Kume, M. Yoshida, A. A. Y. El-Sayed, M. H. Mohamed and M. Abdel-Mottaleb, J. Pharm. Biomed. Anal., 2002, 30, 1087–1092 CrossRef CAS.
- M. P. Boland, PhD Thesis, University of Manchester, 2007 Search PubMed.
- J. H. Davis, K. R. Jeffrey, M. Bloom, M. I. Valic and T. P. Higgs, Chem. Phys. Lett., 1976, 42, 390–394 CrossRef CAS.
- A. E. Bennett, C. M. Rienstra, M. Auger, K. V. Lakshmi and R. G. Griffin, J. Chem. Phys., 1995, 103, 6951–6958 CrossRef.
- M. Hong, J. D. Gross and R. G. Griffin, J. Phys. Chem. B, 1997, 101, 5869–5874 CrossRef CAS.
- M. Auger, H. C. Jarrell and I. C. P. Smith, Biochemistry, 1988, 27, 4660–4667 CrossRef CAS.
- J. Seelig, Q. Rev. Biophys., 1977, 10, 353–418 CrossRef CAS.
- M. P. Boland and D. A. Middleton, Magn. Reson. Chem., 2004, 42, 204–211 CrossRef CAS.
- S. G. Patching, A. R. Brough, R. B. Herbert, J. A. Rajakarier, P. J. F. Henderson and D. A. Middleton, J. Am. Chem. Soc., 2004, 126, 3072–3080 CrossRef CAS.
- M. Auger, Biophys. Chem., 1997, 68, 233–241 CrossRef CAS.
- D. P. Yang, T. Mavromoustakos, K. Beshah and A. Makriyannis, Biochim. Biophys. Acta, 1992, 1103, 25–36.
- G. Grobner, C. Glaubitz and A. Watts, J. Magn. Reson., 1999, 141, 335–339 CrossRef CAS.
- J. J. Buffy, T. Hong, S. Yamaguchi, A. J. Waring, R. I. Lehrer and M. Hong, Biophys. J., 2003, 85, 2363–2373 CrossRef CAS.
- P. A. Luchette, R. S. Prosser and C. R. Sanders, J. Am. Chem. Soc., 2002, 124, 1778–1781 CrossRef CAS.
- M. Lee and W. I. Goldburg, Phys. Rev., 1965, 140, 1261 CAS.
|
| This journal is © the Owner Societies 2008 |
Click here to see how this site uses Cookies. View our privacy policy here. 