A robust conglomerate structure type in salts of cationic organopalladium complexes and non-coordinating anions†‡
Irmgard Kalf, Ruimin Wang and Ulli Englert*
Institute of Inorganic Chemistry, RWTH Aachen University, Landoltweg. 1, 52074 Aachen, Germany. E-mail: ullrich.englert@ac.rwth-aachen.de; Fax: + 49- 241- 8092288; Tel: +49 241 8094666
First published on 15th October 2007
Abstract
Ionic compounds derived from square planar cations of the type [Pd(C6H4CHMeNH2)(tmeda)]+ (tmeda = 1,1,2,2-tetramethylethylenediamine) and the non-coordinating anions BF4–, PF6– and CF3SO3– have been prepared by cyclopalladation of (aryl-substituted) primary amines and subsequent reaction with the chelating tmeda ligand. Six compounds among the tetrafluoroborates and hexafluorophosphates of unsubstituted and methyl, fluoro or chloro substituted cyclopalladated amines are isotypic: They all show conglomerate behaviour, crystallizing in the same space group with similar lattice parameters and closely related packing. Only one moderately strong hydrogen bond per formula unit between an amine H and a counteranion is formed. The stability range of this remarkably robust structure type has been explored by variation of the anion and substitution on the aryl ring of the primary amine. For three related palladium complexes which do not show conglomerate crystallization, the structures of their enantiopure and racemic crystals are reported for comparison.
Introduction
When crystals of a chiral compound precipitate from racemic solution, the outcome will be either (a) racemic crystals, (b) a conglomerate, or (c) a solid solution of the enantiomers. Pallavicini and coworkers have recently reported1 that three carnitinamide salts, namely the chloride, nitrate and sulfate, match these three crystallization alternatives. From a statistical point of view, alternative (a) represents the standard situation, (b) accounts for roughly 10–15% of all cases and (c) is restricted to rare exceptions.2–4 The probability to encounter a conglomerate is higher for ionic than for neutral compounds. 2 With respect to enrichment via entrainment and purification,5,6 conglomerates often represent the more attractive solids, for example in pharmaceutical industry.7 Extensive work on conglomerate formation in coordination compounds is due to Bernal and coworkers.8 We have decided to dedicate a systematic study to the comparison of conglomerates and racemates of organopalladium compounds, and we have recently communicated9 our results on partially racemic solids, i.e. salts containing an enantiopure ion and the racemate of a counteranion in the same unit cell.10 Partially racemic solids compete with diastereomeric salts and hence prevent the routinely performed resolution of a racemate, representing an unwanted crystallization alternative. In the present contribution, we wish to report the positive result of our investigation, namely the formation of a robust conglomerate structure type among salts of cationic organopalladium tmeda (tmeda = 1,1,2,2-tetramethylethylenediamine) complexes. This class of compounds is particularly suited for the intended study: in the more popular non-conglomerate cases, the use of enantiopure amines enforces the formation of homochiral solids which may then be compared to the racemic crystals. Such comparisons may help to identify features which favour or preclude conglomerate formation. From a synthetic point of view, these salts offer variability with respect to the anions and aryl substitution in the amine ligand.Scheme 1 provides a summary of the compounds synthesized and structurally characterized in the context of this work.
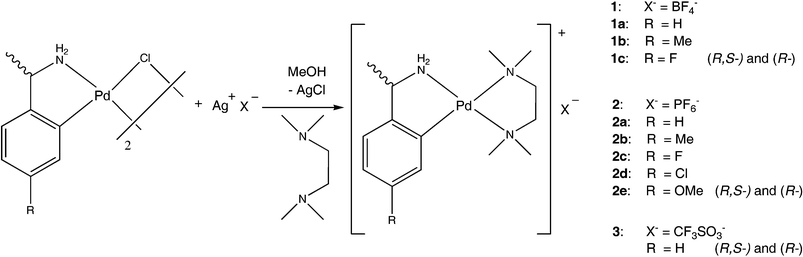 | ||
| Scheme 1 | ||
Experimental
Chemicals and reagents
Silver tetrafluoroborate, hexafluorophosphate and trifluoromethanesulfonate were purchased and used without further purification. The organometallic palladium reagents were obtained via cyclopalladation of the corresponding amines and converted to chloro-bridged dinuclear complexes by well-established methods.11,12Synthesis and characterization
General properties: 1a - R-3 are colourless, air stable solids. They decompose in solution under the formation of a Pd metal, especially in EtOH and at higher temperature. The solids do not melt but decompose between 120 and 195 °C; an exception is R-2e which melts at 120 °C; the heterochiral congener rac-2e decomposes around 170 °C.NMR spectra were recorded on a Varian Mercury 200 (1H: 200 MHz, 13C: 50 MHz), a Varian Unity 500 (1H: 500 MHz, 13C: 125 MHz) and a Bruker Avance II (1H: 400 MHz, 13C: 100 MHz, 19F: 376.5 MHz) at ambient temperature. 1H and 13C resonances are referenced to TMS, 19F spectra to CCl3F. IR spectra were recorded on KBr pellets with a Nicolet Avatar 360 E.S.P. FT instrument; absorption bands are reported in cm–1. Elemental analyses were carried out on a Heraeus CHNO-Rapid apparatus.
In the IR spectra, the following strong bands characteristic of the anions are found: tetrafluoroborates 1a - R-1c, νst(BF) around 1065; hexafluorophosphates 2a - R-2e, νst(PF) around 840; trifluoromethylsulfonates rac-3a and R-3a, νst(CF) around 1270, νst (SO) around 1160 and 1030 cm–1. In the case of compounds 1a, 1b and 2a–2d, the preparation and NMR data refer to racemic solutions from which conglomerates crystallize. In this case, it is commonly assumed that the enantiomers adopt the same or the mirror image structure as individual crystals of the conglomerate.4 In order to confirm this expectation for at least one example, preparation, spectroscopic characterization, crystallization and structure determination of 1a were successfully repeated starting from the enantiomerically pure organometallic precursor [{Pd(µ-Cl)(R-C6H4CHMeNH2)}2].
Synthesis: Products 1a - R-3 were obtained by the same method; as an example, the preparation of 1a is described: 0.200 g (0.382 mmol) of the dinuclear palladium complex [{Pd(µ-Cl)(C6H4CHMeNH2)}2] were dissolved in 60 mL of MeOH at 50 °C. 0.089 g (0.764 mmol) tetramethylethylenediamine dissolved in ca. 1 mL of MeOH and 0.149 g (0.764 mmol) solid AgBF4 were added. The reaction mixture was allowed to cool to room temperature under stirring and was filtered. For several compounds, crystals were directly obtained by slow evaporation of this filtrate. As slow decomposition with formation of metallic palladium competes with crystallization, optimum crystallization conditions are difficult to predict: we therefore conducted the isothermal evaporation in parallel at temperatures of 20 and 40 °C. Alternatively, the solvent may be completely evaporated in vacuum, and the resulting solid can be recrystallized from MeOH. For the latter procedure, yields in the isolated product range from 70 to 76%.
[Pd(C6H4CHMeNH2)tmeda]BF4, 1a, C14H26BF4N3Pd, MW = 429.61 g mol–1, decomp ca. 180 °C. Spectroscopic data 1H-NMR (ppm, DMSO-d6): δ = 1.54 (d, 3H, J = 6.4 Hz, CHMe); 2.61, 2.65, 2.81, 2.84 (4s, 4×3 H, tmeda-Me), 2.70–3.01 (m, 4H, tmeda-CH2), 4.07 (m, 1H, CHMe), 4.70 (br, 1H, NH2), 5.45 (br, 1H, NH2), 6.87–7.14 (m, 4H, aryl-H). 13C{1H}-NMR (ppm, DMSO-d6): δ = 24.74, 47.24, 48.32, 49.31, 50.91, 57.85, 58.57, 63.16, 121.08, 124.14, 125.06, 131.80, 147.53, 156.65. CHN-Analysis (%): Calcd.: C: 39.14, H: 6.10, N: 9.78. Found: C: 38.85, H: 5.89, N: 9.84.
[Pd(p-MeC6H3CHMeNH2)tmeda]BF4, 1b, C15H28BF4N3Pd, MW = 443.63 g mol–1, decomp ca. 180 °C. 1H-NMR (ppm, DMSO-d6): δ = 1.49 (d, 3H, J = 6.4 Hz, CHMe); 2.23 (s, 3H, 4-Me(Ph)); 2.59, 2.63, 2.81, 2.84 (4s, 4×3 H, tmeda-Me), 2.63–3.08 (m, 4H, tmeda-CH2), 4.01 (m, 1H, CHMe), 4.63 (br, 1H, NH2), 5.39 (br, 1H, NH2), 6.76 (br, 2H, aryl-H), 6.87 (br, 1H, aryl-H). 13C{1H}-NMR (ppm, DMSO-d6): δ = 21.16, 24.88, 48.35, 48.34, 49.42, 50.93, 57.87, 58.34, 63.17, 120.76, 124.80, 132.31, 133.71, 147.37, 153.56.
(rac)-[Pd(p-FC6H3CHMeNH2)tmeda]BF4, rac-1c, C14H25BF5N3Pd, MW = 447.58 g mol–1, decomp ca. 160 °C. 1H-NMR (ppm, DMSO-d6): δ = 1.52 (d, 3H, J = 6.4 Hz, CHMe); 2.61, 2.66, 2.80, 2.84 (4s, 4×3 H, tmeda-Me), 2.69–3.01 (m, 4H, tmeda-CH2), 4.07 (m, 1H, CHMe), 4.72 (br, 1H, NH2), 5.47 (br, 1H, NH2), 6.76–6.95 (m, 4H, aryl-H). 13C{1H}-NMR (ppm, DMSO-d6): δ = 24.78, 47.42, 48.34, 49.37, 50.83, 57.99, 58.09, 63.14, 110.60, 118.02, 122.10, 150.42, 152.64, 159.06 (d, J = 244 Hz, CF). 19F–NMR (ppm, DMSO-d6): δ = –116.2 (m, aryl-F), –148.2 (br, BF4–).
(R)-[Pd(p-FC6H3CHMeNH2)tmeda]BF4, R-1c, C14H25BF5N3Pd, MW = 447.58 g mol–1, decomp ca. 170 °C. 1H-NMR (ppm, DMSO-d6): δ = 1.53 (d, 3H, J = 6.3 Hz, CHMe), 2.61, 2.66, 2.81, 2.84 (4s, 4×3 H, tmeda-Me), 2.70–3.02 (m, 4H, tmeda-CH2), 4.07 (m, 1H, CHMe), 4.72 (br, 1H, NH2), 5.48 (br, 1H, NH2), 6.73–6.97 (m, 4H, aryl-H). 13C{1H}-NMR (ppm, DMSO-d6): δ = 24.78, 47.32, 48.35, 49.38, 50.84, 57.98, 58.09, 63.15, 110.60, 118.02, 122.10, 150.41, 152.63, 157.84, 160.28. 19F–NMR (ppm, DMSO-d6): δ = –116.2 (m, aryl-F), –148.2 (br, BF4–). CHN-Analysis (%): Calcd.: C: 37.57, H: 5.63, N: 9.39. Found: C: 37.07, H: 5.76, N: 8.94.
[Pd(C6H4CHMeNH2)tmeda]PF6, 2a: C14H26F6N3PPd, MW = 487.77 g mol–1, decomp ca. 180 °C. 1H-NMR (ppm, DMSO-d6): δ = 1.53 (d, 3H, J = 6.3 Hz, CHMe), 2.60, 2.64, 2.80, 2.83 (4s, 4×3 H, tmeda-Me), 2.68–2.99 (m, 4H, tmeda-CH2), 4.06 (m, 1H, CHMe), 4.67 (br, 1H, NH2), 5.45 (br, 1H, NH2), 6.86–7.13 (m, 4H, aryl-H). 13C{1H}-NMR (ppm, DMSO-d6): δ = 24.75, 47.22, 48.32, 49.28, 50.90, 57.83, 58.57, 63.14, 121.07, 124.14, 125.06, 131.80, 147.56, 156.66. CHN-Analysis (%): Calcd.: C: 34.47, H: 5.37, N: 8.61. Found: C: 34.12, H: 5.44, N: 8.45.
[Pd(p-MeC6H3CHMeNH2)tmeda]PF6, 2b, C15H28F6N3PPd, MW = 501.79 g mol–1, decomp ca. 170 °C. 1H-NMR (ppm, DMSO-d6): δ = 1.50 (d, 3H, J = 6.3 Hz, CHMe), 2.24 (s, 3H, 4-Me(Ph)); 2.60, 2.64, 2.81, 2.85 (4s, 4×3 H, tmeda-Me), 2.72–3.10 (m, 4H, tmeda-CH2), 4.02 (m, 1H, CHMe), 4.64 (br, 1H, NH2), 5.40 (br, 1H, NH2), 6.76 (br, 2H, aryl-H), 6.88 (br, 1H, aryl-H). 13C{1H}-NMR (ppm, DMSO-d6): δ = 21.03, 24.76, 47.28, 48.26, 49.38, 50.87, 57.83, 58.28, 63.13, 120.74, 124.80, 132.31, 133.71, 147.34, 153.58. CHN-Analysis (%): Calcd.: C: 35.90, H: 5.62, N: 8.37. Found: C: 35.85, H: 5.75, N: 8.31.
[Pd(p-FC6H3CHMeNH2)tmeda]PF6, 2c, C14H25F7N3PPd, MW = 505.76 g mol–1, decomp ca. 145 °C. 1H-NMR (ppm, DMSO-d6): δ = 1.52 (d, 3H, J = 6.4 Hz, CHMe), 2.61, 2.66, 2.80, 2.83 (4s, 4×3 H, tmeda-Me), 2.73–3.00 (m, 4H, tmeda-CH2), 4.07 (m, 1H, CHMe), 4.74 (br, 1H, NH2), 5.48 (br, 1H, NH2), 6.76–6.95 (m, 4H, aryl-H). 13C{1H}-NMR (ppm, DMSO-d6): δ = 24.78, 47.38, 48.35, 49.40, 50.85, 58.00, 58.10, 63.17, 110.60, 118.02, 122.10, 150.39, 152.64, 159.06 (d, J = –244 Hz, CF). 19F-NMR (ppm, DMSO-d6): δ = –111.0 (m, aryl-F), –65.0 (d, J = 712 Hz, PF6–). CHN-Analysis (%): Calcd.: C: 33.25, H: 4.98, N: 8.31. Found: C: 32.93, H: 4.90, N: 8.19.
[Pd(p-ClC6H3CHMeNH2)tmeda]PF6, 2d, C14H25ClF6N3PPd, MW = 522.21 g mol–1, decomp ca. 120 °C. 1H-NMR (ppm, DMSO-d6): δ = 1.53 (d, 3H, J = 6.4 Hz, CHMe), 2.61, 2.66, 2.80, 2.83 (4s, 4×3 H, tmeda-Me), 2.69–3.03 (m, 4H, tmeda-CH2), 4.07 (m, 1H, CHMe), 4.74 (br, 1H, NH2), 5.51 (br, 1H, NH2), 6.92–7.05 (m, 4H, aryl-H). 13C{1H}-NMR (ppm, DMSO-d6): δ = 24.57, 47.39, 48.42, 49.37, 50.91, 57.20, 58.10, 63.11, 122.63, 124.02, 129.12, 130.91,150.26, 155.49.
[Pd(p-MeOC6H3CHMeNH2)tmeda]PF6, rac-2e, C15H28F6N3OPPd, MW = 517.79 g mol–1, decomp ca. 170 °C. 1H-NMR (ppm, DMSO-d6): δ = 1.50 (d, 3H, J = 6.5 Hz, CHMe), 2.61, 2.64, 2.81, 2.83 (4s, 4×3 H, tmeda-Me), 2.72–3.10 (m, 4H, tmeda-CH2), 3.70 (s, 3H, 4-MeO), 4.03 (m, 1H, CHMe), 4.67 (br, 1H, NH2), 5.39 (br, 1H, NH2), 6.53–6.59 (m, 2H, aryl-H), 6.78–6.83 (m, 1H, aryl-H). 13C{1H}-NMR (ppm, DMSO-d6): δ = 24.77, 47.38, 48.16, 49.48, 50.73, 54.73 (4-MeO), 57.86, 57.94, 63.08, 108.93, 117.74, 121.40, 148.75, 148.85, 156.00. CHN-Analysis (%): Calcd.: C: 34.79, H: 5.45, N: 8.12. Found: C: 34.57, H: 5.80, N: 8.19.
[Pd(p-MeOC6H3CHMeNH2)tmeda]PF6, R-2e, C15H28F6N3OPPd, MW = 517.79 g mol–1, mp 120 °C. 1H-NMR (ppm, DMSO-d6): δ = 1.50 (d, 3H, J = 6.3 Hz, CHMe), 2.61, 2.64, 2.81, 2.83 (4s, 4×3 H, tmeda-Me), 2.67–3.00 (m, 4H, tmeda-CH2), 3.70 (s, 3H, 4-MeO), 4.02 (m, 1H, CHMe), 4.66 (br, 1H, NH2), 5.38 (br, 1H, NH2), 6.54–6.57 (m, 2H, aryl-H), 6.74–6.82 (m, 1H, aryl-H). 13C{1H}-NMR (ppm, DMSO-d6): δ = 24.78, 47.39, 48.18, 49.47, 50.73, 54.70 (4-MeO), 57.88, 57.95, 63.08, 108.93, 117.76, 121.57, 148.82, 148.89, 156.03.
[Pd(C6H4CHMeNH2)tmeda]CF3SO3, rac-3, C15H26F3N3O3PdS, MW = 491.85 g mol–1, decomp ca. 195 °C. 1H-NMR (ppm, DMSO-d6): δ = 1.54 (d, 3H, J = 6.4 Hz, CHMe); 2.61, 2.65, 2.81, 2.84 (4s, 4×3 H, tmeda-Me), 2.73–3.00 (m, 4H, tmeda-CH2), 4.07 (m, 1H, CHMe), 4.69 (br, 1H, NH2), 5.47 (br, 1H, NH2), 6.88–7.13 (m, 4H, aryl-H). 13C{1H}-NMR (ppm, DMSO-d6): δ = 24.74, 47.23, 48.32, 49.30, 50.92, 57.84, 58.56, 63.14, 120.58 (q, J = –322 Hz, CF3), 121.08, 124.14, 125.06, 131.80, 147.57, 156.65. CHN-Analysis (%):Calcd.: C: 36.63, H: 5.33, N: 8.54. Found: C: 36.32, H: 5.65, N: 8.59.
[Pd(C6H4CHMeNH2)tmeda]CF3SO3, R-3, C15H26F3N3O3PdS, MW = 491.85 g mol–1, decomp ca. 175 °C. 1H-NMR (ppm, DMSO-d6): δ = 1.54 (d, 3H, J = 6.4 Hz, CHMe); 2.61, 2.65, 2.81, 2.84 (4s, 4×3 H, tmeda-Me), 2.73–3.00 (m, 4H, tmeda-CH2), 4.07 (m, 1H, CHMe), 4.69 (br, 1H, NH2), 5.47 (br, 1H, NH2), 6.88–7.13 (m, 4H, aryl-H). 13C{1H}-NMR (ppm, DMSO-d6): δ = 24.74, 47.22, 48.32, 49.29, 50.91, 57.83, 58.56, 63.13, 120.58 (q, J = –322 Hz, CF3), 121.07, 124.14, 125.06, 131.80, 147.57, 156.65. 19F-NMR (ppm, DMSO-d6): δ = –77.7 (s, CF3). CHN-Analysis (%):Calcd.: C: 36.63, H: 5.33, N: 8.54. Found: C: 36.27, H: 5.18, N: 8.53.
Crystallographic studies
Compounds 1a – R-3 have been studied by single crystal X-ray diffraction. For intensity data collection the crystals were mounted on glass fibers or cryo loops and placed directly in a cold stream of dinitrogen; data for rac-2e were collected at room temperature, those for 1b at 278 K.Crystal data, parameters of data collection and convergence results are listed in Tables 1 and 2.
| Compound | 1a | 1b | 2a | 2b | 2c | 2d |
|---|---|---|---|---|---|---|
| Formula cation | C14H26N3Pd | C15H28N3Pd | C14H26N3Pd | C15H28N3Pd | C14H25FN3Pd | C14H25ClN3Pd |
| Anion | BF4 | BF4 | PF6 | PF6 | PF6 | PF6 |
| Formula weight | 429.59 | 443.61 | 487.75 | 501.77 | 505.74 | 522.19 |
| Crystal size/mm | 0.30![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) ×0.27×0.13 ×0.27×0.13 | 0.28×0.12×0.12 | 0.55×0.50×0.45 | 0.40×0.34×0.28 | 0.30×0.30×0.10 | 0.28×0.23×0.22 |
| Habit | Plate | Rod | Prism | Prism | Plate | Rod |
| Crystal system | Orthorhombic | Orthorhombic | Orthorhombic | Orthorhombic | Orthorhombic | Orthorhombic |
| Space group | P212121 | P212121 | P212121 | P212121 | P212121 | P212121 |
| a/Å | 10.9504(13) | 10.824(3) | 10.908(2) | 10.8390(7) | 10.777(3) | 10.7696(9) |
| b/Å | 11.8398(14) | 12.230(3) | 12.578(3) | 12.5518(9) | 12.438(3) | 12.4391(10) |
| c/Å | 13.2335(16) | 14.106(3) | 13.989(3) | 14.3611(10) | 14.353(3) | 14.6194(12) |
| V/Å3 | 1715.7(4) | 1867.3(8) | 1919.3(7) | 1953.8(2) | 1923.9(8) | 1958.5(3) |
| D/g cm–1 | 1.663 | 1.578 | 1.688 | 1.706 | 1.746 | 1.771 |
| Z | 4 | 4 | 4 | 4 | 4 | 4 |
| µ(Mo Kα)/mm–1 | 1.121 | 1.033 | 1.108 | 1.091 | 1.116 | 1.224 |
| F(000) | 872 | 904 | 984 | 1016 | 1016 | 1048 |
| θ range/° | 2.3–27.5 | 2.2–28.0 | 2.2–29.2 | 2.3–30.1 | 2.4–28.5 | 2.4–27.1 |
| Temperature/K | 110 | 278 | 110 | 110 | 130 | 153 |
| Refls. collected | 13938 | 13053 | 27760 | 29134 | 21810 | 24502 |
| Rint | 0.0299 | 0.0705 | 0.0259 | 0.0311 | 0.0440 | 0.0714 |
| Unique refl. in ref. | 3919 | 4514 | 5181 | 5659 | 4822 | 4300 |
| Refl. with I > 2σ(I) | 3874 | 3308 | 5069 | 5626 | 4464 | 4189 |
| Param. refined | 213 | 223 | 213 | 241 | 253 | 222 |
| R1 (2σ(I)) | 0.0215 | 0.0493 | 0.0354 | 0.0221 | 0.0309 | 0.0627 |
| R1 (all data) | 0.0219 | 0.0829 | 0.0361 | 0.0223 | 0.0350 | 0.0649 |
| wR2 | 0.0534 | 0.0873 | 0.0971 | 0.0532 | 0.0749 | 0.1455 |
| Flack param. | –0.02(2) | –0.04(5) | –0.01(4) | 0.006(17) | –0.02(3) | 0.03(7) |
| Chirality of crystal studied | R | S | S | R | R | R |
| Goodness of fit | 1.083 | 1.041 | 1.103 | 1.097 | 1.084 | 1.196 |
| Diff. peak/hole/e Å–3 | 0.44/–0.46 | 0.66/–0.54 | 1.61/–1.33 | 0.87/–0.33 | 0.77/–0.57 | 1.07/–1.81 |
| CCDC | 657272 | 657273 | 657276 | 657277 | 657278 | 657279 |
| Compound | rac-1c | R-1c | rac-2e | R-2e | rac-3 | R-3 |
|---|---|---|---|---|---|---|
| Formula cation | C14H25FN3Pd | C14H25FN3Pd | C15H28N3OPd | C15H28N3OPd | C14H26N3Pd | C14H26N3Pd |
| Anion | BF4 | BF4 | PF6 | PF6 | CF3O3S | CF3O3S |
| Formula weight | 447.58 | 447.58 | 517.77 | 517.77 | 491.85 | 491.85 |
| Crystal size (mm) | 0.22×0.06×0.02 | 0.54×0.06×0.05 | 0.3×0.1×0.1 | 0.39×0.13×0.02 | 0.45×0.25×0.24 | 0.54×0.31×0.10 |
| Habit | Plate | Rod | Rod | Plate | Fragment | Plate |
| Crystal system | Monoclinic | Orthorhombic | Monoclinic | Monoclinic | Monoclinic | Monoclinic |
| Space group | P21/c | P212121 | P21/c | P21 | P21/n | P21 |
| a/Å | 10.3539(10) | 8.9088(9) | 7.724(3) | 7.5980(5) | 11.6744(17) | 11.6452(13) |
| b/Å | 16.9103(16) | 9.5432(10) | 20.015(8) | 20.5971(14) | 14.108(2) | 14.3172(13) |
| c/Å | 11.3615(11) | 20.917(2) | 14.106(6) | 13.8222(9) | 11.9538(18) | 12.0277(13) |
| β/° | 116.461(2) | 102.012(9) | 105.038(2) | 96.137(4) | 96.018(4) | |
| V/Å3 | 1780.9(3) | 1778.3(3) | 2133.0(15) | 2089.1(2) | 1957.5(5) | 1994.3(4) |
| D/g cm–1 | 1.669 | 1.672 | 1.612 | 1.646 | 1.669 | 1.638 |
| Z | 4 | 4 | 4 | 4 | 4 | 4 |
| µ(Mo Kα)/mm–1 | 1.091 | 1.093 | 1.005 | 1.026 | 1.102 | 1.081 |
| F(000) | 904 | 904 | 1048 | 1048 | 1000 | 1000 |
| θ range/° | 2.2–28.3 | 2.3–30.6 | 1.8–25.0 | 2.5–27.6 | 2.2–34.4 | 2.2–26.6 |
| Temperature/K | 100 | 110 | 293 | 130 | 110 | 130 |
| Refls. collected | 23141 | 25367 | 22622 | 20848 | 37702 | 13912 |
| Rint | 0.0697 | 0.0653 | 0.1630 | 0.0458 | 0.0520 | 0.0858 |
| Unique refl. in ref. | 4377 | 5271 | 3757 | 9493 | 7507 | 7804 |
| Refl. with I > 2σ(I) | 3585 | 5038 | 2015 | 8807 | 6870 | 5406 |
| Param. refined | 265 | 222 | 250 | 499 | 240 | 377 |
| R1 (2σ(I)) | 0.0551 | 0.0346 | 0.0614 | 0.0437 | 0.0352 | 0.0602 |
| R1 (all data) | 0.0724 | 0.0370 | 0.1340 | 0.0480 | 0.0386 | 0.0928 |
| wR2 | 0.1039 | 0.0825 | 0.1245 | 0.0956 | 0.0865 | 0.1055 |
| Flack param. | 0.00(3) | –0.03(2) | –0.14(5) | |||
| Goodness of fit | 1.153 | 1.048 | 1.001 | 1.018 | 1.057 | 0.999 |
| Diff. peak/hole/e/Å3 | 0.81/–1.83 | 2.21/–1.26 | 0.63/–0.41 | 1.29/–0.70 | 1.65/–0.87 | 0.67/–0.77 |
| CCDC | 657274 | 657275 | 657280 | 657281 | 657282 | 657283 |
Data for 1b were collected on an ENRAF-NONIUS CAD4 diffractometer (Mo Kα radiation, λ = 0.71073 Å, graphite monochromator); azimuthal scans on selected suitable reflections were used to correct the intensities for absorption.13 All other intensity data collections were performed with a Bruker Smart APEX CCD (Mo Kα radiation, λ = 0.71073 Å, graphite monochromator) area detector on a D8 goniometer in the ω scan mode. The SADABS14 program was used for multi-scan absorption correction. The structures were solved by direct methods (SHELXS97)15 or by replacement with a related model and refined by full matrix least-squares on F2 (SHELXL97).16 Non-hydrogen atoms were assigned anisotropic displacement parameters. H atoms were calculated in idealized positions and refined using a riding model. For the homochiral crystals, a satisfactory Flack17 parameter was obtained; for the crystals derived from enantiomerically pure compounds, the absolute structure matches the central chirality of the starting material (R in all cases). Disorder was encountered in two out of the twelve crystal structures: in rac-1c, the residual electron density in the tetrafluoroborate suggested an alternative minority orientation for the anion; in addition, this compound represents the only example for which (minor) disorder associated with the center of chirality was observed. In 2c, the hexafluorophosphate anion is ordered with respect to a local fourfold axis in the octahedron and shows disorder in the plane perpendicular to this axis. Pseudosymmetry in the homochiral crystals with Z′ > 1, i.e. in R-2e and R-3 deserves a comment: we have already encountered pseudosymmetry between the symmetrically independent constituents of homochiral crystals in the past.18,19 In the present work, the structure of R-3 shows very pronounced pseudosymmetry: the MISSYM algorithm20,21 as implemented in PLATON22 suggests that 96% of the electron density match the higher symmetry of the centrosymmetric supergroup P21/n. This close relationsship is also reflected in very similar lattice parameters for R-3 and rac-3. In R-3 high correlation between the anisotropic displacement parameters of atoms related by pseudo-inversion was encountered; in the final least-squares refinement, the anisotropic displacement parameters of these atom pairs were constrained to be equal.
Calculations concerning space filling properties were performed with PLATON22 and are based on the following van der Waals radii: C 1.70; H 1.20; B 1.63; Cl 1.75; F 1.47; N 1.55; O 1.52; P 1.80; Pd 2.30; S 1.80 Å.
Lattice energy minimizations
Local energy minima for the crystal structures of the closely related pairs of solids rac-2e/R-2e and rac-3/R-3 were obtained using the program PCK83.23 Prior to minimization the experimental distances to H atoms were normalized to the following values: C–H 1.083, N–H 1.009, O–H 0.983 Å. Independent residues were treated as rigid objects in their experimentally observed conformation and were allowed to translate and rotate. The van der Waals interactions were simulated with a Buckingham potential using published parameters.24 Intermolecular hydrogen bonds were modelled by additional attractive exponential functions; the following expressions were used: N–H⋯O, E = –8184.5exp(–2.9rij); N–H⋯F, E = –9100exp(–3.18rij) with E in kJ mol–1 and hydrogen-acceptor distances rij in Å. In view of the very similar arrangement of cations and anions in rac-2e/R-2e and rac-3/R-3, electrostatic contributions were not taken into account. Parameters for the lattice energy minimizations are compiled in the ESI.‡Results and discussion
The ionic compounds studied in the context of this work consist of chiral organopalladium cations and non-coordinating anions. We will first describe their structures with respect to cation geometry, then discuss hydrogen bonding, and finally summarize the most important trends in packing. Although cyclopalladation can be considered a classical reaction,25 the Cambridge Structural Database26 contains only 15 examples of cyclopalladated amines and N,N′-chelating ligands,27 six of them reported in our recent communication about partially racemic solids.9All complex cations described here are essentially square planar; the distance of the metal center to the best plane through the coordinating atoms is most pronounced in the cation of 1b and amounts to 0.0718(4) Å. Bond lengths between palladium and the tmeda nitrogen atoms differ significantly: The Pd–N distance trans to carbon, with values ranging from 2.1523(19) to 2.180(4), is consistently longer than the bond to the nitrogen atom trans to the amino group (2.085(2)–2.122(8) Å). In all compounds under study the conformationally soft28–31 five-membered ring formed by the chelating tmeda ligand and the metal was ordered: neither the displacement ellipsoids associated with these atoms nor the C–C bond distances indicate major static or dynamic disorder. As an example, a displacement ellipsoid plot for 1a is shown in Fig. 1.
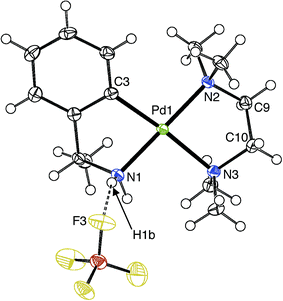 | ||
| Fig. 1 Displacement ellipsoid plot of an ion pair in the crystal structure of 1a; ellipsoids are drawn at 50% probability, H atoms are represented as spheres with an arbitrary radius. Selected distances and angles: Pd1–C3 2.005(2), Pd1–N1 2.0410(19), Pd1–N2 2.085(2), Pd1–N3 2.1523(18), C9–C10 1.500(4), N1–H1b 0.92, H1b–F3 2.09, N1⋯F3 2.966(3) Å; C3–Pd1–N1 80.35(9), N2–Pd1–N3 84.71(8), N1–Pd1–N2 176.01(8), C3–Pd1–N3 174.46(9), N1–H1b⋯F3 158°. | ||
Hydrogen bonding in the structures is limited by the number of potential H donor functions: only the two hydrogen atoms of the primary amine are, in principle, available for classical H bonds whereas potential acceptor groups are more numerous: they comprise the fluorine atoms in the BF4– (1) and PF6– (2) anions and the oxygen atoms of the methoxy substituents (2e) and in the trifluoromethylsulfonate anions (3). All H bonds can be classified as moderately strong:32 The shortest donor⋯acceptor distance amounts to 2.878(10) Å and is encountered for rac-1c. Table 3 compiles data on hydrogen bonding, together with chirality information and packing coefficients for all twelve structures under study.
| Compound | Chirality | No of H bonds per formula unit | Shortest classical H bond | Packing coefficient | |
|---|---|---|---|---|---|
| Type | Donor⋯acceptor/Å | ||||
| a Packing coefficients depend on temperature. Data for 1b and rac-2e are based on experiments at 278 and 293 K, respectively, whereas all other structures were obtained at 100 < T < 150 K, cfTables 1 and 2.b The crystal structures of rac-1c and 2c show disorder; only the majority conformations were taken into account for calculating the packing coefficients. | |||||
| 1a | homochiral; conglomerate | 1 | N–H⋯F | 2.966(3) | 0.726 |
| 1b | homochiral; conglomerate | 1 | N–H⋯F | 2.972(11) | 0.692a |
| rac-1c | heterochiral | 2 | N–H⋯F | 2.878(10) | 0.711b |
| R-1c | homochiral | 2 (1 bifurcated) | N–H⋯F | 2.946(3) | 0.715 |
| 2a | homochiral; conglomerate | 1 | N–H⋯F | 3.137(5) | 0.693 |
| 2b | homochiral; conglomerate | 1 | N–H⋯F | 2.974(2) | 0.714 |
| 2c | homochiral; conglomerate | 1 | N–H⋯F | 3.067(5) | 0.705b |
| 2d | homochiral; conglomerate | 1 | N–H⋯F | 2.960(12) | 0.707 |
| rac-2e | heterochiral | 2 | N–H⋯F | 3.075(12) | 0.658a |
| N–H⋯O | 3.076(8) | ||||
| R-2e | homochiral | 1.5 | N–H⋯F | 3.119(6) | 0.685 |
| N–H⋯O | 3.081(6) | ||||
| rac-3 | heterochiral | 1 | N–H⋯O | 2.981(3) | 0.703 |
| R-3 | homochiral | 1 | N–H⋯O | 2.912(12) | 0.692 |
In the conglomerate structure type, i.e. in 1a, 1b and 2a–2d, one cation and one nearest counteranion interact via a single hydrogen bond; Fig. 1 shows the structure of such an ion pair in the asymmetric unit for 1a. A similar situation is encountered for the triflate salts rac-3 and R-3.
In the structures adopted by the tetrafluoroborates of the fluoro substituted cations, rac-1c and R-1c, both N bonded hydrogen atoms are engaged in classical hydrogen bonds. In the racemic crystals of rac-1c, two cations and two anions in the neighborhood of a crystallographic inversion center interact as depicted in Fig. 2a; this arrangement can be described by the graph set symbol33 R24(8). The enantiopure solid R-1c represents the only structure reported in this communication which is associated with an extended system of hydrogen bonds: Fig. 2b shows how infinite chains of alternating cations and anions interact along the [010] direction; one of the N–H⋯F interactions is bifurcated.
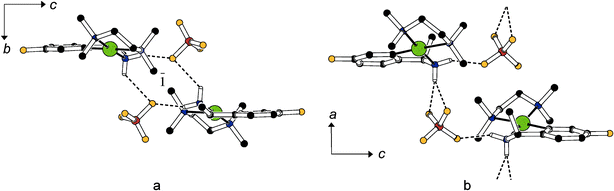 | ||
| Fig. 2 Hydrogen bonding in (a) rac-1c and (b) R-1c. Colour code C black, N blue, Pd green, F yellow, B brown. | ||
In the methoxy substituted derivatives 2e, neighbouring cations are linked by amine N–H donor to methoxy O acceptor hydrogen bonds. In the racemic crystals of rac-2e, two cations form a centrosymmetric pair (Fig. 3a) whereas only one N–H⋯O bond connects two neighbouring cations in the enantiopure solid R-2e (Fig. 3b). If only the number of interactions is taken into account, the hydrogen bonding efficiency is superior for the racemic crystals; this qualitative argument is confirmed by lattice energy calculations, see below. In both rac-2e and R-2e each cation forms a hydrogen bond to a hexafluorophosphate anion via the second amine H.
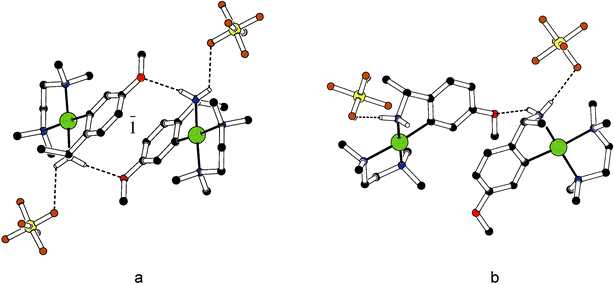 | ||
| Fig. 3 Hydrogen bonding in (a) rac-2e and (b) R-2e. Colour code C black, N blue, O red, Pd green, F orange, P yellow. | ||
Predictions of molecular packing in general represent a major challenge, and this is particularly true in the present case: intermolecular interactions in the solids reported here cover a wide range, from coulomb forces and hydrogen bonds to van der Waals interactions. Despite this inherent complexity, several trends may be perceived. The conglomerate structure type is remarkably stable: it tolerates para substitution on the arene ring of the cyclopalladated amine, the change in the anion and a combination of both modifications. Table 1 shows that the tetrafluoroborates 1a and 1b as well as the four hexafluorophosphates 2a–2d crystallize in the same space group P212121 with closely related lattice parameters. The packing patterns are also very similar: As an example, the crystal structures of 1a and 2d are compared in Fig. 4. In this structure type, one cation and one nearest counteranion interact via hydrogen bonds; no extended networks form. Interhalogen contacts are rather weak and do not have significant impact on packing: both F⋯F (3.055(4) Å in 2c) and F⋯Cl (3.392(12) Å in 2d) interactions are longer than the sum of the van der Waals radii.
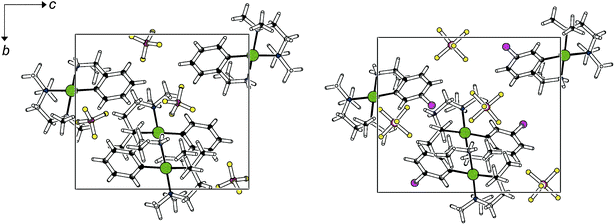 | ||
| Fig. 4 Projection of the unit cells of 1a (left) and 2d (right) in the direction of the crystallographic a axis. Colour code C black, N blue, Pd green, Cl magenta, F yellow, B/P brown. | ||
Among the six conglomerate structures, 1a, i.e. the salt combining the smallest cation (derived from the unsubstituted amine) and the smaller BF4– anion, and 2b, the salt derived from the largest cation (the para methyl substituted amine) and the larger PF6– anion show the most efficient space filling as indicated by their packing coefficients (see Table 3). Our series of related compounds has enabled us to establish the stability limits of this structure type: Although it comprises the tetrafluoroborates of both the smallest and the largest cationic complex, the para fluoro substituted derivative (1c) adopts a different structure. The para substitution of the primary amine with the relatively bulky methoxy group (2e) and exchange of the rather isometric counter anions for trifluoromethylsulfonate (3) also leads to new structure types: all of these are non-conglomerates: from racemic solutions, racemic crystals precipitate. However, a closer inspection of the non-conglomerates (cf.Table 2) reveals different relationships between racemic and enantiopure solids: on the one hand, rac-1c and R-1c differ clearly in terms of space groups, unit cell parameters and hydrogen bonding as shown above; on the other hand, rac-2e/R-2e and rac-3/R-3 exist in pairs of centrosymmetric heterochiral and pseudo-centrosymmetric homochiral solids. Within each pair, lattice parameters are similar; group–subgroup relationships link the centrosymmetric space groups of the racemic and the non-centrosymmetric subgroups of the homochiral crystals. In the latter, the asymmetric units contain two independent molecules. Fig. 5 and 6 underline the common packing characteristics within each pair of solids: Fig. 5 shows the very similar arrangement of cations and anions in rac-2e and R-2e.
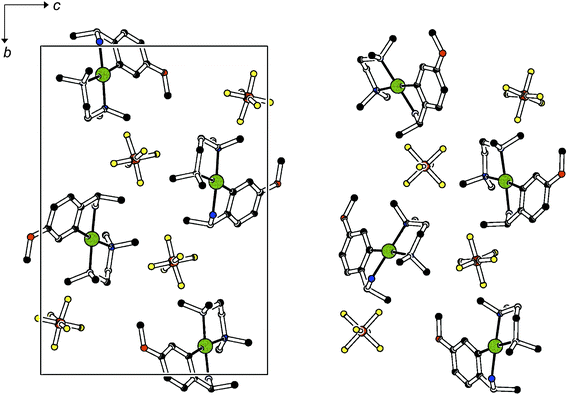 | ||
| Fig. 5 Projection of the unit cells of rac-2e (left) and R-2e (right) in the direction of the crystallographic a axis. Colour code C black, N blue, O red, Pd green, F yellow, P brown. | ||
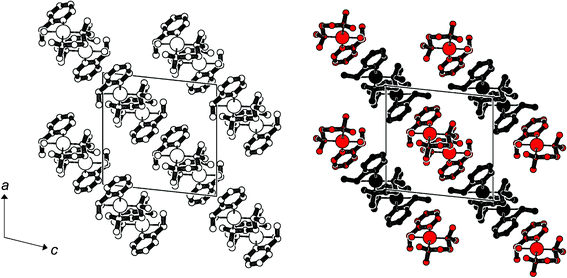 | ||
| Fig. 6 Projection of the unit cells of rac-3 (left) and R-3 (right) in the direction of the crystallographic b axis. Triflate anions and hydrogen atoms have been omitted; the two independent molecules in R-3 are distinguished as black and red residues. | ||
The close relationship between rac-3 and R-3 is reflected in the patterns of the larger cationic residues (Fig. 6).
For 2e packing coefficients are the lowest among the compounds reported here (Table 2). Hydrogen bonding is decisive for the preferred formation of racemic crystals: in rac-2e both H atoms of the amine act as donors, whereas one potential hydrogen bond donor in R-2e is not engaged in a short interaction (Fig. 3, Table 2). The situation for 3 is different: Heterochiral rac-3 and homochiral R-3 share the same hydrogen bond pattern, and the driving force for the precipitation of racemic crystals from racemic solution is more efficient space filling and better van der Waals energy. Lattice energy minimizations corroborate this qualitative interpretation: we have performed computer simulations on intermolecular interactions for the closely related pairs of solids rac-2e/R-2e and rac-3/R-3 with the atom–atom potential method. This approach allows to obtain relative lattice energies and to judge the relevance of their main contributions, i.e. van der Waals and hydrogen bond energies, for the structures under investigation. Table 4 summarizes the results: an essential part of the lower lattice energy of rac-2e with respect to its homochiral congener R-2e is due to more favourable hydrogen bonding, whereas the contribution of H bonds has no significant impact on the relative energies of rac-3 and R-3. Additional details about the minimization results are provided in the ESI.‡
| Compound | rac-2e | R-2e | rac-3 | R-3 |
|---|---|---|---|---|
| a The unit cell volumes after minimization are usually lower than the experimental values because the latter refer to finite temperatures ≫0 K. The difference is therefore most marked for rac-2e.b Lattice energy/ion pair, taking into account both van der Waals interactions and H bonds.c Hydrogen bond energy/ion pair. | ||||
| V experimental/Å3 | 2133.0 | 2089.1 | 1957.5 | 1994.3 |
| T experimenta/K | 293 | 130 | 110 | 130 |
| V minimized/Å3 | 1963.2 | 2064.2 | 1943.0 | 1965.8 |
| E totalb/kJ mol–1 | –183.4 | –166.1 | –167.4 | –148.3 |
| E H bondsc/kJ mol–1 | –34.3 | –24.4 | –18.6 | –20.0 |
Conclusions
The crystal structures of a series of chemically similar complex salts have been studied, and different crystallization alternatives have been encountered: They comprise a remarkably stable conglomerate structure type, an example (1c) for which hetero- and homochiral crystals differ completely, and two cases (2e and 3) with close relationship between racemic and enantiopure solids. In one of the latter pairs, hydrogen bonding does not discriminate between homochiral and heterochiral arrangement: the crystal structures of rac-3 and R-3 are very similar in terms of hydrogen bonds, and the compound comes close to conglomerate formation. We note that the above-mentioned robustness of the conglomerate structure type is not trivial: for comparison with the tmeda complexes reported in this work, a series of 2-aminomethylpyridine derivatives have been synthesized.34 The aminomethyl ligand as well as ethylenediamine9 exhibit potential hydrogen bond donors. Hydrogen bonding in these donor-rich derivatives plays a much more dominant role than in the complexes discussed here—does an extended network of hydrogen bonds preclude conglomerate formation in these cases?Acknowledgements
Support by Deutsche Forschungsgemeinschaft (“Conglomerates”) is gratefully acknowledged. We thank BASF for providing the enantiomerically pure primary amines and an anonymous referee for providing valuable feedback.References
- M. Pallavicini, C. Bolchi, L. Fumagalli, O. Piccolo and E. Valoti, Tetrahedron: Asym., 2007, 18, 906 Search PubMed.
- J. Jacques, M. Leclercq and M. J. Brienne, Tetrahedron, 1981, 37, 1727 CrossRef.
- C. J. Price, Chem. Eng. Prog., 1997, 93, 34 Search PubMed.
- J. Jacques, A. Collet and S. H. Wilen, Enantiomers, Racemates, and Resolutions, Krieger Publishing Company, Malabar, Florida, 1994 Search PubMed.
- A. Collet, M. J. Brienne and J. Jacques, Chem. Rev., 1980, 80, 215 CrossRef CAS.
- E. Ballesteros, M. Gallego and M. Valcarcel, Anal. Chem., 1996, 68, 322 CrossRef CAS.
- W. M. L. Wood, in Chirality in Industry II. ed. A. N. Collins, G. N. Sheldrake and J. Crosby, John Wiley & Sons, Chichester, 1997, pp. 119 Search PubMed.
- Part 57 of the series of publications dealing with conglomerate crystallization is: M. K. Saha, R. Ramanujam, I. Bernal and F. R. Fronczek, Cryst. Growth Des., 2002, 2, 205 Search PubMed.
- I. Kalf, R. Wang and U. Englert, J. Organomet. Chem., 2006, 691, 2277 CrossRef CAS.
- A. Ladenburg, Liebigs Ann. Chem., 1908, 364, 227.
- J. Vicente, I. Saura-Llamas and M. G. Palin, Organometallics, 1997, 16, 826 CrossRef CAS.
- Y. Fuchita, K. Yoshinaga, Y. Ikeda and J. Kinoshita-Kawashima, J. Chem. Soc., Dalton Trans., 1997, 2495 RSC.
- A. C. T. North, D. C. Phillips and F. S. Mathews, Acta Crystallogr., Sect. A, 1968, 24, 351 CrossRef.
- G. M. Sheldrick, “SADABS, Program for Empirical Absorption Correction of Area Detector Data,” University of Göttingen, 1996.
- G. M. Sheldrick, “SHELXS97, Program for Crystal Structure Solution,” University of Göttingen, 1997.
- G. M. Sheldrick, “SHELXL97, Program for Crystal Structure Refinement,” University of Göttingen, 1997.
- H. D. Flack, Acta Crystallogr., Sect. A, 1983, 39, 876 CrossRef.
- U. Englert, A. Häring, C. Hu and I. Kalf, Z. Anorg. Allg. Chem., 2002, 628, 1173 CrossRef CAS.
- B. Calmuschi and U. Englert, Acta Crystallogr., Sect. C, 2003, 58, m545.
- Y. LePage, J. Appl. Crystallogr., 1987, 20, 264 CrossRef CAS.
- Y. LePage, J. Appl. Crystallogr., 1988, 21, 983 CrossRef.
- A. L. Spek, J. Appl. Crystallogr., 2003, 36, 7 CrossRef CAS.
- D. E. Williams, “PCK83, a crystal molecular packing analysis program,” Quantum Chemistry Programs Exchange, 1983.
- U. Englert, in Advances in Molecular Structure Research, Vol. 6, ed. M. Hargittai and I. Hargittai, JAI Press Inc., Greenwich, CT, 2000, pp. 49 Search PubMed.
- A. C. Cope and E. C. Friedrich, J. Am. Chem. Soc., 1968, 90, 909 CrossRef CAS.
- F. Allen, Acta Crystallogr., Sect. B, 2002, B58, 380 CrossRef.
- Error-free entries without disorder for which 3D coordinates were available, CSD Version 5.28, including update of January 2007.
- W. E. Rhine, J. H. Davis and G. Stucky, J. Am. Chem. Soc., 1975, 97, 2079 CrossRef CAS.
- W. E. Rhine, J. H. Davis and G. Stucky, J. Organomet. Chem., 1977, 134, 139 CrossRef CAS.
- M. F. Lappert, A. Singh, L. M. Englehardt and A. H. White, J. Organomet. Chem., 1984, 262, 271 CrossRef CAS.
- M. H. Wang, U. Englert and U. Koelle, J. Organomet. Chem., 1993, 453, 127 CrossRef CAS.
- G. Gilli, in Fundamentals of Crystallography, ed. G. Giacovazzo, Oxford University Press, Oxford, 2002, pp. 585 Search PubMed.
- M. C. Etter, J. C. MacDonald and J. Bernstein, Acta Crystallogr., Sect. B, 1990, 46, 256 CrossRef.
- I. Kalf and U. Englert, unpublished results. Microanalytical data and preliminary X-ray results suggest solvate formation for several of these complexes. No conglomerates and no dominant structure type were encountered: exchange of the counteranion and/or substitution on the arene ring resulted in different packing patterns even for closely related compounds. These largely unrelated structures will be reported elsewhere as structure communications.
Footnotes |
| † CCDC reference numbers 657272–657283. For crystallographic data in CIF or other electronic format see DOI: 10.1039/b712641g |
| ‡ Electronic supplementary information (ESI) available: Parameters for lattice energy minimizations and details of the minimization results. See DOI: 10.1039/b712641g |
| This journal is © The Royal Society of Chemistry 2008 |