Neutral and ionic complexes of C60 with (ZnOEP)2·BPy coordination dimers†‡
Dmitri V.
Konarev
*a,
Salavat S.
Khasanov
b,
Yury L.
Slovokhotov
c,
Gunzi
Saito
*d and
Rimma N.
Lyubovskaya
a
aInstitute of Problems of Chemical Physics RAS, Chernogolovka, Moscow region, 142432 Russia. E-mail: konarev@icp.ac.ru; Fax: +007-49652-21852
bInstitute of Solid State Physics RAS, Chernogolovka, Moscow region 142432, Russia
cInstitute of Organoelement Compounds RAS, 28 Vavilov St., 119991 Moscow, Russia
dDivision of Chemistry, Graduate School of Science, Kyoto University, Sakyo-ku, Kyoto, 606-8502 Japan. E-mail: saito@kuchem.kyoto-u.ac.jp; Fax: +81-75-753-40-35
First published on 5th September 2007
Abstract
Coordination (ZnOEP)2·BPy dimers formed by zinc octaethylporphyrin and 4,4′-bipyridine co-crystallize with neutral C60 and the (TDAE˙+)·(C60˙–) ion-radical salt to form molecular and ionic multi-component complexes: {(ZnOEP)2·BPy}·(C60)2·(CHCl3)2 (1) and (TDAE˙+)2·{(ZnOEP)2·BPy}·(C60˙–)2·(C6H6)0.75·(C6H4Cl2)0.25 (2). The C60 molecules are arranged in layers in 1 with the interfullerene center-to-center distances of 10.04–10.17 Å and in linear chains in 2 with the uniform interfullerene center-to-center distance of 10.61 Å. EPR measurements show exchange interaction between C60˙– and TDAE˙+ ion-radicals in 2, which results in the appearance of one signal with g = 2.0027 and ΔHpp = 19.8 G at 295 K rather than two signals from individual ion-radicals. The EPR signal from 2 is split into two components below 30 K and its integral intensity decreases below 9.6 K, which is evidence of antiferromagnetic interactions of spins.
Introduction
Fullerene complexes evoked great interest due to promising magnetic, conducting and optical properties.1–3 Up to now a wide variety of organic and organometallic compounds were used for the preparation of these complexes, namely, aromatic hydrocarbons,4,5 amines,6metallocenes,7 metalloporphyrins8–11 and metal dithiocarbamates.12 The design of functional complexes based on fullerenes requires both the search for new classes of donor molecules able to form complexes with fullerenes and the development of synthetic approaches, which allow one to modify known molecular complexes (D1·fullerene) and obtain multi-component neutral {(D1·L)·fullerene} and ionic {(D2+)·(D1)·(fullerene–)} complexes.13–19The first approach uses relatively weak coordination bonding and allows the design of fullerene complexes with coordination assemblies of metalloporphyrins and metal (II) dithiocarbamates. For example, zinc tetraphenylporphyrin forms monomers, dimers and pentamers ({ZnTPP}x·L, x = 1, 2, and 4; L = pyridine, tetramethylethylenediamine, pyrazine, 4,4′-bipyridine, and tetrapyridylporphyrin), which co-crystallize with fullerenes to form complexes with one-dimensional, layered and paired arrangement of fullerene molecules.13,14 Similarly, coordination monomers and dimers of metal (II) dithiocarbamates with diazabicyclooctane, N,N′-dimethylpyperazine and hexamethylenetetramine as L form complexes with C60, which have layered and 3D packing of fullerene molecules.15
The second approach uses D1 molecules, which can produce large cavities when packing with spherical fullerene molecules to accommodate a third component, which can be either a strong donor or a cation (D2+).16–19 This approach allows one to vary (D2+) cations with different spin states within series of {(D2+)·(D1)·(fullerene–)} complexes to affect their properties.18,19
In this work we combined the two above mentioned approaches. We used bidentate 4,4′-bipyridine ligand to assemble coordination dimers with zinc octaethylporphyrin, (ZnOEP)2·BPy, which were then co-crystallized with C60 to form molecular complex 1: {(ZnOEP)2·BPy}·(C60)2·(CHCl3)2. The packing of (ZnOEP)2·BPy dimers and C60 can produce large cavities, which were used to accommodate a strong TDAE donor to form ionic multi-component complex 2: (TDAE˙+)2·{(ZnOEP)2·BPy}·(C60˙–)2·(C6H6)0.75·(C6H4Cl2)0.25. Crystal structures of both complexes were solved, their UV-vis-NIR and IR spectra were analyzed, and magnetic properties were studied.
Results and discussion
1. Synthesis
Both complexes were obtained by the diffusion method. For the preparation of 1, a CHCl3 solution containing ZnOEP and an excess of BPy diffused into a benzene solution of C60. The crystals of 1 were formed after one month. For the preparation of 2, a hexane solution diffused into a dichlorobenze-benzene solution containing (TDAE˙+)·(C60˙–) salt, ZnOEP and an excess of BPy. The crystals of 2 were formed after one month and were free of (TDAE˙+)·(C60˙–) admixture.2. IR- and UV-vis-NIR spectra
The IR spectra of 1 and 2 are superpositions of the spectra of starting components: ZnOEP, BPy, C60, TDAE and solvent molecules (see ESI‡). There are four IR active bands from C60 at 527, 577, 1182 and 1429 cm–1 (F1u(1–4) modes, respectively). F1u(4) mode of C60 (which is most sensitive to charge transfer to the C60 molecule)20 is positioned in the spectrum of 1 at 1424 cm–1 that can justify only weak charge transfer to C60. In contrast to 1, C60 has –1 charged state in 2 since the position of F1u(4) mode of C60 in the spectrum of 2 (1390 cm–1) is very close to the position of this mode in the spectra of other ion-radical salts with approximate –1 charged state of C60.20,21 The C–N stretching mode of TDAE is very sensitive to its charged state and shifts in the following order: 1340 cm–1 for neutral TDAE; 1518 cm–1 for (TDAE˙+)·(C60˙–) with approximate +1 charge on TDAE; and 1659, 1675 cm–1 for TDAE(Cl2) with +2 charge on TDAE.22 The position of this mode in the spectrum of 2 at 1516 cm–1 clearly indicates the formation of TDAE˙+ radical cations.The UV-visible-NIR spectra of starting ZnOEP, 1 and 2 are shown in Fig. 1. The position of the Soret band for ZnOEP is at 409 nm and those of the Q-bands are at 542 and 586 nm (Fig. 1a). The formation of 1 and 2 noticeably shifts (by 9–10 nm) the position of the Soret band to the red side (418 and 419 nm, respectively). The positions of the Q-bands are shifted insignificantly (542 and 576 nm for 1 and 545 and 574 nm for 2). These shifts are a result of the coordination of the BPy ligand to ZnOEP. Similar shifts are observed at the formation of coordination dimers of ZnTPP with N-containing ligands in the complexes with C60.13,14 Complex 1 manifests a relatively intense broad charge transfer (CT) band in the visible-NIR range with the maximum at about 800 nm (Fig. 1b). Such bands are characteristic of C60 complexes with metalloporphyrins, which acquire a concave shape as a result of coordination of additional ligands.14 Complexes of fullerenes with planar porphyrins manifest only weak or even no CT bands.11 Absorption at 1070 nm characteristic of C60˙– is not observed in the spectrum of 1 indicating its neutral ground state. Complex 2 manifests intense bands at 930 and 1076 nm characteristic of C60˙– (Fig. 1c). It was shown that the charged state of C60 in (TDAE˙+)·(C60˙–) is also close to –1.22
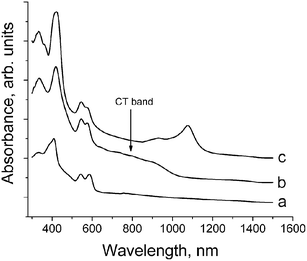 | ||
| Fig. 1 UV-visible-NIR spectra of starting ZnOEP (a); complexes 1 (b) and 2 (c) in KBr pellets in the 300–1500 nm range. | ||
3. Crystal structures
{(ZnOEP2·BPy}·(C60)2·(CHCl3)2 (1). The complex crystallizes in a triclinic unit cell. Each (ZnOEP)2·BPy dimer forms vdW contacts (Fig. 2) with two fullerene molecules, and two solvent CHCl3 molecules occupy vacancies between the ZnOEP planes (Fig. 3). The length of the Zn⋯N(BPy) coordination bond is 2.173(3) Å that is noticeably longer than the averaged length of the Zn⋯N(OEP) bonds (2.080 Å). Porphyrin macrocycles in the dimer are arranged nearly parallel to each other and do not rotate. All ethyl substituents of ZnOEP are directed toward fullerene molecules. The ZnOEP macrocycle has a concave shape with the rms deviations of atoms by 0.176 Å from the mean porphyrin plane and the displacement of the zinc atom from this plane toward the BPy ligand by 0.539 Å. As a result, the shape of the ZnOEP macrocycle conforms well to the spherical shape of C60 and allows the formation of multiple vdW contacts between them (Fig. 2). The C⋯C contacts lie in the 3.28–3.39 Å range and the N⋯C contacts lie in the 3.09–3.21 Å range for both orientations of C60. Though C60 is surrounded by eight ethyl groups of ZnOEP, no shortened H(ethyl of ZnOEP)⋯C(C60) contacts are formed. This could be a reason for C60 disorder in this complex. In previously studied C60 complexes with metal (II) octaethylporphyrins the C60 molecules were usually ordered to form multiple H(MOEP)⋯C(C60) contacts.8 The shortest Zn⋯C(C60) contacts of 3.101 and 3.369 Å for one C60 orientation and 3.150 and 3.268 Å for the other C60 orientation are formed with the 6-6 bond of C60 (Fig. 2) The C60 molecules form layers in 1 (Fig. 3). The interfullerene center-to-center distances between C60 molecules in the layer are 10.041, 10.080 and 10.166 Å. These distances are rather close to the center-to-center distances in pure C60 at 153 K (9.94 Å23).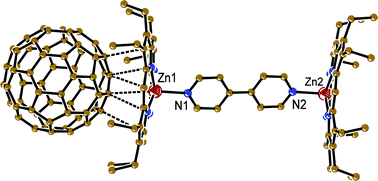 | ||
| Fig. 2 Van der Waals contacts between C60 molecule and coordination ZnOEP2·BPy dimers in 1 (dashed lines). Only one most occupied orientation was shown for disordered C60. | ||
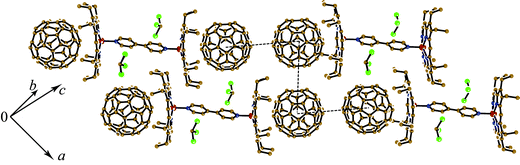 | ||
| Fig. 3 Fragment of crystal structure of 1. C60 layers are shown by dashed lines. Only one most occupied orientation was shown for disordered C60 and CHCl3 molecules. | ||
Complex 2 has an orthorhombic unit cell. The C60˙–radical anions rotate even at 90 K. Probably this rotation is not free because of the non-uniform distribution of 120 carbon atoms over the fullerene sphere. However, the attempts to approximate fullerene disorder by several orientations showed that there are more than three orientations of C60˙–. The TDAE˙+ radical cations are disordered between two orientations, whereas the BPy ligand is disordered between four orientations. In 2 there is one crystallographically independent (ZnOEP)2·BPy dimer, two C60˙– and TDAE˙+ ion-radicals and one vacancy, which is shared by solvent C6H6 and C6H4Cl2 molecules.
TDAE˙+ and C60˙– ion-radicals form ionic TDAE˙+ - C60˙– layers. Fig. 4a shows the projection on this layer and Fig. 4b shows the view along these layers. The ionic layers alternate with the neutral layers composed of closely packed ZnOEP molecules. Within ionic layers one-dimensional chains formed by C60˙– can be outlined along the b axis with a uniform interfullerene center-to-center distance of 10.61 Å. This distance is longer than that in the antiferromagnetic phase of (TDAE˙+)·(C60˙–) (9.915 Å, the shortened vdW C⋯C contacts are 3.081–3.379 Å25) and prevents the formation of shortened vdW C⋯C contacts between C60˙– in 2 (all C⋯C contacts are longer than 3.45 Å). The chains of C60˙– are separated by the chains formed by alternating (solvent–TDAE˙+–BPy–TDAE˙+) units (Fig. 4a). The TDAE˙+ radical cations are isolated in the chains but form vdW contacts with solvent molecules (Cl⋯C contacts), BPy (C and N⋯C contacts) and C60˙–. Therefore, TDAE˙+ and C60˙– ion-radicals have rather loose packing in 2.
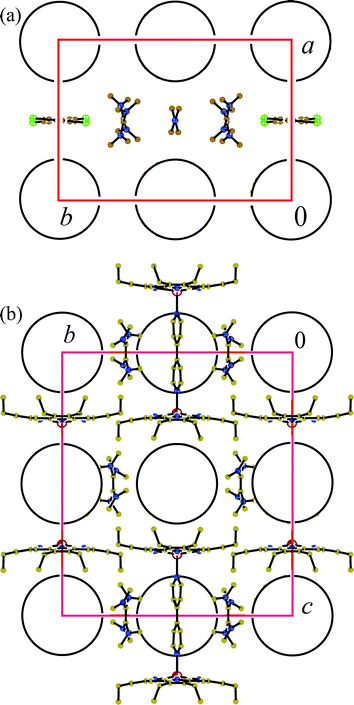 | ||
| Fig. 4 View of the crystal structure of 2 along crystallographic c- (a) and a-axes (b). C60˙– are shown by circles. Only one most occupied orientation is shown for TDAE˙+. In (b) only one orientation of BPy is shown and solvent molecules are not depicted for clarity. | ||
The geometry of the (ZnOEP)2·BPy dimer in 2 is slightly different from that in 1. Porphyrin macrocycles are arranged parallel to each other and rotate by an angle of 3.38°. The length of the Zn⋯N(BPy) coordination bond is noticeably shorter in 2 (2.112(1) Å) than in 1 and even close to the length of the averaged Zn⋯N(OEP) bonds (2.0705 Å). The ZnOEP macrocycles have the rms deviations of atoms 0.1477 Å from the mean porphyrin plane and the displacement of zinc atom by 0.462 Å from this plane toward the BPy ligand. One of two C60˙– is located in the cage formed by two bowl-shaped ZnOEP molecules (Fig. 4b), which conform well with the spherical shape of C60 to provide the formation of vdW N, C(ZnOEP)⋯C(C60˙–) and Zn⋯C(C60˙–) contacts, the Zn⋯C(C60˙–) distances being longer than 3 Å. The other C60˙–radical anion is positioned in the voids formed by two (ZnOEP)2·BPy dimers (this space is limited by planes of the four ZnOEP macrocycles and two BPy ligands) (Fig. 4b). Though there is the essential difference in the environment of two crystallographically independent C60˙– in 2, they are disordered in both positions. To understand why C60˙– is disordered in the cage formed by ZnOEP molecules one should take into account that neither shortened H(ZnOEP)⋯C(C60˙–) contacts nor the coordination of zinc atom to the fullerene cage are found in this case (similarly to 1). Most probably good complementarity between the shapes of ZnOEP macrocycles and C60 spheres and the formation of shortened N, C(ZnOEP)⋯C(C60˙–) contacts is still not enough to fix C60 rotation.
4. Magnetic properties of 2
The two-component (TDAE˙+)·(C60˙–) complex is a well-known ferromagnet with the highest transition temperature (Tc = 16 K) for metal free compounds.24 The antiferromagnetic phase of (TDAE˙+)·(C60˙–) is also known.25 In both antiferro- and ferromagnetic phases the C60˙–radical anions form closely packed one-dimensional chains and the difference between the two phases is only in the orientation of C60˙–.26 We studied the (TDAE˙+)·(TBPDA)2·(C60˙–) complex, in which TDAE˙+ and C60˙– ion-radicals were isolated by bulky TBPDA molecules. Nevertheless, antiferromagnetic interaction of spins found in this complex resulted in the decrease in magnetic moment of the complex even below 100 K.18,19 In 2 we constructed one-dimensional chains from C60˙–, which are similar to the C60˙– chains in (TDAE˙+)·(C60˙–), but have longer distances between C60˙–.The magnetic behaviour of 2 was studied on a polycrystalline sample by EPR spectroscopy from room temperature (RT = 295 K) down to 4 K. At RT the complex manifests a single Lorentzian line with g-factor equal to 2.0027 and a linewidth (ΔHpp) of 19.8 G. The g-factor value is an intermediate one between those characteristic of TDAE˙+ (g = 2.0035)27 and C60˙– (1.9996–2.0000)28. The appearance of one signal instead of two signals from individual ion-radicals indicates the existence of exchange interaction between TDAE˙+ and C60˙– in 2. It should be noted that only one signal was found in other TDAE containing fullerene complexes (g = 2.0003 and ΔHpp = 22 G in (TDAE˙+)·(C60˙–)29 and g = 2.0009 and ΔHpp = 29.3 G in (TDAE˙+)·(TBPDA)2·(C60˙–)18,19). Magnetic exchange can be realized either by direct interaction between two ion-radicals as in (TDAE˙+)·(C60˙–) or indirectly through the diamagnetic TBPDA molecules as in (TDAE˙+)·(TBPDA)2·(C60˙–)19. Most probably, exchange interaction is realized in 2 in both ways: directly due to the presence of vdW contacts between TDAE˙+ and C60˙– and indirectly through the diamagnetic solvent molecules and BPy.
The EPR signal noticeably narrows with the temperature decrease (Fig. 5, inset) that is characteristic of the EPR signal from C60˙–.28 Below 30 K the signal splits into two components, which become slightly wider with the temperature decrease and the drop in integral intensity below 9.6 K (Fig. 5) is evidence for the antiferromagnetic interaction of the spins. Similar splitting of the EPR signal into two components was observed previously in several ionic C60 complexes, which also manifest antiferromagnetic interaction of spins.18,19,30 The magnetic behaviour of 2 is similar to that of the antiferromagnetic phase of (TDAE˙+)·(C60˙–).31 However, due to more loose packing of C60˙– in the chains of 2, the antiferromagnetic ordering manifests itself at lower temperatures. The (intensity × T) value decreases in (TDAE˙+)·(C60˙–) even below 60 K,31 whereas in 2 the decrease in the (intensity × T) value is observed only below 20 K.
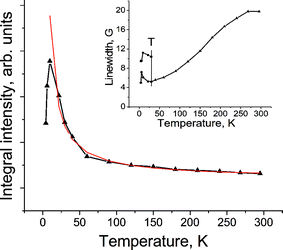 | ||
| Fig. 5 Temperature dependence of integral intensity of the EPR signal in 2. Red line shows the Curie curve calculated for 2. Inset shows the temperature dependence of linewidth of the EPR signals. “T” marks the temperature of the splitting of the EPR signal into two components. | ||
Conclusion
The multi-component approach allows one to design neutral and ionic complexes of ZnOEP with C60. Bidentate BPy coordinates to two ZnOEP molecules to form coordination dimers with the Zn⋯N(BPy) bonds in the 2.11–2.17 Å range. These dimers efficiently pack with C60 molecules to form molecular complex 1: {(ZnOEP)2·BPy}·(C60)2·(CHCl3)2 The reason for this is the slightly concave shape of the ZnOEP macrocycles, which conforms well with the spherical shape of C60.The packing of the (ZnOEP)2·BPy dimers and C60 has large cavities. Such packing allows additional donors or cations of small size to be introduced to form multi-component ionic complexes. In this work TDAE was used as an additional donor. Since TDAE reduces C60 to an anion-radical state, (TDAE˙+)2·{(ZnOEP)2·BPy}·(C60˙–)2·(C6H6)0.75·(C6H4Cl2)0.25 (2) with an ionic ground state is formed. In 2 the ionic TDAE˙+ - C60˙– layers alternate with the neutral ZnOEP layers. Complex 2 manifests antiferromagnetic interaction of spins at low temperatures (below 30 K), similarly to the antiferromagnetic phase of (TDAE˙+)·(C60˙–). However, the magnetic interactions in 2 are weaker due to longer distances between C60˙– in one-dimensional chains and between TDAE˙+ and C60˙– ion-radicals. Nevertheless, the multi-component approach is a useful tool to trace how changes in the crystal structure can affect magnetic properties. It also makes it possible to vary cations (D2+) or neutral metalloporphyrin molecules (for example, using MnIIOEP or CoIIOEP) within a series of {(D2+)2·{(MIIOEP)2·BPy}·(C60˙–)2·solvent} complexes. Such variations can essentially affect magnetic properties of the complexes. This work is now in progress.
Experimental
Materials
Zinc octaethylporphyrin (ZnTPP), 4,4′-bipyridine (BPy) and tetrakis(dimethylamino)ethylene (TDAE) were purchased from Aldrich. C60 of 99.98% purity was received from MTR Ltd. Chloroform (CHCl3) was used as received. Other solvents were purified in an argon atmosphere. o-Dichlorobenzene (C6H4Cl2) was distilled over CaH2 under reduced pressure, benzene (C6H6) and hexane were distilled over Na/benzophenone. The solvents were degassed and stored in a glove box. All manipulations for the synthesis of 2 were carried out in a MBraun 150B-G glove box with controlled atmosphere and the content of H2O and O2 was less than 1 ppm. The crystals were stored in a glove box and sealed in 2 mm quartz tubes for EPR measurements under 10–5 Torr. KBr pellets for IR- and UV-visible-NIR measurements were prepared in a glove box.Synthesis
The crystals of (ZnOEP2·BPy)·(C60)2·(CHCl3)2 (1) were obtained by diffusion. The CHCl3 solution (20 mL) containing ZnOEP (20 mg, 0.0335 mmol) and an excess of BPy (80 mg, 0.51 mmol) diffused in the C6H6 solution (20 mL) containing C60 (20 mg, 0.0277 mmol). After 1 month the crystals were formed on the walls of the diffusion tube. The solvent was decanted and the crystals were washed with acetone to give black prisms with characteristic blue luster (20 mg, 48% yield).The crystals of (TDAE˙+)2·{(ZnOEP)2·BPy}· (C60˙–)2·(C6H6)0.75·(C6H4Cl2)0.25 (2) were obtained by diffusion. ZnOEP (20 mg, 0.0335 mmol), an excess of BPy (80 mg, 0.51 mmol) and C60 (25 mg, 0.0347 mmol) were dissolved upon stirring in 20 mL of the C6H4Cl2–C6H6 (4 : 1) mixture during 2 h at 60 °C. The solution was cooled down to room temperature and 1 mL of TDAE was added. The solution was stirred for one more hour, filtered in the tube for diffusion (45 mL volume), and hexane (22 mL) was carefully layered over. After 1 month the solvent was decanted from the crystals precipitated and they were washed with hexane to give 14 mg (25% yield) of black prisms with characteristic blue luster.
The composition of 1 and 2 was determined from X-ray diffraction data for a single crystal. The unit cell parameters of several selected crystals from one synthesis were the same.
General
FT-IR spectra were measured in KBr pellets with a Perkin-Elmer 1000 Series spectrometer (400–7800 cm–1). UV-visible-NIR spectra were measured in KBr pellets on a Shimadzu-3100 spectrometer in the 220–1600 nm range. EPR spectra of 2 were recorded from 295 down to 4 K with a JEOL JES-TE 200 X-band ESR spectrometer.X-Ray crystal structure determination
Crystal data of 1 at 110(2) K: C204H98Cl6N10Zn2, Mr = 3032.36 g mol–1, black prisms, triclinic, P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) , a = 13.847(9), b = 14.011(11), c = 19.795(16) Å, α = 78.50(3), β = 76.35(3), γ = 63.27(2)°, V = 3314(4) Å3, Z = 2, dcalc = 1.519 g cm–3, µ = 0.555 mm–1, F(000) = 1554, max. 2θmax = 60.08°. In total, 26 298 reflections were measured, 18 271 of which were independent. The least-squares refinement on F2 was done to R1 [F > 2σ(F)] = 0.0917 for 10 828 observed reflections, wR2 = 0.2453 (all data), R1 = 0.1366 for all 18 271 observed reflections with 1238 parameters and 8840 restraints; final GoF = 0.951. CCDC reference number is 648658.
, a = 13.847(9), b = 14.011(11), c = 19.795(16) Å, α = 78.50(3), β = 76.35(3), γ = 63.27(2)°, V = 3314(4) Å3, Z = 2, dcalc = 1.519 g cm–3, µ = 0.555 mm–1, F(000) = 1554, max. 2θmax = 60.08°. In total, 26 298 reflections were measured, 18 271 of which were independent. The least-squares refinement on F2 was done to R1 [F > 2σ(F)] = 0.0917 for 10 828 observed reflections, wR2 = 0.2453 (all data), R1 = 0.1366 for all 18 271 observed reflections with 1238 parameters and 8840 restraints; final GoF = 0.951. CCDC reference number is 648658.
Crystal data of 2 at 90(2) K: C228H149.5Cl0.5N18Zn2, Mr = 3289.6 g mol–1, black prisms, orthorhombic, I 222, a = 14.8555(4), b = 21.2150(7), c = 24.2022(6) Å, V = 7627.6(4) Å3, Z = 2, dcalc = 1.436 g cm–3, µ = 0.397 mm–1, F(000) = 3440, max. 2θmax = 69°. In total, 124 312 reflections were measured, 15 596 of which were independent. The least-squares refinement on F2 was done to R1 [F > 2σ(F)] = 0.0496 for 13 620 observed reflections, wR2 = 0.1466 (all data), R1 = 0.0571 for all 15 605 observed reflections with 930 parameters and 1934 restraints; final GoF = 1.077. CCDC reference number is 648659.
X-Ray diffraction data for 1 were collected using a Bruker SMART CCD diffractometer (sealed tube, Mo Kα radiation, λ = 0.71073 Å, µ = 0.08 mm–1) and for 2 using a Bruker SMART1000 CCD diffractometer installed at a rotating anode source (Mo Kα radiation, λ = 0.71073 Å). Both diffractometers were equipped with an Oxford Cryosystems nitrogen gas-flow apparatus. For 1 a series of three ω scans with a 0.3° frame width was collected. For 2 the data were collected by the rotation method with 0.3° frame-width (ω scan) and 10 s exposure time per frame. Four sets of data (600 frames in each set) were collected, nominally covering half of the reciprocal space. Both structures were solved by direct methods using the SHELXTL program package32 and refined by full-matrix least squares on F2. The ordered non-hydrogen atoms were refined anisotropically. Positions of hydrogen atoms were calculated geometrically. Subsequently, the positions of H atoms were refined by the “riding” model with Uiso = 1.2Ueq of the connected non-hydrogen atom or as ideal CH3groups with Uiso = 1.5Ueq.
Disorder
Fullerenes are disordered in 1 between two orientations with the 0.512/0.488 occupancies. These orientations are linked by the rotation of fullerene molecules about threefold non-crystallographic axis. Chloroform molecules are disordered between three positions with the 0.658/0.237/0.105 occupancies.Both crystallographically independent fullerene molecules rotate almost freely in 2. The attempts to approximate them by several orientations show that there are more than three orientations for each C60˙–. In this case disordered fullerene molecules were approximated with 120 carbon atoms with the 0.5 occupancy assigned to electron density peaks after using direct method and consequent Fourier syntheses. Atomic coordinates and anisotropic thermal parameters for fullerene atoms were refined by a SHELXL conjugate-gradient least square procedure at the early stages and full-matrix least square procedure in final refinement, with thermal parameters fixed. The BPy ligand is disordered between four orientations with the 0.30/0.26/0.22/0.22 occupancies, which are linked by its rotation about the axis passing through two nitrogen atoms. The TDAE˙+ radical cations are disordered between two positions with 0.698/0.302 occupancies. The position of solvent molecules is shared by benzene and dichlorobenzene molecules with the 0.75/0.25 occupancies.
Acknowledgements
The work was supported by Grant-in-Aid Scientific Research from the Ministry of Education, Culture, Sports, Science and Technology, Japan (152005019, 21st Century COE), the Russian Science Support Foundation, INTAS YSF 05-109-4653 and RFBR grant N 06-03-32824 and 06-03-91361 and Japan–Russia Research Cooperative Program.References
- M. J. Rosseinsky, J. Mater. Chem., 1995, 5, 1497 RSC.
- B. Gotschy, Fullerene Sci. Technol., 1996, 4, 677 CAS.
- D. V. Konarev and R. N. Lyubovskaya, Russ. Chem. Rev., 1999, 68, 19 RSC.
- D. V. Konarev, Yu. V. Zubavichus, E. F. Valeev, Yu. L. Slovokhotov, Yu. M. Shul'ga and R. N. Lyubovskaya, Synth. Met., 1999, 103, 2364 CrossRef CAS.
- A. L. Litvinov, D. V. Konarev, A. Yu. Kovalevsky, I. S. Neretin, Yu. L. Slovokhotov, P. Coppens and R. N. Lyubovskaya, CrystEngComm, 2002, 4, 618 RSC.
- D. V. Konarev, A. Yu. Kovalevsky, A. L. Litvinov, N. V. Drichko, B. P. Tarasov, P. Coppens and R. N. Lyubovskaya, J. Solid State Chem., 2002, 168, 474 CrossRef CAS.
- A. L. Balch and M. M. Olmstead, Chem. Rev., 1998, 98, 2123 CrossRef CAS.
- M. M. Olmstead, D. A. Costa, K. Maitra, B. C. Noll, S. L. Phillips, P. M. Van Calcar and A. L. Balch, J. Am. Chem. Soc., 1999, 121, 7090 CrossRef CAS.
- T. Ishii, N. Aizawa, M. Yamashita, H. Matsuzaka, T. Kodama, K. Kikuchi, I. Ikemoto and Y. Iwasa, J. Chem. Soc., Dalton Trans., 2000, 4407 RSC.
- P. D. W. Boyd, M. C. Hodgson, C. E. F. Rickard, A. G. Oliver, L. Chaker, P. J. Brothers, R. D. Bolskar, F. S. Tham and C. A. Reed, J. Am. Chem. Soc., 1999, 121, 10487 CrossRef CAS.
- D. V. Konarev, I. S. Neretin, Yu. L. Slovokhotov, E. I. Yudanova, N. V. Drichko, Yu. M. Shul'ga, B. P. Tarasov, L. L. Gumanov, A. S. Batsanov, J. A. K. Howard and R. N. Lyubovskaya, Chem.–Eur. J., 2001, 7, 2605 CrossRef CAS.
- D. V. Konarev, A. Yu. Kovalevsky, S. S. Khasanov, G. Saito, D. V. Lopatin, A. V. Umrikhin, A. Otsuka and R. N. Lyubovskaya, Eur. J. Inorg. Chem., 2006, 1881 CrossRef CAS.
- D. V. Konarev, I. S. Neretin, A. L. Litvinov, N. V. Drichko, Yu. L. Slovokhotov, R. N. Lyubovskaya, J. A. K. Howard and D. S. Yufit, Cryst. Growth Des., 2004, 4, 643 CrossRef CAS.
- A. L. Litvinov, D. V. Konarev, A. Yu. Kovalevsky, P. Coppens and R. N. Lyubovskaya, Cryst. Growth Des., 2005, 5, 1807 CrossRef CAS.
- D. V. Konarev, S. S. Khasanov, D. V. Lopatin, V. V. Rodaev, G. Saito and R. N. Lyubovskaya, Russ. Chem. Bull., 2007, in press Search PubMed.
- D. V. Konarev, A. Yu. Kovalevsky, A. Otsuka, G. Saito and R. N. Lyubovskaya, Inorg. Chem., 2005, 44, 9547 CrossRef CAS.
- D. V. Konarev, S. S. Khasanov, G. Saito, A. Otsuka and R. N. Lyubovskaya, J. Mater. Chem., 2007 Search PubMed in press.
- D. V. Konarev, I. S. Neretin, G. Saito, Yu. L. Slovokhotov, A. Otsuka and R. N. Lyubovskaya, Dalton Trans., 2003, 3886 RSC.
- D. V. Konarev, A. Yu. Kovalevsky, S. S. Khasanov, G. Saito, A. Otsuka and R. N. Lyubovskaya, Eur. J. Inorg. Chem., 2005, 4822 CrossRef CAS.
- T. Picher, R. Winkler and H. Kuzmany, Phys. Rev. B, 1994, 49, 15879 CrossRef.
- V. N. Semkin, N. G. Spitsina, S. Krol and A. Graja, Chem. Phys. Lett., 1996, 256, 616 CrossRef CAS.
- K. Pokhodnia, J. Papavassiliou, P. Umek, A. Omerzu and D. Mihailovič, J. Phys. Chem., 1999, 110, 3606 Search PubMed.
- H.-B. Bürgi, E. Blanc, D. Schwarzenbach, S. Liu, Y. Lu, M. M. Kappes and J. A. Ibers, Angew. Chem., Int. Ed. Engl., 1992, 31, 640 CrossRef.
- P.-M. Allemand, K. C. Khemani, A. Koch, F. Wudl, K. Holczer, S. Donovan, G. Grüner and J. D. Thompson, Science, 1991, 253, 301 CrossRef CAS.
- B. Narymbetov, H. Kobayashi, M. Tokumoto, A. Omerzu and D. Mihailovič, Chem. Commun., 1999, 1511 RSC.
- B. Narymbetov, A. Omerzu, V. V. Kabanov, M. Tokumoto, H. Kobayashi and D. Mihailovič, Nature, 2000, 407, 883 CrossRef CAS.
- K. Kuwata and D. H. Geske, J. Am. Chem. Soc., 1964, 86, 2101–2105 CrossRef CAS.
- C. A. Reed and R. D. Bolskar, Chem. Rev., 2000, 100, 1075 CrossRef CAS.
- K. Tanaka, A. A. Zakhidov, K. Yoshizawa, K. Okahara, T. Yamabe, K. Yakushi, K. Kikuchi, S. Suzuku, L. Ikemoto and Y. Achiba, Phys. Rev. B, 1993, 47, 7554 CrossRef CAS.
- D. V. Konarev, S. S. Khasanov, A. Otsuka, G. Saito and R. N. Lyubovskaya, Inorg. Chem., 2007, 46, 2261 CrossRef CAS.
- D. Arčon, R. Blink, D. Mikhailovič, A. Omerzu and P. Cevc, Europhys. Lett., 1999, 46, 667 CrossRef CAS.
- G. M. Sheldrick, SHELX97, University of Göttingen, Germany, 1999.
Footnotes |
| † CCDC reference numbers 648658 and 648659. For crystallographic data in CIF or other electronic format see DOI: 10.1039/b708100f |
| ‡ Electronic supplementary information (ESI) available: IR spectra of starting compounds and complexes 1 and 2, EPR spectra of 2 and temperature dependencies of the parameters of EPR signal in 2. See DOI: 10.1039/b708100f |
| This journal is © The Royal Society of Chemistry 2008 |