A catemer-to-dimer structural transformation in cyheptamide†‡
A. J.
Florence
*a,
K.
Shankland
b,
T.
Gelbrich
c,
M. B.
Hursthouse
c,
N.
Shankland
a,
A.
Johnston
a,
P.
Fernandes
a and
C. K.
Leech
b
aSolid-State Research Group, Strathclyde Institute of Pharmacy and Biomedical Sciences, University of Strathclyde, 27 Taylor St, Glasgow, UK G4 0NR. E-mail: alastair.florence@strath.ac.uk; Fax: +44 141 552 2562; Tel: +44 141 548 4877
bISIS Facility, Rutherford Appleton Laboratory, Chilton, Didcot, Oxon, UK OX11 0QX
cSchool of Chemistry, University of Southampton Highfield, Southampton, UK SO17 1BJ
First published on 9th November 2007
Abstract
A catemeric crystal structure of cyheptamide undergoes a transformation in the solid-state upon heating to produce a dimer-based form whose structure has been determined from laboratory X-ray powder diffraction (XRPD) data, thereby providing the first conclusive evidence of a carbamazepine analogue crystallising in both hydrogen bonded motifs.
Cyheptamide (CYH) and dihydrocarbamazepine (DHC) are analogues of carbamazepine (CBZ; Fig. 1), a dibenzazepine drug used to control seizures. CBZ crystallises in 4 known polymorphic forms, all dimer-based,1 whereas the previously reported polymorphs of CYH (form I)2 and DHC (forms I–III)3 all adopt catemeric crystal structures (see, for example, Fig. 2 in ref. 2). This communication details a catemer → dimer transformation of CYH, the significance of which is that there has been no prior experimental report of pure CBZ, or one of its analogues, crystallising in both hydrogen bonded motifs. The molecular packing arrangements in CYH forms I and II, plus CBZ form I and DHC form I, are then compared using the XPac program.4
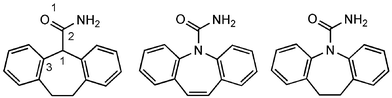 | ||
| Fig. 1 Left to right: CYH, CBZ and DHC. Torsion angle O1–C2–C1–C3 in CYH determines the orientation of the amide group in the rigid-body Rietveld refinement reported here. | ||
CYH form I was sourced from Sigma–Aldrich and the transformation, form I → II, was identified as a DSC endotherm with Tonset = 441.6 K (see ESI).‡ The transformation was then effected in a rotating 0.7 mm borosilicate glass capillary, mounted on Bruker AXS D8 Discover TXS diffractometer, by heating to 458 K. The sample of form II was then cooled to 100 K and held at that temperature for the duration of the XRPD data collection.§
The diffraction pattern indexed to a triclinic unit cell with dimensions sufficiently similar to CBZ form I to suggest that the former is essentially isostructural with the latter.¶ This was confirmed by a rigid-body Rietveld refinement7 of CYH form II, in the program TOPAS,8 from a starting structure generated using simulated annealing.|| The final refinement included a total of 76 parameters (27 profile, 6 cell, 1 scale, 14 preferred orientation, 4 torsion angles, 12 position, 12 rotation), yielding Rwp = 4.6 (Fig. 2).
![Final observed (points), calculated (line) and difference [(yobs – ycalc)/σ(yobs)] profiles for the Rietveld refinement of CYH form II.](/image/article/2008/CE/b712547j/b712547j-f2.gif) | ||
| Fig. 2 Final observed (points), calculated (line) and difference [(yobs – ycalc)/σ(yobs)] profiles for the Rietveld refinement of CYH form II. | ||
The resulting structure was further scrutinised by allowing all fractional coordinates to refine freely (444 parameters, Rwp = 1.5). As expected, the improved Rwp came at the expense of some chemical sense (e.g. H-atoms moving to nonsensical positions), but otherwise, the geometry of each of the 4 independent molecules was well preserved (Fig. 3), confirming the correctness of the rigid-body refined crystal structure.
 | ||
| Fig. 3 The 4 independent molecules of the rigid-body refined crystal structure of CYH form II (blue), with the freely refined equivalent overlaid in red. | ||
The XPac program4a was used to determine how the molecular packing arrangements of CYH forms I2 and II, CBZ form I1a and DHC form I3a are related to each other. The procedure was carried out as previously described for a study of 25 CBZ-related structures.4b The current investigation confirmed that CYH form I is isostructural with DHC form I and that CYH form II is isostructural with CBZ form I. Hence, CYH form II exhibits the same approximate non-crystallographic symmetry elements (local centres of inversion and local 31 symmetry) that have been discussed for CBZ form I, which in turn resembles the R![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) structure of CBZ form II.4b Moreover, there is a single common packing motif which occurs in all four structures that were investigated here—the one-dimensional (1D) stack of molecules depicted in Fig. 4 and 5.
structure of CBZ form II.4b Moreover, there is a single common packing motif which occurs in all four structures that were investigated here—the one-dimensional (1D) stack of molecules depicted in Fig. 4 and 5.
 | ||
| Fig. 4 Structural similarity exhibited by CYH forms I and II. From top to bottom: (1) a common 1D stack of molecules viewed along the translation vector (parallel to the a-axis of each form); (2) adjacent stacks linked by hydrogen bonds to give either a catemer (form I, left) or a dimer (form II, right); (3) the unit cells. | ||
 | ||
| Fig. 5 Representation of the unit cells of CYH forms I and II, with just one example of their common 1D stack of molecules in a similar orientation. | ||
This stack is identical with one out of two mutually exclusive packing arrangements, the other being a dimeric “handshake” motif, which dominate the crystal structures formed by CBZ and related molecules.4b Space-filling arguments were employed to explain the frequent occurrence of this 1D stack (referred to as “supramolecular construct A” in ref. 4b) and its finite counterpart.4b In CBZ structures, these stacks are associated with translation periods of 4.92 to 5.46 Å (Table 3, column 3 in ref. 4b). The corresponding values are elongated to 5.65 Å in the case of CYH and DHC, following the volume increase due to the different structure of their central seven-member rings (Fig. 1). Fig. 4 illustrates that the CYH I → II transition is associated with a catemer → dimer change in hydrogen bonding, thus involving the most prominent intermolecular interactions in these structures. At the same time, the 1D stacking of CYH molecules along the respective a-axis is preserved (Fig. 5), despite the absence of prominent intermolecular interactions within the resulting stack, confirming the importance of space-filling effects as a driving force in the formation of crystals.
Amongst the many reports on the solid-state diversity of CBZ and its analogues, one fact continues to intrigue—CYH and DHC crystallise readily in the C(4) catemeric motif, but there is only one report of catemeric CBZ (CBZ![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :DHC 1:1 solid solution).11 This is noteworthy, given the molecular similarities and the fact that CBZ crystal structure predictions12 show the catemeric form to be energetically competitive with the widely observed R22(8) dimer. The present study offers further encouragement in the pursuit of a pure C(4) form of CBZ by showing that the similarity between CBZ and CYH is not merely molecular—it is apparent at the level of crystal structure, with a strong structural correspondence between CBZ form I and CYH form II.
:DHC 1:1 solid solution).11 This is noteworthy, given the molecular similarities and the fact that CBZ crystal structure predictions12 show the catemeric form to be energetically competitive with the widely observed R22(8) dimer. The present study offers further encouragement in the pursuit of a pure C(4) form of CBZ by showing that the similarity between CBZ and CYH is not merely molecular—it is apparent at the level of crystal structure, with a strong structural correspondence between CBZ form I and CYH form II.
Acknowledgements
We thank the Basic Technology programme of the UK Research Councils for funding this work under the project Control and Prediction of the Organic Solid State (http://www.cposs.org.uk).Notes and references
- (a) S. G. Fleischman, S. S. Kuduva, J. A. McMahon, B. Moulton, R. D. B. Walsh, N. Rodriguez-Hornedo and M. J. Zaworotko, Cryst. Growth Des., 2003, 3, 909 CrossRef CAS; (b) A. L. Grezsiak, M. Lang, K. Kim and A. J. Matzger, J. Pharm. Sci., 2003, 92, 2260 CrossRef CAS; (c) M. Lang, J. W. Kampf and A. J. Matzger, J. Pharm. Sci., 2002, 91, 1186 CrossRef CAS; (d) C. Rustichelli, G. Gamberini, V. Ferioli, M. C. Gamberini, R. Ficarra and S. Tommasini, J. Pharm. Biomed. Anal., 2000, 23, 41 CrossRef CAS.
- C. K. Leech, A. J. Florence, K. Shankland, N. Shankland and A. Johnston, Acta Crystallogr., Sect. E, 2007, 63, o205.
- (a) G. Bandoli, M. Nicolini, A. Ongaro, G. Volpe and A. Rubello, J. Crystallogr. Spectrosc. Res., 1992, 22, 177 CrossRef CAS; (b) W. T. A. Harrison, H. S. Yathirajan and H. G. Anilkumar, Acta Crystallogr., Sect. C, 2006, 62, o240 CrossRef; (c) C. K. Leech, A. J. Florence, K. Shankland, N. Shankland and A. Johnston, Acta Crystallogr., Sect. E, 2007, 63, o675 CrossRef.
- (a) T. Gelbrich and M. B. Hursthouse, CrystEngComm, 2005, 7, 324 RSC; (b) T. Gelbrich and M. B. Hursthouse, CrystEngComm, 2006, 8, 448 Search PubMed.
- I. C. Madsen and R. J. Hill, J. Appl. Crystallogr., 1994, 27, 385 CrossRef CAS; K. Shankland, W. I. F. David and D. S. Sivia, J. Mater. Chem., 1997, 7, 569 RSC.
- P. Fernandes, K. Shankland, A. J. Florence, N. Shankland and A. Johnston, J. Pharm. Sci., 2007, 96, 1192 CrossRef CAS.
- H. M. Rietveld, J. Appl. Crystallogr., 1969, 2, 65 CrossRef CAS.
- A. A. Coelho, TOPAS User Manual, 2003, v3.1. Bruker AXS GmbH, Karlsruhe, Germany.
- G. S. Pawley, J. Appl. Crystallogr., 1981, 14, 357 CrossRef CAS.
- W. I. F. David, K. Shankland and N. Shankland, Chem. Commun., 1998, 931 RSC; W. I. F. David, K. Shankland, J. van de Streek, E. Pidcock, W. D. S. Motherwell and J. C. Cole, J. Appl. Crystallogr., 2006, 39, 910 CrossRef CAS.
- A. J. Florence, C. K. Leech, N. Shankland, K. Shankland and A. Johnston, CrystEngComm, 2006, 8, 746 RSC.
- A. J. Florence, A. Johnston, S. L. Price, H. Nowell, A. R. Kennedy and N. Shankland, J. Pharm. Sci., 2006, 95, 1918 CrossRef; A. J. Cruz Cabeza, G. M. Day, W. D. S. Motherwell and W. Jones, Cryst. Growth Des., 2007, 7, 100 CrossRef.
Footnotes |
| † CCDC reference number 656948. For crystallographic data in CIF or other electronic format see DOI: 10.1039/b712547j |
| ‡ Electronic supplementary information (ESI) available: Differential scanning calorimetry analysis (Fig. S1). See DOI: 10.1039/b712547j |
| § XRPD data were collected over the range 3–70° 2θ (12 kW; Cu Kα1, 1.54056 Å; step size 0.017° 2θ), using a variable count time scheme.5 The diffractometer was equipped with a Bruker Vantec PSD and an Oxford Cryosystems Cryostream Plus temperature control device. |
¶ Refined lattice parameters for CYH form II (100 K) and, in square brackets, CBZ form I (160 K):6a = 5.6491(1) [5.18561(9)], b = 19.5639(4) [20.57575(29)], c = 22.0741(5) [22.24106(30)] Å, α = 84.2178(13) [84.1942(8)], β = 88.4073(14) [87.9756(7)], γ = 83.6001(13) [85.1053(8)]°, V = 2411.72(9) [2351.44(7)] Å3 (both structures P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) , Z′ = 4). , Z′ = 4). |
| || The data set was background subtracted and truncated to 30.6° 2θ for Pawley fitting9 (Pawley χ2 = 9.55). The simulated annealing (SA) component of DASH10 was used to optimise the CYH form II crystal structure (internal coordinate description derived from the single-crystal structure of CYH form I;2 7 degrees of freedom for each of the 4 molecules) against the diffraction data (194 reflections) to a favourable χ2 ratio of ca. 8 (profile χ2/Pawley χ2). A TOPAS-type rigid-body description of the CYH molecule was then constructed using the single-crystal structure of form I.2 X–H distances were normalised to typical X-ray values and torsion angles lying very close to 0/180° were set to 0 or 180°, respectively, with O1–C2–C1–C3 (Fig. 1) defined as variable. Four separate rigid-body descriptions were then mapped onto the 4 independent molecules of the SA global minimum, using a distance minimisation procedure within TOPAS. Thereafter, the SA coordinates were deleted and structural refinement proceeded in terms of the rigid bodies only. Assignment of the correct O1–C2–C1–C3 torsion angles was checked by permutation, noting the effect of rotations upon Rwp. The maximum e.s.u. for the rigid-body positional and orientational parameters was small, at 0.0008 and 0.3°, respectively. For the rigid-body refined fractional coordinates, see CCDC reference number 656948.† |
| This journal is © The Royal Society of Chemistry 2008 |