An open-channel architecture assembly from the [Os3(CO)8{µ-η3-ON![[double bond, length as m-dash]](https://www.rsc.org/images/entities/h2_char_e001.gif) CPh(NC5H4)}2] cluster†‡
CPh(NC5H4)}2] cluster†‡
Janet Shuk-Yee
Wong
,
Yan-Juan
Gu
,
Lap
Szeto
and
Wing-Tak
Wong
*
Department of Chemistry, University of Hong Kong, Pokfulam Road, Hong Kong, China. E-mail: wtwong@hkucc.hku.hk; Fax: (852)25472933; Tel: (852)28592157
First published on 10th October 2007
Abstract
A novel osmium cluster with an open-channel structure has been synthesised and characterised by X-ray diffraction.
The design of crystalline molecular porous materials has become an attractive prospect because of their potential applications as molecular sieves, sensors, ion-exchangers and catalysts.1 In the last few years, various approaches to the design of channel-like supramolecular structures have been used. The coordination of transition metal ions by different polyfunctional organic ligands has yielded a large variety of extended open-framework structures.2 Strong (O–H⋯O) and weak (C–H⋯O) hydrogen bonds, halogen bond (C–X⋯O) and weak C–H⋯π have been well characterised and exploited to playing an important role in crystal engineering and organometallic architecture.3 In addition to the above mentioned interactions, a C
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O⋯CO interaction was also observed in the crystal structures deposited, which is relatively uncommon.4Hydroxyamine or oxime containing metal complexes display very rich structural diversity as these ligands can act as bi- or multidentate ligands that are attached to a single metal atom.5 In this communication, we report an unusual Os cluster with two different polymorphous shapes that precipitated from the same solvent. The architecture self-assembled from the cluster by means of an interesting interplay of carbonyl C
O⋯CO interaction was also observed in the crystal structures deposited, which is relatively uncommon.4Hydroxyamine or oxime containing metal complexes display very rich structural diversity as these ligands can act as bi- or multidentate ligands that are attached to a single metal atom.5 In this communication, we report an unusual Os cluster with two different polymorphous shapes that precipitated from the same solvent. The architecture self-assembled from the cluster by means of an interesting interplay of carbonyl C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) O⋯OC interactions, weak hydrogen bond (C–H⋯O) and C–H⋯π interactions, giving rise to a hexagonal channel structure. To the best of our knowledge, molecular metal clusters with channel structures are very rare.
O⋯OC interactions, weak hydrogen bond (C–H⋯O) and C–H⋯π interactions, giving rise to a hexagonal channel structure. To the best of our knowledge, molecular metal clusters with channel structures are very rare.
Treatment of [Os3(CO)11(NCMe)] with one equivalent amount of phenyl 2-pyridyl ketoxime at room temperature afforded complex [Os3(CO)8{µ-η3-ONCPh(NC5H4)}2]§1 (which is crystal b in this article) as previously reported.6 The reaction is shown in Scheme S1.‡ A red, needle-shaped crystal a was obtained by diffusion of n-hexane into a CH2Cl2 solution at –20 °C. The molecular structure was compared to a block-shaped crystal b by X-ray crystallographic analysis. We also found that only crystal b could be observed when the crystallisation process was carried out at RT, which suggests that crystal a is the kinetic product and crystal b is the thermodynamic product. X-Ray diffraction analysis on these two crystals showed that they have the same molecular structure with different cell parameters and crystal packings. The selected bond lengths and angles of the two kinds of crystals are summarised in Table S1.‡ As the two structures highly resemble each other, our discussion mainly focuses on crystal a. Fig. 1 shows the perspective view of the molecular configuration of crystal a. As previously reported, crystal a consists of an open triosmium triangle [Os1–Os2 2.8232(7) Å] with two oximato ligands spanning the open Os1⋯Os1viii edge [3.5861(7) Å; symmetry code: (viii) y, x, ½ – z] via the N–O groups in a µ-η2 fashion. This is similar to the [Ru3(µ3-NPh)2(µ-η2-ONPh)2(CO)7] cluster.7 In the molecules of crystal a, there is a 2-fold rotation axis passing through Os2 and bisecting the open triangular metal framework. The molecule of a contains two oximato ligands that chelate to the metal core in a µ-η3 manner. The ligand is bound to the metal core by coordination of the pyridine nitrogen lone pair [Os1–N1 2.147(6) Å] and the oximato oxygen and nitrogen atoms [Os1viii–O5 2.120(5), Os1–N2 2.133(6) Å]. The oximato oxygen atoms span to the neighbouring metal centre over the open metal–metal edge. The N2–O5 bond distance is 1.347(8) Å.
![ORTEP drawing of [Os3(CO)8{µ-η3-ONCPh(NC5H4)}2] (a), with a labelling scheme and 50% probability thermal ellipsoids, showing a two-fold axis passing through Os2. (Symmetry code *: y, x, 1/2 – z.)](/image/article/2008/CE/b708080h/b708080h-f1.gif) | ||
| Fig. 1 ORTEP drawing of [Os3(CO)8{µ-η3-ONCPh(NC5H4)}2] (a), with a labelling scheme and 50% probability thermal ellipsoids, showing a two-fold axis passing through Os2. (Symmetry code *: y, x, 1/2 – z.) | ||
The crystal information for the two forms is summarised in Table 1. The crystal a is centrosymmetric, the molecules are arranged to give channels descending along the c-axis in the unit cell. Moreover, the complex molecules are arranged in pairs, 6 of which form a hexagonal ring with a void in the centre (see Fig. 2). Because the adjacent channel shares a pair of molecules, each unit cell contains only 18 complex molecules. For the cavity, a diagonal distance of ca. 14 Å was observed. Using the PLATON software,8 it was found that each channel in one unit cell has an approximate van der Waals volume of ∼2658 Å3. Three channels were observed per unit cell, the centres of which were located at (0, 0, z), (1/3, 2/3, z) and (2/3, 1/3, z). A total of 37.5% of solvent accessible volume for crystal a was found. There were peaks with residual electron densities of range –0.948 to 3.03 e–/Å3 inside the channel. Unfortunately, attempts to identify and further refine these peaks failed, suggesting that the solvates in the channels were disordered. The solvent contribution was modelled by the SQUEEZE procedure of PLATON, from which an estimate of the voids (each about 2640 Å3) and the approximate number of electrons contained in them (∼1592 electron counts) was found. As only n-hexane and CH2Cl2 were used in the recrystallisation, as a consequence the void is supposed to be filled by molecules of these two solvents, which could be in any combination and ratio. By taking into consideration both of the electron counts and the size of the void volume found from above, one possibility is that there may be as many as about 7.5 molecules of n-hexane and 29 molecules of CH2Cl2 solvates in each void. While possibilities of other combinations could not be ruled out, the intensity contribution of the disordered solvates was thus subtracted from the full data set. The new data set, with the solvent contribution removed, was used in the final refinement.
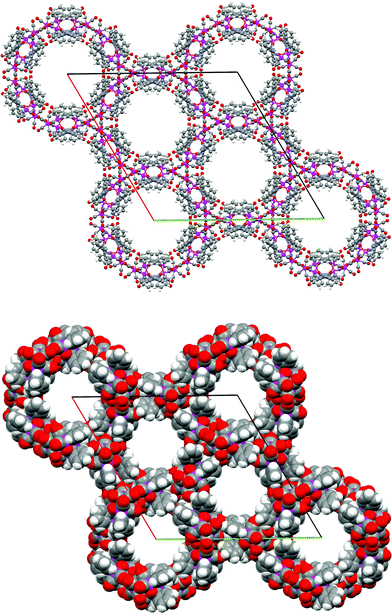 | ||
| Fig. 2 Packing diagram (upper) and space-filling (lower) of crystal a viewed down the c-axis showing the hexagonal channels (Os atoms are green, C gray, H white, O red). | ||
| Complex | [Os3(CO)8{µ-η3-ONCPh(NC5H4)}2·1.25(C6 H14)· 4.83(CH2Cl2] a |
[Os3(CO)8{µ-η3-ONCPh(NC5H4)}2] b |
|---|---|---|
| Formula | C44.83H45.17Cl9.67N4O10Os3 | C32H18N4O10Os3 |
| Formula weight | 1707.43 | 1189.10 |
| Space group | R-3c(h) | P43212 |
| a/Å | 37.703 (8) | 12.955(5) |
| b/Å | 37.703 (8) | 12.955(5) |
| c/Å | 17.320(5) | 19.352(6) |
| α/° | 90 | 90 |
| β/° | 90 | 90 |
| γ/° | 120 | 90 |
| V/Å3 | 21321(9) | 3248.0(19) |
| Z | 18 | 4 |
| D calc/g cm–3 | 2.392 | 2.432 |
| Crystal size/mm | 0.06 × 0.08 × 0.45 | 0.16 × 0.21 × 0.27 |
| Reflections collected | 42081 | 20116 |
| Independent reflections | 5653 (Rint = 0.0534) | 3658 (Rint = 0.0495) |
| Data/restraints/parameters | 5653/0/223 | 3658/0/224 |
| Goodness-of-fit on F2 | 1.098 | 1.020 |
| Final R indices [I > 2σ(I)] | R 1 = 0.0501, wR2 = 0.1310 | R 1 = 0.0248, wR2 = 0.0509 |
| Largest difference peak and hole/e Å–3 | 3.028 and –0.948 | 1.457 and –0.576 |
An X-ray crystallographic study revealed that crystal a is a trigonal lattice with a R-3c space group which is centrosymmetric. The molecular structure and detailed bonding in crystal a are shown in Fig. 3. Intriguingly, the enantiomers, which form a unit of hexagonal ring, were depicted in Fig. 3a. In Fig. 3b, the green, blue and red molecules have the same handedness conformation. The interactions between these molecules are shown by dotted lines and summarized in Table 2. Hydrogen bonding is observed between one of the H atoms of pyridine and the O atom of the oximato ligand (C–H⋯O–N) of adjacent enantiomers (Fig. 3a) [C6–H6⋯O5i (C⋯O 3.276(11) Å) (i): 1 – y, 1 – x, –1/2 + z]. This type of C–H⋯O–N interaction is not commonly observed in all types of C–H⋯X hydrogen bonds.5b Additionally, the intermolecular C–H⋯π interaction between one of the H atoms of pyridine and the phenyl ring of these enantiomers [C5–H5⋯centroid of C11i –C16i (2.933 Å)] occurs along the channel direction. Two additional types of bonding are responsible for the crystal architecture of the framework. The first is a hydrogen bonding (C–H⋯OC) of the H atom of pyridine (red molecule) and the O atom of the carbonyl group of the neighbouring molecule (blue molecule) [C8–H8⋯O1ii (C⋯O 3.297(9) Å) at (ii): 2/3 + x – y, 1/3 + x, 1/3 – z]. The distance is consistent with the weak hydrogen bond acceptor (Fig. 4) nature of the CO ligand.9 The second type of interaction is the carbonyl–carbonyl interaction between adjacent molecules, which has seldom been reported. Two pairs of carbonyl O⋯O short contacts are observed between the O3 and O3iii (2.632 Å; (iii): 1/3 + x – y, 2/3 – y, 1/6 – z), O4 and O4iii (2.939 Å) of the two adjacent molecules (red and green molecules), respectively. This interaction is also important in stabilising the channel architecture.
 | ||
| Fig. 3 (a) The enantiomers in crystal a: showing the H-bond interactions: C6–H6⋯O5i (C⋯O 3.276(11) Å) and the complementary intermolecular C–H⋯π interactions: C5–H5⋯CgC11–C16 (2.933 Å). (The cyan dot represents the centroid of the C11i–C16i ring and the yellow dot: the centroid of the C11viii–C16viii ring). Symmetry code: (i) 1 – y, 1 – x, –1/2 + z; (viii) y, x, 1/2 – z; (ix) 1 – x, 1 – y, –z. (b) Intermolecular interactions in crystal a: the network was constructed by hydrogen bonds C8–H8⋯O1ii (C⋯O 3.297(9) Å) and carbonyl O⋯O short contact: O3⋯O3iii (2.632 Å) and O4⋯O4iii (2.939 Å). The molecules depicted in red, green and blue have the same handedness conformation (Os atoms are magenta). Symmetry codes: (ii) 2/3 + x – y, 1/3 + x, 1/3 – z. (iii) 1/3 + x – y, 2/3 – y, 1/6 – z; (viii) y, x, 1/2 – z; (x) 2/3 – x + y, 1/3 + y, –1/6 + z; (xi) 1/3 – x + y, 2/3 – x, –1/3 + z. | ||
 | ||
| Fig. 4 The packing diagram of crystal b viewed down the a-axis. | ||
| D–H⋯A | D–H/Å | H⋯A/Å | D⋯A/Å | ∠(D–H⋯A)/° |
|---|---|---|---|---|
| a Symmetry operators are (i) 1 – y, 1 – x, –1/2 + z; (ii) 2/3 + x – y, 1/3 + x, 1/3 – z; (iii) 1/3 + x – y, 2/3 – y, 1/6 – z. b Cg refers to the centroids of the phenyl rings formed by carbon atoms C11, C12, C13, C14, C15 and C16. | ||||
| C6–H6⋯O5i | 0.93 | 2.42 | 3.276(11) | 154 |
| C8–H8⋯O1ii | 0.93 | 2.55 | 3.297(9) | 138 |
| C5–H5⋯Cgi | 2.933 | |||
| O3⋯O3iii | 2.632 | |||
| O4⋯O4iii | 2.939 |
Single crystal X-ray analysis of crystal b revealed a tetragonal crystal lattice with a P43212 space group. The three-dimensional packing of crystal b in the unit cell is shown in Fig. 4. Crystal b only contains complex molecules of the same handedness. The interactions between these molecules are shown by dotted lines in Fig. 5. The intermolecular interactions of crystal b are listed in Table 3. The molecules are connected by weak C–H⋯O intermolecular hydrogen bonds between one of the pyridyl H atom and the O atom of oximato ligand [C7–H7⋯O4iv 3.161(8) Å, (iv) –1/2 + x, 3/2 – y, 1/4 – z]. This H atom also interacts with the carbonyl O atom [C7–H7⋯O5v 3.430(8) Å, (v) 1/2 – y, 1/2 + x, –1/4 + z] to form another intermolecular hydrogen bond. Furthermore, there is a pair of intramolecular C–H⋯π interactions between the pyridyl ring (N1, C5–C9) of the ligand and the atom H16 (C16–H16⋯centroid of N1vi, C5vi–C9vi ring; (vi): 1 – y, 1 – x, 1/2 – z) with an H-to-centroid distance of 3.280 Å and an H-to-plane distance of 3.153 Å. Interestingly, there is also a close intermolecular O3⋯O4vii (2.887 Å; (vii): 3/2 – y, –1/2 + x, –1/4 + z) interaction between the carbonyl ligands of the adjacent molecules. By inspecting the packing of crystal b in Fig. S1,‡ it appears that only a 3D network without a channel can be formed.
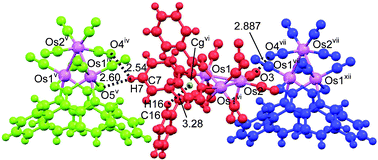 | ||
| Fig. 5 The intermolecular interactions in crystal b: the network was constructed by weak hydrogen bonds C7–H7⋯O4iv (C⋯O 3.161(8) Å) and C7–H7⋯O5v (C⋯O 3.430(8) Å); an intra-molecular C–H⋯π interaction C16–H16⋯Cgvi (3.280 Å) (Cg: centroid of N1/C5–C9: yellow dot); carbonyl O⋯O short contact: O3⋯O4vii (2.887 Å). The molecules depicted in green, red and blue have the same handedness conformation (Os atoms are shown in magenta). Symmetry codes: (iv) –1/2 + x, 3/2 – y, 1/4 – z; (v) 1/2 – y, 1/2 + x, –1/4 + z; (vi) 1 – y, 1 – x, 1/2 – z, (vii) 3/2 – y, –1/2 + x, –1/4 + z, (xii) 1/2 + x, 1/2 – y, 1/4 – z. | ||
| D–H⋯A | D–H/Å | H⋯A/Å | D⋯A/Å | ∠( D–H⋯A)/° |
|---|---|---|---|---|
| a Symmetry operators are (iv) –1/2 + x, 3/2 – y, 1/4 – z, (v) 1/2 – y, 1/2 + x, –1/4 + z; (vi) 1 – y, 1 – x, 1/2 – z, (vii) 3/2 – y, –1/2 + x, –1/4 + z, b Cg refers to the centroid of the pyridine ring formed by the N1, C5, C6, C7, C8, C9. | ||||
| C7–H7⋯O4iv | 0.93 | 2.54 | 3.161(8) | 124 |
| C7–H7⋯O5v | 0.93 | 2.60 | 3.430(8) | 148 |
| C16–H16⋯Cgvi | 3.280 | |||
| O3⋯O4vii | 2.887 |
In crystals a and b, the molecules are linked together to form 3D frameworks by C–H⋯π, C–H⋯OC and CO⋯OC interactions. However, there are two major differences between the structures of these two crystals. One is the molecular conformation. In the case of a, two enantiomers can embed into each other and form a dimer viahydrogen bonding and C–H⋯π bonding. These dimers are the actual building blocks, and are brought together viahydrogen bonds and O⋯O short contact. In contrast, only one-handedness molecules are present in crystal b. Every molecule is connected to two different molecules by hydrogen bonding and a O⋯O short contact. Another structural feature differentiating the two forms is the C–H⋯π interaction. The molecule in crystal a exhibits a pair of C–H⋯π interactions between enantiomers, whereas in crystal b, the molecule exhibits only one C–H⋯π intramolecular interaction. All these results demonstrate that the distinct morphologies of self-assembled supramolecular structures are intrinsically related to the molecular structures. The different interactions caused by the isomeric molecular structures are most likely responsible for the different morphological evolution.10
In conclusion, we have shown that two different kinds of crystals were assembled from the [Os3(CO)8{µ-η3-ONCPh(NC5H4)}2] complex. Despite the similarity in the molecular structures, the crystal packing of the needle-shaped and block-shaped crystals are, however, quite different with an open-channel hexagonal cage in the former case and a 3-D coordination network in the latter. It is interesting that the different crystal packing structures can be mainly ascribed to the presence of both enantiomers in the needle-shaped crystal.
We thank the Hong Kong Research Grants Council and the University of Hong Kong for financial support. J. S.-Y. Wong and Y. J. Gu acknowledge the receipt of postgraduate studentships administered by the University of Hong Kong. We also thank X. M. Chen, Sun-Yat Sen University, for assistance with collecting crystal data.
Notes and references
- (a) P. J. Langley and J. Hulliger, Chem. Soc. Rev., 1999, 28, 279 RSC; (b) R. Robson, J. Chem. Soc., Dalton Trans., 2000, 3735 RSC.
- (a) Z. Q. Qin, M. C. Jennings, R. J. Puddephatt and K. W. Muir, CrystEngComm, 2000, 1, 73 RSC; (b) J. Do and A. J. Jacobson, Inorg. Chem., 2001, 40, 2468 CrossRef CAS; (c) B. F. Abrahams, M. Moylan, S. D. Orchard and R. Robson, CrystEngComm, 2003, 5, 313 RSC; (d) D. Maspoch, D. Ruiz-Molina, K. Wurst, G. Vaughan, N. Domingo, J. Tejada, C. Rovira and J. Veciana, CrystEngComm, 2004, 6, 573 RSC.
- (a) U. Rychlewska and B. Warzajtis, Acta Crystallogr., 2000, B56, 833 CAS; (b) B. Venkataramanan, M.-A. Saifudin, V. Jagadese J. and V. Suresh, CrystEngComm, 2004, 6, 284 RSC; (c) J. Bernstein, R. E. Davis, L. Shimoni and N.-L. Chang, Angew. Chem., Int. Ed. Engl., 1995, 34, 1555 CrossRef CAS; (d) A. M. Beatty, CrystEngComm, 2001, 3, 243 RSC; (e) C.-F. Wang, Z.-Y. Zhu, Z.-X. Zhang, Z.-X. Chen and X.-G. Zhou, CrystEngComm, 2007, 9, 35 RSC.
- (a) W. Bolton, Nature, 1964, 201, 987 CAS; (b) R. G. Teller, R. G. Finke, J. P. Collman, H. B. Chin and R. Bau, J. Am. Chem. Soc., 1977, 99, 1104 CrossRef CAS; (c) F. H. Allen, C. A. Baalham, J. P. M. Lommerse and P.R. Raithby, Acta Crystallogr., Sect. B, 1998, 54, 320 CrossRef; (d) G. Bhattacharjya, G. Savitha and G. Ramanathan, CrystEngComm, 2004, 6, 233 RSC; (e) H. A. Sparkes, P. R. Raithby, E. Clot, G. P. Shields, J. A. Chisholm and F. H. Allen, CrystEngComm, 2006, 8, 563 RSC.
- (a) A. J. Deeming, D. W. Owen and N. I. Powell, J. Organomet. Chem., 1990, 398, 299 CrossRef CAS; (b) M. H. Chao, S. Kumaresan, Y. S. Wen, S. C. Lin, J. R. Hwu and K. L. Lu, Organometallics, 2000, 19, 714 CrossRef CAS.
- J. S. Y. Wong and W. T. Wong, New J. Chem., 2002, 26, 94 RSC.
- K. K. H. Lee and W. T. Wong, J. Chem. Soc., Dalton Trans., 1996, 3911 RSC.
- A. L. Spek, Acta Crystallogr., Sect. A, 1990, 46, C34.
- (a) D. Braga and F. Grepioni, Acc. Chem. Res., 1997, 30, 81 CrossRef CAS; (b) S. E. Gibson, H. Ibrahim and J. W. Steed, J. Am. Chem. Soc., 2002, 124, 5109 CrossRef.
- (a) Y. Wang, H. Fu, A. Peng, Y. Zhao, J. Ma, Y. Ma and J. Yao, Chem. Commun., 2007, 1623 RSC; (b) S. Sharma and T. P. Radhakrishnan, ChemPhysChem, 2003, 4, 67 CrossRef CAS.
Footnotes |
| † CCDC reference numbers 645290 and 645291. For crystallographic data in CIF or other electronic format see DOI: 10.1039/b708080h |
‡ Electronic supplementary information (ESI) available![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) : Reaction of [Os3(CO)11(NCMe)] with phenyl 2-pyridyl ketoxime (Scheme S1); selected bond distances and angles of crystals a and b (Table S1); unit cell of packing diagram in ball and stick form (upper) and space-filling (lower) of crystal b viewed down the c-axis (Fig. S1); IR spectrum of compound a in solution (Fig. S2); powder XRD of compound a using copper Kα1 radiation, 0–50° (Fig. S3); simulated powder XRD of compound a using copper Kα1 radiation (Fig. S4); simulated powder XRD of compound b using copper Kα1 radiation from 0–50° (Fig. S5); . See DOI: 10.1039/b708080h : Reaction of [Os3(CO)11(NCMe)] with phenyl 2-pyridyl ketoxime (Scheme S1); selected bond distances and angles of crystals a and b (Table S1); unit cell of packing diagram in ball and stick form (upper) and space-filling (lower) of crystal b viewed down the c-axis (Fig. S1); IR spectrum of compound a in solution (Fig. S2); powder XRD of compound a using copper Kα1 radiation, 0–50° (Fig. S3); simulated powder XRD of compound a using copper Kα1 radiation (Fig. S4); simulated powder XRD of compound b using copper Kα1 radiation from 0–50° (Fig. S5); . See DOI: 10.1039/b708080h |
| § A solution of [Os3(CO)11(NCMe)] (100 mg, 0.109 mmol) in CH2Cl2 was stirred with 1 equivalent of phenyl 2-pyridyl ketoxime (21.6 mg, 0.109 mmol) at room temperature. The reaction mixture gradually changed from yellow to deep yellow. Reactions were completed after stirring for 5 h, as monitored by TLC and IR. The reaction solution was concentrated in vacuo and separated by TLC using n-hexane-CH2Cl2 (1:6, v/v) as the eluent to afford pale orange [Os3(CO)8{µ-η3-ONCPh(NC5H4)}2] complex, 1 (Rf ≈ 0.50, 50 mg, 0.004 mmol, 40%). Elemental Anal. For cluster 1: Calc: C, 32.3; H, 1.5; N, 4.7%. Found: C, 32.2; H, 1.5; N, 4.5%. IR adsorptions (νmax/cm–1) of carbonyl: 2072(m), 1995(vs), 1925(w), 1922(w),1914(w); δH 8.86 (d, J 6.7, 4H, Py) and 7.81(td, J 6.7, 1.3, 4H, Py). Accurate mass m/z = 1189. X-Ray data were collected on a Bruker APEX CCD diffractometer. |
| This journal is © The Royal Society of Chemistry 2008 |