Targeted crystallisation of novel carbamazepine solvates based on a retrospective Random Forest classification†‡
Andrea
Johnston
a,
Blair F.
Johnston
a,
Alan R.
Kennedy
b and
Alastair J.
Florence
*a
aStrathclyde Institute of Pharmacy and Biomedical Sciences, University of Strathclyde, 27 Taylor Street, Glasgow, Scotland G4 0NR. E-mail: alastair.florence@strath.ac.uk; Fax: +44 (0) 141 552 2562; Tel: +44 (0) 141 548 4877
bWestCHEM, Department of Pure and Applied Chemistry, University of Strathclyde, 295 Cathedral Street, Glasgow, Scotland G1 1XL
First published on 2nd November 2007
Abstract
Three novel crystalline solvates of the antiepileptic compound carbamazepine were obtained by targeted crystallisation from solvents identified by a Random Forest classification of solvent properties, experimental conditions and known crystallisation outcomes.
Random Forest (RF)1 has been successfully applied to a range of classification and/or regression problems in physical chemistry, biology and process manufacturing.2 The basis of the method has been detailed elsewhere,1,2 but it is worth highlighting particular advantages (compared to other multivariate data analysis tools, such as principal components analysis and neural networks) that make it well suited to the analysis of solvate formation i.e. it cannot be overtrained and it provides a measure of the relative importance of descriptors used in the classification. This communication reports a retrospective RF classification using the solvent properties, experimental conditions and crystallisation outcomes from a previous automated parallel crystallisation3 study of carbamazepine4 (CBZ; Fig. 1). The results of the analysis provided a rational basis for the subsequent targeted crystallisation of three novel CBZ solvates (see ESI‡).
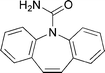 | ||
| Fig. 1 Molecular structure of carbamazepine (CBZ). | ||
The RF classification was performed using the ‘randomForest’ library package,1 in statistical computing environment ‘R’ v2.4.1.5 The training data set (see ESI‡) comprised 15 numerical physicochemical solvent descriptors and 3 categorical variables describing experimental conditions (T = High/Med/Low; Vacuum = On/Off; Vortex stirring = On/Off). Six known experimental outcomes input to the training data set were defined as: zero, where no recrystallised sample was obtained; one, two or three relating to CBZ polymorphs I, II and III;6four a mixture of one or more of these polymorphs and five, a solvate. The RF classification model was trained using all 18 descriptors plus crystallisation outcomes using the following parameters: seed = 45, mtry = 3, ntree = 10000.§ The multi-dimensional scaling (MDS) plot using two dimensions (Dim1 and Dim2, Fig. 2) obtained from the RF classification proximity matrix represents the crystallisations as two orthogonal clusters. Points with Dim1 < 0 correspond to crystallisations that yielded exclusively non-solvated forms or no sample, whilst the majority of points with Dim1 > 0 correspond with solvates. That said, amongst the Dim1 > 0 cluster it is significant that there are three groups of points that correspond with crystallisations that produced non-solvated CBZ. These crystallisations utilised the following solvents—nitromethane (NM), N,N-dimethylacetamide (DMA) and N-methylpyrrolidone (NMP) (Fig. 3).
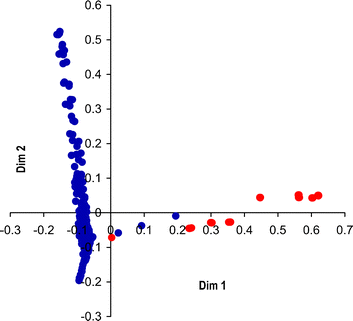 | ||
| Fig. 2 The MDS plot obtained from the RF classification proximity matrix. Each of the 326 points represents one crystallisation from the previous study4 and is coloured according to outcome; non-solvated forms or no sample (blue) and solvates (red). Note that data from several crystallisations (multiple conditions) from each solvent were used and consequently these points may appear as coincident on the plot. | ||
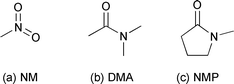 | ||
| Fig. 3 Molecular structures of solvents that produced non-solvated CBZ forms in the previous crystallisation study, yet were classified by the RF analysis alongside all other solvate forming solvents. | ||
Removal from the training set of all records of crystallisation outcomes from these three solvents, followed by regrowth of the forest and subsequent prediction¶ of their respective experimental outcomes, classified each as being most likely to form a solvate compared with any other outcome. The predicted probabilities|| for solvate formation were 50%, 58% and 45% for NM, DMA and NMP respectively (see ESI‡).
With specific reference to solvate formation, the RF classification also provides a rank dependence of CBZ solvate formation on the solvent descriptors. The variable dependency plots indicate that CBZ solvate formation is favoured for solvents with AlogP98 < 0.5; dielectric constant ≥ 31.5; phi (a measure of molecular flexibility) ≤ 1.75; surface tension ≥ 32 mN m–1; accessible surface area < 150 Å2; molecular volume < 99 cm3 mol–1 (see ESI‡ for full details). The relevant properties of all three solvents shown in Fig. 3 fall within these ranges. Notably, a previous principal components analysis failed to identify any reliable correlations between solvent properties and outcomes for CBZ crystallisation.4
In essence, the RF analysis points to the solvate-forming potential of NM, DMA and NMP, yet multiple CBZ crystallisations from these three solvents in our previous study4 yielded no evidence of solvates. Accordingly, in this work, the NM, DMA and NMP crystallisations were revisited, this time employing the logical tactic of crystallising at a lower temperature (2–5 °C) than was used previously. Novel 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 CBZ solvates of NM, DMA and NMP were indeed produced at the new, lower temperature and identified initially by multi-sample foil-transmission X-ray powder diffraction7 (see ESI‡). All three solvated forms were observed to start desolvating upon removal from solution or storage at room temperature. Single crystals of the three solvates were subsequently grown by slow solvent evaporation at 2–5 °C** and the crystal structures determined†† using SHELXS-97.8
:1 CBZ solvates of NM, DMA and NMP were indeed produced at the new, lower temperature and identified initially by multi-sample foil-transmission X-ray powder diffraction7 (see ESI‡). All three solvated forms were observed to start desolvating upon removal from solution or storage at room temperature. Single crystals of the three solvates were subsequently grown by slow solvent evaporation at 2–5 °C** and the crystal structures determined†† using SHELXS-97.8
All three structures display packing motifs previously described for several CBZ solvates4,9 in which CBZ molecules form an R22 (8)10 dimer and the solvent molecules H-bond with the anti-orientated N–H of the carboxamide group (Fig. 4). The CBZ:NMP and CBZ:DMA crystal structures are isostructural and both contain disordered solvent molecules. It has been noted previously that the observed frequency of organic crystalline solvate formation for solvent molecules within the Cambridge Structural Database11 is related to their extent of use as crystallisation solvents.12 As such, it is not straightforward to identify the actual probabilities with which any given solvent will crystallise as an organic solvate, based on such an analysis, but it is noteworthy that the three solvents of interest here have relatively low frequencies of occurrence (NM = 0.56% of organic structures; DMA = 0.22%; NMP = not listed;12cf.water = ca. 5–18% and dimethylsulfoxide = ca. 2%, for example). Hence, by identifying the actual distribution of CBZ solvate formation within the solvent library used, RF analysis has made a valuable contribution by correctly identifying the solvate-forming potential of NM, DMA and NMP, leading to 3 new CBZ crystal structures.
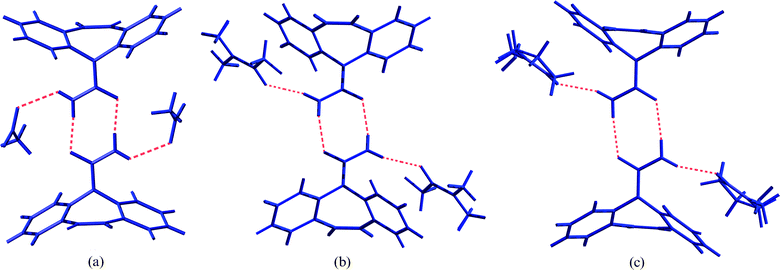 | ||
| Fig. 4 Illustration of the hydrogen bonded (dashed lines) contacts between CBZ R22 (8) dimers and solvent molecules in the crystal structures of CBZ solvates of (a) NM, (b) DMA and (c) NMP. | ||
Whilst the analysis does not provide specific directions as to how the crystallisation conditions should be varied in order to produce the solvates, the decision to recrystallise at a lower temperature was an obvious one that proved the veracity of the predictions. It might, of course, have been possible to crystallise the novel solvates, without recourse to RF analysis, by simply having implemented a more extensive crystallisation search in the first place. However, the larger the search, the larger the experimental/analytical overhead and the more time-consuming the exercise becomes, with no guarantee of finding novel solvates. Furthermore, solvate structures can be missed in a search, if the sample is labile and desolvates prior to analysis. Thus, where the emphasis is on maximising the number of physical forms discovered, retrospective RF analysis offers an efficient and effective strategy for assessing the completeness of the search. Crucially, the application of RF analysis to large quantities of data incorporating both numerical and categorical descriptors is straightforward to implement without the need for any preprocessing of input parameters.
This discovery of new solvated forms of CBZ by applying an alternative analysis method to results from an extensive prior experimental study, highlights the value of both storing such data in accessible electronic database formats and making it available for retrospective analysis by other groups. Such a repository, combined with effective data mining tools, provides new opportunities for researchers concerned with the identification of relationships between solute, solvent, crystallisation conditions and crystalline form.
Acknowledgements
The authors thank the Basic Technology programme of The Research Council's UK for funding this work under the project Control and Prediction of the Organic Solid State (http://www.cposs.org.uk) and gratefully acknowledge the input received from Dr Kenneth Shankland and Dr Norman Shankland.Notes and references
- A. Liaw and M. Wiener, R News, 2002, 2(3), 18–22 Search PubMed.
- D. S. Palmer, N. M. O'Boyle, R. C. Glen and J. B. O. Mitchell, J. Chem. Inf. Model., 2007, 47, 150–158 Search PubMed; Q.-Y. Zhang and J. Aires-de-Sousa, J. Chem. Inf. Model., 2007, 47, 1–8 Search PubMed; R. L. Lawrence, S. D. Wood and R. L. Sheley, Remote Sensing Environ., 2006, 100(3), 356–362 Search PubMed; F. Li, G. C. Runger and E. Tuv, Int. J. Pharm. Prod. Res., 2006, 44(14), 2853–2868 Search PubMed.
- A. J. Florence, A. Johnston, P. Fernandes, N. Shankland and K. Shankland, J. Appl. Crystallogr., 2006, 39, 922–924 CrossRef CAS.
- A. J. Florence, A. Johnston, S. L. Price, H. Nowell, A. R. Kennedy and N. Shankland, J. Pharm. Sci., 2006, 95(9), 1918–1930 CrossRef.
- R Development Core Team, R Foundation for Statistical Computing, Vienna, Austria, 2006, http://www.R-project.org.
- A. L. Grezsiak, M. Lang, K. Kim and A. J. Matzger, J. Pharm. Sci., 2003, 92, 2260 CrossRef CAS; M. Lang, J. W. Kampf and A. J. Matzger, J. Pharm. Sci., 2002, 91, 1186 CrossRef CAS; C. Rustichelli, G. Gamberini, V. Ferioli, M. C. Gamberini, R. Ficarra and S. Tommasini, J. Pharm. Biomed. Anal., 2000, 23, 41 CrossRef CAS.
- A. J. Florence, B. Baumgartner, C. Weston, N. Shankland, A. R. Kennedy, K. Shankland and W. I. F. David, J. Pharm. Sci., 2003, 92(9), 1930–1938 CrossRef CAS.
- G. M. Sheldrick, SHELX97 and SHELXL97, University of Gottingen, Germany, 1997 Search PubMed.
- S. G. Fleischman, S. S. Kuduva, J. A. McMahon, B. Moulton, R. D. B. Walsh, N. Rodriguez-Hornedo and M. J. Zaworotko, Cryst. Growth Des., 2003, 3, 909 CrossRef CAS.
- M. C. Etter, Acc. Chem. Res., 1990, 23, 120–126 CrossRef CAS.
- F. H. Allen, Acta Crystallogr., Sect. B, 2002, 58, 380–388 CrossRef.
- C. H. Gorbitz and H.-P. Hersleth, Acta Crystallogr., Sect. B, 2000, 56, 526–534 CrossRef.
Footnotes |
| † CCDC reference numbers 643264–643266. For crystallographic data in CIF or other electronic format see DOI: 10.1039/b713373a |
| ‡ Data set used in the Random Forest classification; resultant plot of RF classification and associated confusion matrix; probabilities of solvate formation; XRPD data of CBZ:NM, CBZ:DMA and CBZ:NMP solvates. See DOI: 10.1039/b713373a |
| § seed is arbitrarily set to give reproducibility in the random numbers required by the RF; the default value of mtry was used and based upon the number of input descriptors; ntree, the number of trees grown, was increased incrementally until no further improvement was observed in the model (see ESI‡). |
| ¶ Prediction is achieved by entering the individual descriptors for each of the three solvents, without specifying an outcome. The package is instructed to predict an outcome based on the RF classification model (see ESI‡), with the predictions encompassing no sample, novel solvates and CBZ forms I, II and III (pure form and mixtures). |
| || The probabilities reported correspond with the percentage votes for CBZ crystallising as each outcome from a given solvent. For each solvent the RF predict function yields a distribution of percentage votes for each defined outcome that totals 100%. |
| ** All chemicals were purchased from Sigma Aldrich and were used as supplied. |
| †† Crystal data† for: (a) CBZ:NM: C15H12N2O·CH3NO2, M = 297.31; needle crystallised from saturated NM solution, 0.3 × 0.08 × 0.05 mm; monoclininc, P21/n, a = 10.924(11) Å, b = 5.1617(5) Å, c = 26.309(3) Å, β = 100.104(2)°, V = 1460.5(3) Å3, Z = 4; T = 123 K; µ(Mo-Kα1) = 0.096 mm–1; λ = 0.71073 Å; max. sinθ/λ = 0.617 Å–1; 15958 reflections measured; 2888 unique reflections used in refinements with 215 refineable parameters and 0 restraints; final wR(F) = 0.0902 (all data), R(F) = 0.0516 (F2 > 2σF2). CCDC reference number 643266. (b) CBZ:DMA: C15H12N2O·C4H9NO, M 323.29; needle crystallised from saturated DMA solution, 0.25 × 0.20 × 0.18 mm; monoclininc, P21/c, a = 7.505(7) Å, b = 19.506(2) Å, c = 11.781(13) Å, β = 96.6(8)°, V = 1713.1(3) Å3, Z = 4; T = 123 K; µ(Mo-Kα1) = 0.083 mm–1; λ = 0.71073 Å; max. sinθ/λ = 0.595 Å–1; 9611 reflections measured; 2664 unique reflections used in refinements with 257 refineable parameters and 0 restraints; final wR(F) = 0.1435 (all data), R(F) = 0.0676 (F2 > 2σF2). CCDC reference number 643264. (c) CBZ:NMP: C15H12N2O·C5H9NO, M 335.40; needle crystallised from saturated NMP solution, 0.30 × 0.25 × 0.15 mm; monoclininc, P21/c, a = 7.545(4) Å, b = 19.512(10) Å, c = 11.878(6) Å, β = 98.013(3)°, V = 1731.5(15) Å3, Z = 4; T = 123 K; µ(Mo-Kα1) = 0.085 mm–1; λ = 0.71073 Å; max. sinθ/λ = 0.649 Å–1; 22709 reflections measured; 3952 unique reflections used in refinements with 261 refineable parameters and 0 restraints; final wR(F) = 0.1 (all data), R(F) = 0.0545 (F2 > 2σF2). CCDC reference number 643265. |
| This journal is © The Royal Society of Chemistry 2008 |