DOI:
10.1039/B712182B
(Paper)
CrystEngComm, 2008,
10, 117-124
Hydrothermal synthesis of divalent metal pyromellitate/dipyridylamine complexes: From an unprecedented supramolecular three-dimensional topology to an anionic coordination polymer framework with large incipient cation-bearing voids†‡
Received
8th August 2007
, Accepted 17th September 2007
First published on 24th September 2007
Abstract
Hydrothermal synthesis has afforded two divalent metal coordination complexes incorporating both fully deprotonated pyromellitate (pyro) anions and the kinked dipodal organodiimine 4,4′-dipyridylamine (dpa), {[Co(H2O)4(Hdpa)2][pyro]} (1) and {[H2dpa][Zn(pyro)]·2H2O} (2). Both new complexes were characterized by single crystal X-ray diffraction and spectral and thermogravimetric analyses. Complex 1 possesses discrete [Co(H2O)4(Hdpa)2]4+ cations linked into supramolecular 2-D (4,4) rhomboid grid layer motifs by hydrogen bonding between the unligated pyro anions and the aquo ligands. The pendant, monodentate Hdpa+ cations also engage in charge-separated hydrogen bonding to pyro anions in neighboring layers, resulting in a supramolecular 3-D structure with an unprecedented 6-connected 446108 topology. In contrast, 2 is a true coordination polymer, with divalent Zn atoms linked by ligated pyro anions into a binodal 4-connected PtS-type anionic network (4284 topology), which encapsulates H2dpa2+ cations and unligated water molecules within large incipient pores coursing through the structure. Irradiation of a solid sample of 2 with ultraviolet light resulted in blue-light emission, likely indicative of π–π* transitions within the orbital manifolds of both the pyro anions and unligated H2dpa2+ cations.
Introduction
The synthesis and characterization of metal–organic framework (MOF) materials continues to be a productive research area, sparked by these materials' significant potential in diverse industrially relevant applications such as hydrogen storage,1 molecular separations,2 ion exchange,3 catalysis,4 non-linear optics,5 and luminescence.6 Divalent metal 1,3,5-benzenetricarboxylate coordination polymers have proven to be an especially intriguing and well-explored MOF system, displaying properties including microporosity,7 reversible phase transitions,8 functional-group selective adsorption8,9 and photoluminescence.10 The significant structural diversity uncovered in benzenetricarboxylate coordination polymer systems is predicated on the wide variety of accessible carboxylate binding and bridging modes, acting synergistically with the coordination geometry preferences at the cationic metal nodes.7–10
Several researchers have extended this work into pyromellitate (pyro, 1,2,4,5-benzenetetracarboxylate) 1-D, 2-D and 3-D coordination polymers, taking advantage of this ligand's ability to undergo different levels of deprotonation and to connect from one to ten cationic metal nodes.11–18 For instance, [M2(pyro)] (M = Co, Fe, Mn) materials exhibit isomorphous 3-D structures, where each pyro subunit forms twelve M–O bonds to ten different M atoms. A variety of magnetic properties are observed, with antiferromagnetic (Fe, Mn) or ferromagnetic (Co) interactions evident at short-range, with consistent long-range antiferromagnetic interactions occurring throughout the series. The Co congener also manifests complex field-dependent metamagnetic behavior.11 The luminescent material [Zn(pyro)0.5] based on tetrahedrally coordinated zinc has a novel (4,8)-connected 2-D layered structure, where each pyro tetraanion binds to eight different Zn atoms.12 In systems containing both pyromellitate and various organoimines such as 4,4′-bipyridine (4,4′-bpy), different binding preferences can be promoted depending on the metal, reaction pH, and specific ligands used. While {[Co(H2pyro)(H2O)4]·4-aminopyridine·(H2O)2} possesses 1-D coordination polymer chains formed by the junction of Co2+ ions through bis-bridging bis-monodentate H2pyro dianions, {[Co2(4,4′-bpy)2(H2O)8]·(pyro)·(H2O)2} displays inclined polycatenated 1-D [Co(H2O)4(4,4′-bpy)]n ribbons held into a 3-D supramolecular arrangement by extensive hydrogen bonding patterns mediated by unligated pyro tetraanions.13 Shorter reaction times and higher temperatures resulted in the 2-D rhomboid grid coordination polymer {[H24,4′-bpy][Co(pyro)(4,4′-bpy)(H2O)2]}.14
Recently we have taken advantage of the kinked donor disposition and the hydrogen bonding capability of 4,4′-dipyridylamine (dpa) to prepare several dicarboxylate or pseudohalide coordination polymers with unique structural topologies, in most cases substantially divergent from their 4,4′-bipyridine analogues. For example, {[Ni(dpa)2(succinate)0.5]Cl} displayed a rare self-penetrated structure with the first regular 5-connected 610 network topology.19 [Ni(adipate)(dpa)(H2O)] represented the first example of a triply-interpenetrated PtS-type network (pts, 4284 topology).20 In these and other materials,21–24 the kinked orientation of the nitrogen donor atoms of the dpa and the hydrogen bonding imparted by the central N–H group act in tandem as structure-directing agents during coordination polymer self-assembly. Herein we report our first efforts to obtain supramolecular solids or coordination polymers containing both the kinked dipodal organodiimine dpa and pyromellitate ions.
Results and discussion
Synthesis and spectral characterization
Compounds 1 and 2 were prepared cleanly as single phase crystalline products under hydrothermal conditions via combination of the appropriate metal salt, 4,4′-dipyridylamine, and pyromellitic acid. The infrared spectra of 1 and 2 were consistent with their single crystal structures. Sharp, medium intensity bands in the range of ∼1600 cm–1 to ∼1200 cm–1 correspond to stretching modes of the aromatic rings of the dpa and pyro moieties.25 Features corresponding to the puckering of the pyridyl and phenyl rings are evident in the region between 820 cm–1 and 600 cm–1. Asymmetric and symmetric C–O stretching modes of the fully deprotonated pyro4– anions were evidenced by strong, slightly broadened bands at ∼1590 cm–1 and ∼1350 cm–1, respectively. The absence of any bands in the region of 1700 cm–1 is diagnostic of full deprotonation of the original pyromellitic acid. Broad bands in the region of ∼3400 cm–1 to ∼3200 cm–1 in all cases represent N–H stretching modes within the dpa ligands and O–H stretching modes within the water molecules of crystallization in 1 and 2, and the aquo ligands in 1. The broadness of these higher energy spectral features is caused by the hydrogen bonding mechanisms prevalent in both 1 and 2 (see below).
Structural description of {[Co(H2O)4(Hdpa)2][pyro]} (1)
Compound 1 crystallized in the monoclinic P21/n space group with an asymmetric unit consisting of one cobalt atom sited on a crystallographic inversion center, one Hdpa+ ligand bound at its unprotonated pyridyl nitrogen atom, two ligated water molecules, and half of an unligated pyromellitate tetraanion. Operation of the inversion center reveals the presence of an individual [Co(H2O)4(Hdpa)2]4+ cation, exhibiting a [CoN2O4] octahedral coordination sphere with two trans-disposed nitrogen donor atoms (belonging to two different Hdpa+ cations) and four bound water molecules. A complex with similar monodentate, singly protonated Hdpa+ ligands, [Cu(SO4)2(H2O)2(Hdpa)2], was previously reported by Hanton et al.26 Bond lengths and angles within 1 are standard for octahedral cobalt complexes (Table 1). Charge balance within 1 is provided by one fully deprotonated pyromellitate unit. A neutral cation/anion pair, defining a salt with a formula of {[Co(H2O)4(Hdpa)2][pyro]}, is shown with thermal ellipsoids in Fig. 1.
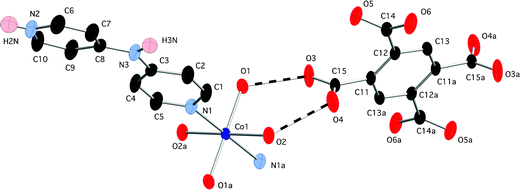 |
| | Fig. 1 Expanded asymmetric unit in 1, with thermal ellipsoids at 50% probability and atom numbering scheme. Most hydrogen atoms have been omitted for clarity. Hydrogen bonding interactions are shown as dashed lines. Atoms denoted with the label a are generated through crystallographic inversion centers. | |
Table 1 Selected bond distance (Å) and angle (°) data for 1
|
Symmetry transformation to generate equivalent atoms: #1 –x + 1, – y, z + 1/2.
|
| Co1–O1 (×2) |
1.986(2) |
O4–C15 (×2) |
1.226(3) |
| Co1–O2 (×2) |
2.1649(17) |
O5–C16 (×2) |
1.254(3) |
| Co1–N1 (×2) |
2.203(2) |
O6–C16 (×2) |
1.241(3) |
| O3–C15 (×2) |
1.253(3) |
|
|
| O1–Co1–O1#1 |
180.0 |
O1#1–Co1–N1 |
93.23(9) |
| O1–Co1–O2#1 |
92.03(8) |
O2#1–Co1–N1 |
89.07(8) |
| O1–Co1–O2 |
87.97(8) |
O2–Co1–N1 |
90.93(8) |
| O2#1–Co1–O2 |
180.0 |
N1#1–Co1–N1 |
180.0 |
| O1–Co1–N1 |
86.77(9) |
|
|
Examination of the supramolecular hydrogen bonding patterns reveals the connection of one type of aquo ligand (denoted by O1) to two different pyro units to form a R24(8) type27 circuit (O3⋯H1A–O1–H1B⋯O3⋯H1A–O1–H1B⋯). Additionally, the other type of aquo ligand (denoted by O2) engages in hydrogen bonding to two different pyro units, this time forming a short discrete chain pattern (O5⋯H2B–O2–H2A⋯O4) with graph set notation D22(5). Thus, each pyro tetraanion is conjoined to four cobalt atoms through hydrogen bonding interactions provided by the bound water molecules (Fig. 2). In order to optimize the supramolecular interactions, the carboxylate groups twist by ∼43° and ∼71° relative to the plane of the pyromellitate aromatic ring.
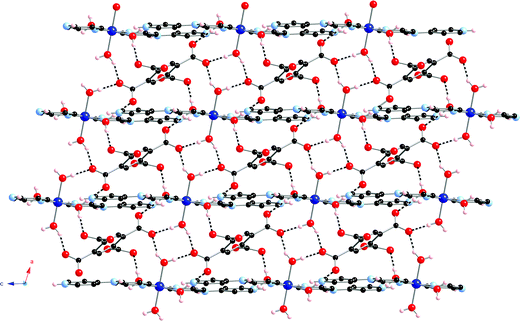 |
| | Fig. 2 A supramolecular (4,4) rhomboid grid layer motif within 1. Hydrogen bonding is shown as dashed lines. | |
Considering both cobalt atoms and the pyromellitate ring centroids as 4-connected nodes, a 2-D (4,4) rhomboid grid can be invoked, which is situated coincident with the ac crystal plane. The Co–centroid distances within the supramolecular grid-like layer measure 5.440 and 7.192 Å. The Co–Co and centroid–centroid through-space distances are 10.768 and 7.268 Å, respectively. The angles within the rhomboid cavities subtend ∼69.1 and ∼110.9°. The aforementioned rhomboid grids are connected into a supramolecular three-dimensional (3-D) network through additional hydrogen bonding interactions. The distal protonated imine units (N2–H2N) of Hdpa+ cations project above and below the (4,4) grid motifs, and engage in charge-separated hydrogen bonding28 with oxygen atoms of type O6 within pyromellitate groups in two neighboring layers. As a result, a 3-D 6-connected framework can be invoked, where each Co atom is linked to four pyro anions within the ac plane and to two pyro anions along the +b and –b crystal directions. Evaluation of this 3-D network with TOPOS software29 uncovered an unprecedented 446108 6-connected topology. This network is shown in Fig. 3 with four-, six-, and eight-membered circuits highlighted in different colors. This structural morphology varies from the commonly encountered 41263 primitive cubic (pcu) 6-connected topology because of the offset layer stacking pattern enabled by the kinked nature of the pendant Hdpa+ cations. Further stabilization of the supramolecular network is provided by hydrogen bonding donation from the central dpa amine subunit to a proximate pyromellitate carboxylate group (at O5). Metrical parameters for the extensive hydrogen bonding patterns in 1 are given in Table 2.
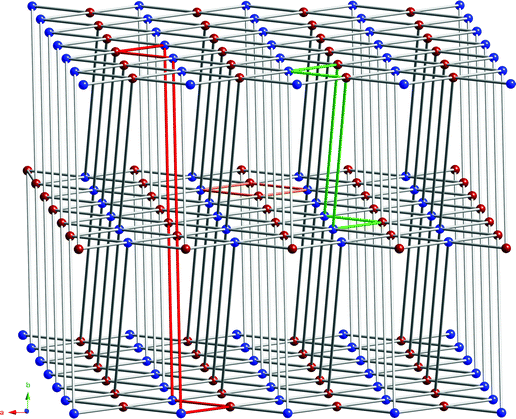 |
| | Fig. 3 A view of the unprecedented 446108 6-connected 3-D supramolecular topology in 1. Cobalt atoms and pyromellitate ring centroids are depicted in blue and maroon, respectively. Four-, six-, and eight-membered circuits are shown in orange, green, and red, respectively. | |
Table 2
Hydrogen bonding distance (Å) and angle (°) data for 1 and 2
| D–H⋯A |
d(H⋯A)/Å |
<DHA/° |
d(D⋯A)/Å |
Symmetry transformation for A |
|
1
|
| O1–H1A⋯O3 |
1.703(17) |
175(3) |
2.550(2) |
|
| O1–H1B⋯O3 |
1.808(19) |
158(3) |
2.597(2) |
–x + 2, – y, –z + 1 |
| O2–H2A⋯O4 |
1.917(16) |
176(2) |
2.758(2) |
|
| O2–H2B⋯O5 |
2.030(16) |
173(2) |
2.862(2) |
x – 1, y, z |
| N2–H2N⋯O6 |
1.786(17) |
171(2) |
2.668(2) |
x – 1/2, – y + 1/2, z + 3/2 |
| N3–H3N⋯O5 |
1.957(16) |
172(2) |
2.824(2) |
x – 1/2, – y + 1/2, z + 1/2 |
|
2
|
| O1W–H1WA⋯O6 |
1.945(16) |
176(3) |
2.750(2) |
– x + 5/2, y –1/2, –z + 1/2 |
| O1W–H1WB⋯O2 |
2.172(17) |
168(2) |
2.987(2) |
|
| O2W–H2WA⋯O1 |
1.983(17) |
174(3) |
2.817(2) |
|
| O2W–H2WB⋯O6 |
2.476(19) |
156(3) |
3.238(2) |
– x + 5/2, y –1/2, –z + 3/2 |
| N1–H1N⋯O8 |
1.816(16) |
174(2) |
2.690(2) |
– x + 5/2, y –1/2, –z + 1/2 |
| N2–H2N⋯O3 |
1.779(17) |
172(3) |
2.648(2) |
x + 3/2, – y –1/2, z + 3/2 |
| N3–H3N⋯O1W |
2.108(18) |
153(2) |
2.900(2) |
x – 1/2, – y –1/2, z + 1/2 |
Structural description of {[H2dpa][Zn(pyro)]·2H2O} (2)
Compound 2 crystallized in the monoclinic P21/n space group with an asymmetric unit containing one zinc atom, one fully deprotonated pyromellitate ligand, one doubly protonated H2dpa2+ cation and two water molecules of crystallization (Fig. 4). The coordination environment is a simple [ZnO4] distorted tetrahedron, where all four oxygen donor atoms belong to different pyromellitate ligands. Bond lengths and angles within 2 are standard for tetrahedral divalent zinc coordination complexes. Selected geometrical information is given in Table 3.
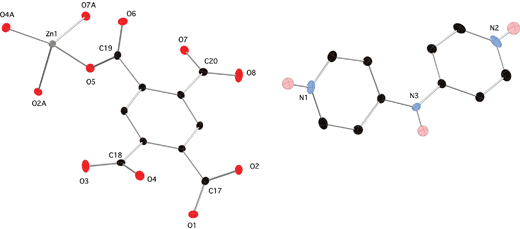 |
| | Fig. 4 Asymmetric unit of 2 with thermal ellipsoids at 50% probability and partial atom numbering scheme. Most hydrogen atoms have been omitted for clarity. The water molecules of crystallization are not shown. Atoms denoted with the label A are generated through crystallographic inversion centers. | |
Table 3 Selected bond distance (Å) and angle (°) data for 2
|
Symmetry transformation to generate equivalent atoms: #1 x – 1/2, –y +1/2, z + 1/2; #2 –x + 5/2, y –1/2, –z + 3/2; #3 x + 3/2, –y – 1/2, z + 1/2.
|
| Zn1–O4 |
1.9510(13) |
O3–C18 |
1.243(2) |
|
Zn1–O7#1 |
1.9675(13) |
O4–C18 |
1.270(2) |
|
Zn1–O5#2 |
1.9683(13) |
O5–C19 |
1.276(2) |
|
Zn1–O2#3 |
1.9816(13) |
O6–C19 |
1.235(2) |
| O1–C17 |
1.232(2) |
O7–C20 |
1.270(2) |
| O2–C17 |
1.289(2) |
O8–C20 |
1.239(2) |
| O4–Zn1–O7#1 |
131.01(6) |
O4–Zn1–O2#3 |
106.08(6) |
| O4–Zn1–O5#2 |
112.07(6) |
O7#1–Zn1–O2#3 |
104.51(6) |
| O7#1–Zn1–O5#2 |
101.64(5) |
O5#2–Zn1–O2#3 |
95.98(6) |
Expansion of this structure through the exotetradentate pyromellitate anions with peripheral monodentate carboxylate groups generates an anionic [Zn(pyro)]n2n– 3-D coordination polymer network, with unligated water molecules and H2dpa2+ cations occupying diamond-shaped channels coursing through the structure (Fig. 5). According to a calculation performed with PLATON,30 these incipient voids occupy 52.9% of the unit cell volume of 2. Due to its coordination environment, each Zn atom acts as a 4-connected “tetrahedral” node. In turn the exotetradentate pyromellitate anion can be considered a 4-connected “square planar” connecting node. Thus, the 3-D topology of the anionic framework constructs a binodal 4284 cooperite-type (pts) network (Fig. 6), as verified with the TOPOS software package. A similar [Zn(pyro)]n2n– framework was observed in {K2[Zn(pyro)]·8H2O},15 which crystallized in a C-centered monoclinic space group and was reported to adopt a tenorite (CuO) structure type. However, a topological analysis with TOPOS indicates a 4284pts lattice for {K2[Zn(pyro)]·8H2O}, identical to that of 2. Nevertheless the structure of CuO appears to be simply a more symmetrical version of that of PtS. It is most likely that the different supramolecular environment in 2 subtly alters the binodal tetrahedral/square planar connectivity between the zinc atoms and the pyromellitate rings, resulting in a lowering of symmetry to a primitive monoclinic space group. The smaller number of unligated water molecules in 2 is reflective of the larger girth of the H2dpa2+ iminium cations as opposed to the alkali metal cations. An attempt to promote ion exchange and convert 2 into {K2[Zn(pyro)]·8H2O} by immersion in a saturated KCl solution resulted in complete dissolution of 2, indicating likely destruction of the anionic framework.
![View of the anionic [Zn(pyro)]n2n– 3-D network in 2 with H2dpa2+ cations and water molecules of crystallization within the large incipient voids.](/image/article/2008/CE/b712182b/b712182b-f5.gif) |
| | Fig. 5 View of the anionic [Zn(pyro)]n2n– 3-D network in 2 with H2dpa2+ cations and water molecules of crystallization within the large incipient voids. | |
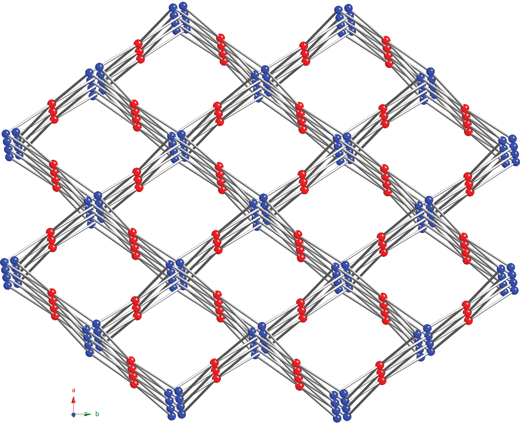 |
| | Fig. 6 A simplified perspective of the pts lattice in 2. Zn atoms and pyromellitate ring centroids are shown in blue and red, respectively. | |
Hydrogen bonding between water molecules of crystallization and unligated oxygen atoms of the pyromellitate carboxylate groups serve to secure the solvent molecules in their respective positions. Each H2dpa2+ cation is also held in the incipient 1-D cavities within the structure of 2 by hydrogen bonding at one terminal imine and the central amine to unligated oxygen atoms of pyromellitate carboxylate groups and a water of crystallization. The other terminal protonated imine of the H2dpa2+ donates a hydrogen bond to a “free” water molecule (O1W). Geometrical parameters for the hydrogen bonding interactions in 2 are given above in Table 2.
Compounds 1 and 2 were subjected to thermogravimetric analysis under flowing N2 to probe their dehydration and decomposition behavior. TGA traces for these compounds are shown in Fig. S1 and S2.‡ The decomposition of compound 1 began at ∼150 °C, with likely elimination of all aquo ligands (8.8% calculated, 9.5% observed). Expulsion of all organic components was complete by ∼800 °C, with a mass remnant of ∼10% (11% expected) consistent with the deposition of CoO. Dehydration of compound 2 was completed by ∼210 °C, showing a mass loss of 7.0%, consistent with the loss of two equivalents of water (6.9% calculated). Decomposition was completed by ∼880 °C, resulting in a mass remnant of 13%, corresponding roughly to the deposition of ZnO (15.5% expected).
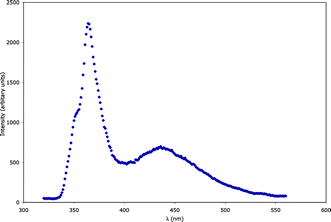
Luminescent behavior of 2
Irradiation of complex 2 with ultraviolet light (λ = 300 nm) in the solid state resulted in blue visible light emission (Fig. 7). By comparison with other Zn- and Cd-based coordination polymers,31 this property is likely a result of ligand-centered π–π* electronic transitions within the molecular orbital manifolds of the aromatic ring systems of the pyromellitate ligands or the enclathrated H2dpa2+ cations. The broad lower energy emission band (λmax ∼ 440 nm) can likely be ascribed to π–π* transitions within the H2dpa2+ cations, as crystals of pure dpa undergo weak luminescence in a similar spectral region (λmax ∼ 420 nm).20 The breadth of this emission feature is attributed to intensity-diminishing facile vibrational relaxation modes within the unligated imine cations. The higher energy, sharper, and more intense spectral feature in the emission spectrum of 2 presumably arises from π–π* transitions within the pyromellitate ligands. As these are rigidly held within the 3-D coordination polymer matrix, radiative energy loss due to molecular vibration is minimized.
Conclusions
Hydrothermal combination of pyromellitic acid, 4,4′-dipyridylamine and cobalt chloride afforded a salt wherein [Co(H2O)4(Hdpa)2]4+ cations link with unligated pyromellitate anions into (4,4) grid-like layers and subsequently a 3-D supramolecular arrangement with a novel 6-connected network, continuing the trend of new topologies predicated on the kinked geometry and hydrogen bonding capability of dpa. Protonation of the distal dpa termini, resulting in uncommonly encountered monoprotonated pendant Hdpa+ ligands, prevents the formation of [Co(H2O)4L]-type 1-D coordination polymer chains, instead promoting the new 446108 3-D supramolecular morphology. Utilizing zinc nitrate in the synthesis produced a luminescent crystalline solid containing a 3-D [Zn(pyro)]n2– coordination polymer network which adopted the pts lattice framework. Within this network lie large incipient voids occupied by water molecules and charge-balancing H2dpa2+ dications. Even though these cations are unligated, they play an important structure-stabilizing role due to hydrogen bonding patterns, as attempts to displace them resulted in degradation of the anionic coordination polymer framework. Efforts to investigate dpa as an ancillary ligand towards the synthesis of new dicarboxylate coordination polymers continue in our research group, and will be reported in due course.
Experimental
General considerations
Metal salts were obtained from Fisher, and pyromellitic acid was purchased from Aldrich. 4,4′-Dipyridylamine (dpa) was prepared via a published procedure.32Water was deionized above 3 MΩ in-house. Thermogravimetric analysis was performed on a TA Instruments TGA 2050 Thermogravimetric Analyzer with a heating rate of 10 °C min–1 up to 900 °C. Elemental analysis was carried out using a Perkin Elmer 2400 Series II CHNS/O Analyzer. IR spectra were recorded on powdered samples using a Perkin Elmer Spectrum One instrument. Luminescence spectra were obtained with a Hitachi F-4500 Fluorescence Spectrometer on solid crystalline samples anchored to quartz microscope slides with Rexon Corporation RX-22P ultraviolet-transparent epoxy adhesive.
Preparation of {[Co(H2O)4(Hdpa)2][pyro]} (1)
CoCl2·6H2O (88 mg, 0.37 mmol), dpa (127 mg, 0.74 mmol) and pyromellitic acid (94 mg, 0.37 mmol), were placed into 10 mL distilled H2O in a 23 mL Teflon-lined Parr acid digestion bomb. The bomb was sealed and heated at 120 °C for 48 h, whereupon it was cooled slowly to 25 °C. Large pink blocks of 1 (67 mg, 25% yield based on Co) were isolated after washing with distilled water, ethanol, and acetone and drying in air. Anal. calc. for C30H30CoN6O121: C, 49.7; H, 4.2; N, 11.6%. Found: C, 46.9; H, 3.9; N, 11.0%. Values for carbon were consistently low, likely due to incomplete combustion. IR (cm–1): 3139 m, 3040 m, 2918 m, 2849 m, 2060 w, 1972 w, 1610 s, 1560 s, 1514 s, 1488 s, 1411 s, 1377 s, 1324 s, 1240 m, 1209 s, 1141 m, 1080 m, 1049 m, 1011 m, 878 m, 836 s, 809 s, 676 m, 657 m, 638 m, 577 m, 546 s, 516 m, 439 m.
Preparation of {[H2dpa][Zn(pyro)]·2H2O} (2)
An identical preparative procedure to that for 1 was followed, except for the use of Zn(NO3)2 (110 mg, 0.37 mmol) as the metal source. Large colorless blocks of 2 (156 mg, 80% yield based on Zn) were obtained. Anal. calc. for C20H17N3O10Zn 2: C, 45.8; H, 3.3; N, 8.0%. Found: C, 46.1; H, 3.0; N, 10.1%. IR (cm–1): 3532 w, 3239 m, 3109 m, 3044 m, 2880 m, 1625 s, 1511 s, 1408 s, 1355 s, 1324 s, 1210 s, 1137 m, 1057 w, 1027 w, 981 w, 954 w, 928 w, 901 w, 863 m, 828 s, 813 s, 775 w, 672 w, 607 m, 573 m, 546 w, 515 w, 478 w.
A pink cube of 1 (with dimensions 0.40 mm × 0.40 mm × 0.40 mm) and a colorless block of 2 (0.32 mm × 0.18 mm × 0.14 mm) were subjected to single crystal X-ray diffraction using a Bruker-AXS SMART 1k CCD instrument. Reflection data were acquired using graphite-monochromated Mo-Kα radiation (λ = 0.71073 Å). The data were integrated viaSAINT.33 Lorentz and polarization effects and empirical absorption corrections were applied with SADABS.34 The structures were solved using direct methods and refined on F2 using SHELXTL.35 All non-hydrogen atoms were refined anisotropically. Hydrogen atoms bound to carbon atoms were placed in calculated positions and refined isotropically with a riding model. The hydrogen atoms bound to the nitrogen atoms of the dpa moieties and any water molecules were found via Fourier difference maps, then restrained at fixed positions and refined isotropically. While a higher symmetry orthorhombic cell could be found for 2, acceptable crystallographic solution was only possible in the monoclinic space groupP21/n. Relevant crystallographic data for 1 and 2 are listed in Table 4.
Table 4 Crystal and structure refinement data for 1 and 2
| Data |
1
|
2
|
| Empirical formula |
C30H30CoN6O12 |
C20H17N3O10Zn |
| Formula weight |
725.53 |
524.74 |
| Collection T/K |
293(2) |
173(2) |
| Crystal system |
Monoclinic |
Monoclinic |
| Space group |
P21/n |
P21/n |
|
a/Å |
7.268 (3) |
11.5836 (13) |
|
b/Å |
20.930 (8) |
14.7422 (17) |
|
c/Å |
10.365 (4) |
11.6194 (13) |
|
α/° |
90 |
90 |
|
β/° |
108.032 (5) |
102.319 (2) |
|
γ/° |
90 |
90 |
|
V/Å3 |
1499.2 (10) |
1938.5 (4) |
|
Z
|
2 |
4 |
|
D
calc/g cm–3 |
1.607 |
1.798 |
|
µ/mm–1 |
0.651 |
1.338 |
| Min./max. T |
0.898 |
0.883 |
|
hkl ranges |
–9 ≤ h ≤ 9, –27 ≤ k ≤ 27, –13 ≤ l ≤ 13 |
–15 ≤ h ≤ 15, –19 ≤ k ≤ 19, –15 ≤ l ≤ 15 |
| Total reflections |
16275 |
23298 |
| Unique reflections |
3423 |
4730 |
|
R(int)
|
0.0267 |
0.0357 |
| Parameters/restraints |
241/8 |
328/9 |
|
R
1 (all data) |
0.0405 |
0.0403 |
|
R
1 (I > 2σ(I)) |
0.0347 |
0.0320 |
|
wR
2 (all data) |
0.0863 |
0.0778 |
|
wR
2 (I > 2σ(I)) |
0.0832 |
0.0746 |
| Max, min residual/e Å3 |
0.472, –0.380 |
0.342, –0.615 |
|
G.O.F. |
1.031 |
1.099 |
Acknowledgements
Funding for this work was provided by Michigan State University. M. A. B. is grateful for the MSU Quality Fund Undergraduate Research Program for financial support. We thank Dr Rui Huang for performing the elemental analyses and Dr Kathryn Severin for use of the fluorimeter. We thank Dr Ronald Supowski of King’s College (Wilkes-Barre, PA) for performing the thermogravimetric analyses.
References
-
(a) H. Li, M. Eddaoudi, M. O'Keeffe and O. M. Yaghi, Nature, 1999, 402, 276–279 CrossRef CAS
 ;
(b) R. Matsuda, R. Kitaura, S. Kitagawa, Y. Kubota, R. U. Belosludov, T. C. Kobayashi, H. Sakamoto, T. Chiba, M. Takata, Y. Kawazoe and Y. Mita, Nature, 2005, 436, 238–241 CrossRef CAS ;
(c) L. Pan, D. Holson, L. R. Ciemnolonski, R. Heddy and J. Li, Angew. Chem., Int. Ed., 2006, 45, 616–619 CrossRef CAS ;
(d) N. L. Rosi, J. Eckert, M. Eddaoudi, D. J. Vodak, J. Kim, M. O'Keeffe and O. M. Yaghi, Science, 2003, 300, 1127–1129 CrossRef CAS ;
(e) M. Dinca, A. F. Yu and J. R. Long, J. Am. Chem. Soc., 2006, 128, 8904–8913 CrossRef CAS ;
(f) G. Ferey, M. Latroche, C. Serre, F. Millange, T. Loiseau and A. Percheron-Guegan, Chem. Commun., 2003, 2976–2977 RSC ;
(g) X. Zhao, B. Xiao, A. J. Fletcher, K. M. Thomas, D. Bradshaw and M. J. Rosseinsky, Science, 2004, 306, 1012–1015 CrossRef CAS .
;
(b) R. Matsuda, R. Kitaura, S. Kitagawa, Y. Kubota, R. U. Belosludov, T. C. Kobayashi, H. Sakamoto, T. Chiba, M. Takata, Y. Kawazoe and Y. Mita, Nature, 2005, 436, 238–241 CrossRef CAS ;
(c) L. Pan, D. Holson, L. R. Ciemnolonski, R. Heddy and J. Li, Angew. Chem., Int. Ed., 2006, 45, 616–619 CrossRef CAS ;
(d) N. L. Rosi, J. Eckert, M. Eddaoudi, D. J. Vodak, J. Kim, M. O'Keeffe and O. M. Yaghi, Science, 2003, 300, 1127–1129 CrossRef CAS ;
(e) M. Dinca, A. F. Yu and J. R. Long, J. Am. Chem. Soc., 2006, 128, 8904–8913 CrossRef CAS ;
(f) G. Ferey, M. Latroche, C. Serre, F. Millange, T. Loiseau and A. Percheron-Guegan, Chem. Commun., 2003, 2976–2977 RSC ;
(g) X. Zhao, B. Xiao, A. J. Fletcher, K. M. Thomas, D. Bradshaw and M. J. Rosseinsky, Science, 2004, 306, 1012–1015 CrossRef CAS .
-
(a) J. S. Seo, D. Whang, H. Lee, S. I. Jun, J. Oh, Y. J. Jeon and K. Kim, Nature, 2000, 404, 982–986 CrossRef CAS ;
(b) B. Chen, C. Liang, J. Yang, D. S. Contreras, Y. L. Clancy, E. B. Lobkovsky, O. M. Yaghi and S. Dai, Angew. Chem., Int. Ed., 2006, 45, 1390–1393 CrossRef CAS .
-
(a) Q.-R. Fang, G.-S. Zhu, M. Xue, J.-Y. Sun and S.-L. Qiu, Dalton Trans., 2006, 2399–2402 RSC ;
(b) X.-M. Zhang, M.-L. Tong, H. K. Lee and X.-M. Chen, J. Solid State Chem., 2001, 160, 118–122 CrossRef CAS ;
(c) O. M. Yaghi, H. Li and T. L. Groy, Inorg. Chem., 1997, 36, 4292–4293 CrossRef CAS .
-
(a) N. Guillou, P. M. Forster, Q. Gao, J. S. Chang, M. Nogues, S.-E. Park, A. K. Cheetham and G. Ferey, Angew. Chem., Int. Ed., 2001, 40, 2831–2834 CrossRef CAS ;
(b) C.-D. Wu, A. Hu, L. Zhang and W. Lin, J. Am. Chem. Soc., 2005, 127, 8940–8941 CrossRef CAS ;
(c) H. Han, S. Zhang, H. Hou, Y. Fan and Y. Zhu, Eur. J. Inorg. Chem., 2006, 1594–1600 CrossRef CAS ;
(d) W. Mori, S. Takamizawa, C. N. Kato, T. Ohmura and T. Sato, Microporous Mesoporous Mater., 2004, 73, 15–30 CrossRef CAS .
-
(a) S. Zang, Y. Su, Y. Li, Z. Ni and Q. Meng, Inorg. Chem., 2006, 45, 174–180 CrossRef CAS ;
(b) L. Wang, M. Yang, G. Li, Z. Shi and S. Feng, Inorg. Chem., 2006, 45, 2474–2478 CrossRef CAS ;
(c) S. Wang, Y. Hou, E. Wang, Y. Li, L. Xu, J. Peng, S. Liu and C. Hu, New J. Chem., 2003, 27, 1144–1147 RSC .
-
(a) L. G. Beauvais, M. P. Shores and J. R. Long, J. Am. Chem. Soc., 2000, 122, 2763–2772 CrossRef CAS ;
(b) H. Jianghua, Y. Jihong, Z. Yuetao, P. Qinhe and X. Ruren, Inorg. Chem., 2005, 44, 9279–9282 CrossRef CAS .
- M. Eddaoudi, H. Li and O. M. Yaghi, J. Am. Chem. Soc., 2000, 122, 1391–1397 CrossRef CAS .
- O. M. Yaghi, H. Li and T. L. Groy, J. Am. Chem. Soc., 1996, 118, 9096–9101 CrossRef CAS .
- O. M. Yaghi, C. E. Davis, G. Li and H. Li, J. Am. Chem. Soc., 1997, 119, 2861–2868 CrossRef CAS .
- Q. Fang, G. Zhu, M. Xue, J. Sun, F. Sun and S. Qiu, Inorg. Chem., 2006, 45, 3582–3587 CrossRef CAS .
- H. Kumagai, K. W. Chapman, C. J. Kepert and M. Kurmoo, Polyhedron, 2003, 22, 1921–1927 CrossRef CAS .
- Y.-H. Wen, Q.-W. Zhang, Y.-H. He and Y.-L. Feng, Inorg. Chem. Commun., 2007, 10, 543–546 CrossRef CAS .
- A. Majumber, V. Gramlich, G. M. Rosair, S. R. Batten, J. D. Masuda, M. S. El Fallah, J. Ribas, J.-P. Sutter, C. Desplanches and S. Mitra, Cryst. Growth Des., 2006, 6, 2355–2368 CrossRef CAS .
- C. Ruiz-Perez, P. Lorenzo-Luis, M. Hernandez-Molina, M. M. Laz, F. S. Delgado, P. Gili and M. Julve, Eur. J. Inorg. Chem., 2004, 3873–3879 CrossRef CAS .
- K.-L. Lu, Y.-F. Chen, Y.-H. Liu, Y.-W. Cheng, R.-T. Liao and Y.-S. Wen, Cryst. Growth Des., 2005, 5, 403–405 CrossRef CAS .
- J. Yang, J.-F. Ma, Y.-Y. Liu, S.-L. Li and G.-L. Zheng, Eur. J. Inorg. Chem., 2005, 2174–2180 CrossRef CAS .
- S. V. Ganesan, P. Lightfoot and S. Natarajan, Solid State Sci., 2004, 6, 757–762 CrossRef CAS .
- H. Kumagai, C. J. Kepert and M. Kurmoo, Inorg. Chem., 2002, 41, 3410–3422 CrossRef CAS .
- M. R. Montney, S. Mallika Krishnan, N. M. Patel, R. M. Supkowksi and R. L. LaDuca, Cryst. Growth Des., 2007, 7, 1145–1153 CrossRef CAS .
- M. R. Montney, S. Mallika Krishnan, R. M. Supkowski and R. L. LaDuca, Inorg. Chem., 2007, 46, 7362–7370 CrossRef CAS .
- M. A. Braverman, R. M. Supkowski and R. L. LaDuca, J. Solid State Chem., 2007, 180, 1852–1862 CrossRef CAS .
- M. A. Braverman, R. M. Supkowski and R. L. LaDuca, Inorg. Chim. Acta, 2007, 360, 2353–2362 CrossRef CAS .
- D. P. Martin, R. M. Supkowski and R. L. LaDuca, Inorg. Chem., 2007, 46, 7917–7922 CrossRef CAS .
- L. L. Johnston, A. J. Ursini, N. P. Oien, R. M. Supkowski and R. L. LaDuca, Inorg. Chim. Acta, 2007, 360, 3619–3625 CrossRef CAS .
- M. Kurmoo, C. Estournes, Y. Oka, H. Kumagai and K. Inoue, Inorg. Chem., 2005, 44, 217–224 CrossRef CAS .
- D. B. Cordes, L. R. Hanton and M. D. Spicer, J. Mol. Struct., 2006, 796, 146–159 CrossRef CAS .
- M. C. Etter, J. C. MacDonald and J. Bernstein, Acta Crystallogr., Sect. B, 1990, 46, 256–262 CrossRef .
- T. Steiner, Angew. Chem., Int. Ed., 2002, 41, 48–76 CrossRef CAS .
- V. A. Blatov, A. P. Shevchenko and V. N. Serezhkin, J. Appl. Crystallogr., 2000, 33, 1193 CrossRef CAS .
-
A. L. Spek, PLATON, A Multipurpose Crystallographic Tool, Utrecht University, Utrecht, The Netherlands, 1998 Search PubMed .
-
(a) J. Tao, J. X. Shi, M. L. Tong, X. X. Zhang and X. M. Chen, Inorg. Chem., 2001, 40, 6328–6330 CrossRef CAS ;
(b) J. Tao, M. L. Tong, J. X. Shi, X. M. Chen and S. W. Ng, Chem. Commun., 2000, 2043–2044 RSC ;
(c) J. C. Dai, X. T. Wu, Z. Y. Fu, C. P. Cui, S. M. Hu, W. X. Du, L. M. Wu, H. H. Zhang and R. Q. Sun, Inorg. Chem., 2002, 41, 1391–1396 CrossRef CAS ;
(d) W. Chen, J. Y. Wang, C. Chen, Q. Yue, H. M. Yuan, J. S. Chen and S. N. Wang, Inorg. Chem., 2003, 42, 944–946 CrossRef CAS ;
(e) N. Hao, E. Shen, Y. B. Li, E. B. Wang, C. W. Hu and L. Xu, Eur. J. Inorg. Chem., 2004, 4102–4107 CrossRef CAS .
- P. J. Zapf, R. L. LaDuca, R. S. Rarig, K. M. Johnson and J. Zubieta, Inorg. Chem., 1998, 37, 3411–3414 CrossRef CAS .
-
SAINT, Software for Data Extraction and Reduction, Version 6.02, Bruker AXS, Inc., Madison, WI, 2002 Search PubMed .
-
SADABS, Software for Empirical Absorption Correction, Version 2.03, Bruker AXS, Inc., Madison, WI, 2002 Search PubMed .
-
G. M. Sheldrick, SHELXTL, Program for Crystal Structure Refinement, University of Göttingen, Göttingen, Germany, 1997 Search PubMed .
|
| This journal is © The Royal Society of Chemistry 2008 |
Click here to see how this site uses Cookies. View our privacy policy here.