Five-fold interpenetrated strongly hydrogen bonded rhomboid grid layers constructed from the aggregation of neutral coordination complexes with long pendant aromatic dicarboxylate and organodiimine ligands†‡
Matthew R.
Montney
a,
Ronald M.
Supkowski
b and
Robert L.
LaDuca
*a
aLyman Briggs College and Department of Chemistry, E-30 Holmes Hall, Michigan State University, East Lansing, MI 48825, USA. E-mail: laduca@msu.edu
bDepartment of Chemistry and Physics, King's College, Wilkes-Barre, PA 18711, USA
First published on 19th September 2007
Abstract
Combination of cobalt or zinc chloride, 4,4′-dipyridylamine (dpa) and 4,4′-biphenyldicarboxylic acid (H2BPDC) under hydrothermal conditions has afforded the neutral coordination complexes {[Co(Hdpa)2(BPDC)2]·2H2O} (1) and [Zn(dpa)2(HBPDC)2] (2). The pendant cationic termini of the Hdpa ligands and anionic uncoordinated BPDC carboxylate groups allow each molecular unit of 1 to engage in strong charge-separated N–H⋯O hydrogen bonding interactions to four neighboring molecules, thereby creating a two-dimensional (2-D) hydrogen bonded (4,4) rhomboid grid motif with extremely large ∼42 Å × ∼25 Å apertures. Intra-layer void space is minimized by the interpenetration of four other identical hydrogen bonded rhomboid grids. Although the pendant carboxylate groups in 2 are protonated, strong O–H⋯N hydrogen bonding occurs, resulting in a supramolecular interaction pattern virtually identical to that of 1. To the best of our knowledge complexes 1 and 2 represent the first observations of a five-fold interpenetrated 2-D hydrogen bonded rhomboid grid supramolecular architecture. Adjacent layers further connect into three dimensions (3-D) by either weaker hydrogen bonding mechanisms mediated by water molecules of crystallization (1) or crystal packing forces (2).
Introduction
Mutual entanglement of structural motifs is a very commonly encountered feature in coordination polymer chemistry.1 Three-dimensional (3-D) coordination polymers can exhibit varying degrees of entanglement, with higher degrees promoted by long-spanning tethering ligands that maximize void spaces within a single covalently connected framework.2 Levels of parallel interpenetration within two-dimensional (2-D) coordination polymer networks tend to be lower due to smaller incipient voids within a single structural motif and possible lower crystal stability caused by the lack of 3-D covalent connectivity.1–3 In comparison with covalently connected metal–organic materials, interpenetrated or polycatenated hydrogen bonded networks of coordination complexes have received less attention.4 The best approach towards their synthesis involves pendant ligands with hydrogen bonding capable termini, usually protonated or deprotonated carboxylate moieties.5,6 For example, the coordination complex {[Pt(isonicotinic acid)2(isonicotinate)2]·2H2O} aggregates into a 3-D structure comprised of three orthogonally polycatenated hydrogen bonded (4,4) rhomboid grid layers.5 An interpenetrated (4,4) hydrogen bonded layer structure with larger grid apertures was observed in a zinc tetra(4-carboxyphenyl)porphyrin system.6For some time we have been interested in the synthesis and characterization of coordination polymers containing the organodiimine 4,4′-dipyridylamine (dpa). Unlike the more commonly used tethering ligand 4,4′-bipyridine (4,4′-bpy), dpa possesses an angular disposition of its terminal nitrogen donor atoms because of its central secondary amine kink, which also provides a supramolecular hydrogen bonding locus. At times one of the nitrogen termini of dpa is protonated under hydrothermal conditions, resulting in a monodentate cationic Hdpa ligand with a pendant pyridinium moiety that can promote strong charge-separated hydrogen bonding mechanisms. The structures of several metal oxides incorporating monodentate Hdpa ligands show the effectiveness of hydrogen bonding imparted by the terminal pyridinium cation in provoking supramolecular aggregation of coordination polymers into higher dimensions. For example, the metal oxide material [Mo4O13(Hdpa)2] possesses interdigitated 1-D metal oxide chains that conjoin into 2-D layers by hydrogen bonding between the pyridinium portion of Hdpa ligands and terminal molybdate oxygen atoms.7 This material was shown to selectively intercalate primary and secondary amines. The isomorphous 2-D layered oxide phases [MV4O12(Hdpa)2] (M = Co, Ni) link into a 3-D lattice via a similar Npyridinium–H⋯O![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) V hydrogen bonding pattern.8
V hydrogen bonding pattern.8
Hanton and co-workers have recently reported supramolecular aggregation into higher dimensionality by “zero-dimensional” discrete neutral coordination complexes bearing monodentate Hdpa ligands.9 Individual [Cd(Hdpa)2(SO4)2(H2O)2] complexes connect into 1-D chains by intermolecular hydrogen bonding between the aquo and sulfato ligands, which in turn form 2-D layers by hydrogen bonding to unligated sulfate oxygen atoms mediated by the pendant pyridinium moieties of the ligated Hdpa. By eschewing oxoanions in favor of the long 4,4′-biphenyldicarboxylate (BPDC) ligand in a similar cobalt/Hdpa system, we have been able to prepare the neutral coordination complexes {[Co(Hdpa)2(BPDC)2]·2H2O} (1) and [Zn(dpa)2(HBPDC)2] (2). The linear extent of the termini of pendant ligands promotes the formation of incipient rhomboid voids within hydrogen bonded 2-D layers. These contain large apertures filled with four other identical hydrogen bonded grids, formed through hydrogen bonding mechanisms between the pendant ligands. These materials therefore exhibit parallel five-fold interpenetrating hydrogen bonded (4,4) layers, a structural motif not previously reported to the best of our knowledge.
Results and discussion
Synthesis and spectral characterization
Hydrothermal treatment of a solution of the requisite metal chloride, 4,4′-dipyridylamine (dpa), and 4,4′-biphenyldicarboxylic acid (H2BPDC) afforded magenta rhombs of {[Co(Hdpa)2(BPDC)2]·2H2O} (1) or colorless blocks of [Zn(Hdpa)2(BPDC)2] as single-phase products (as verified by powder XRD). The infrared spectra of 1 and 2 were consistent with their crystal structures (vide infra). Sharp, medium intensity bands in the range of ∼1600 cm–1 to ∼1200 cm–1 were ascribed to stretching modes of the aromatic rings of the dpa and BPDC moieties.10 Features corresponding to aromatic ring flexing modes were evident in the region between 820 cm–1 and 600 cm–1. Asymmetric and symmetric C–O stretching modes of the carboxylate moieties were observed at ∼1600 cm–1 and ∼1350 cm–1 in both cases. A broad band in the region of ∼3400 cm–1 represents N–H stretching modes within the Hdpa ligands and O–H stretching modes within water molecules of crystallization in 1. The broadness of these latter spectral features is caused by the hydrogen bonding interactions (vide infra). Weak bands at ∼3290 cm–1 and ∼3180 cm–1 in the IR spectrum of 2 corresponds to the O–H bond of the protonated carboxylate and the N–H group within the dpa ligands.Structural description of {[Co(Hdpa)2(BPDC)2]·2H2O} (1)
The neutral complex 1 crystallizes in the centrosymmetric monoclinic space groupC2/c with its asymmetric unit comprising a cobalt atom located on a crystallographic two-fold rotation axis, one monodentate monoprotonated Hdpa ligand, and one dianionic 4,4′-biphenyldicarboxylate (BPDC) ligand that chelates to cobalt at one terminus. The pendant BPDC carboxylate locus is unprotonated yet non-coordinating. Protonation at the pendant, unligated site of the Hdpa ligand was corroborated crystallographically (vide infra). An uncoordinated water molecule is also evident within the asymmetric unit. Operation of the crystallographic two-fold rotation axis results in generation of a single neutral molecular unit of the coordination complex {[Co(Hdpa)2(BPDC)2]·2H2O} (Fig. 1). The coordination sphere is best described as a doubly chelated distorted octahedral [CoO4N2] chromophore, wherein the nitrogen donors belonging to the Hdpa ligands in 1 are disposed in a cis fashion. The carboxylate ligand in 1 engages in asymmetric chelation, with the Co1–O2 bond spanning a distance ∼0.35 Å longer than the corresponding Co1–O1 bond length. Selected bond distances and angles, consistent with a doubly chelated distorted octahedral [CoO4N2] coordination sphere, are given in Table 1.![A complete [Co(Hdpa)2(BPDC)2] coordination complex within 1 with thermal ellipsoids (50% probability) and atom numbering scheme. Hydrogen atoms bound to C are omitted for clarity. Hydrogen bonding interactions to uncoordinated water molecules are shown as dashed lines.](/image/article/2008/CE/b705987f/b705987f-f1.gif) | ||
| Fig. 1 A complete [Co(Hdpa)2(BPDC)2] coordination complex within 1 with thermal ellipsoids (50% probability) and atom numbering scheme. Hydrogen atoms bound to C are omitted for clarity. Hydrogen bonding interactions to uncoordinated water molecules are shown as dashed lines. | ||
| a Symmetry transformations to generate equivalent atoms: #1) –x + 1, y, –z + ½. | |||
|---|---|---|---|
| Co1–O1 | 2.037(3) | O1#1–Co1–O1 | 132.21(15) |
| Co1–N1 | 2.071(3) | O1#1–Co1–N1 | 96.27(11) |
| Co1–O2 | 2.383(3) | O1–Co1–N1 | 115.39(10) |
| O1–C23 | 1.257(4) | N1–Co1–N1#1 | 96.79(16) |
| O2–C23 | 1.263(4) | O1#1–Co1–O2 | 86.89(9) |
| O3–C24 | 1.284(4) | O1–Co1–O2 | 58.72(9) |
| O4–C24 | 1.241(4) | N1–Co1–O2 | 92.04(12) |
| N1#1–Co1–O2 | 154.78(9) | ||
| O2–Co1–O2#1 | 89.85(13) |
The inter-ring torsion angle within the Hdpa ligand in 2 is 94.6°. Within the doubly anionic BPDC ligand, the chelating carboxylate unit is twisted by ∼46° relative to the benzene ring to which it is attached, while the comparable torsion angle (∼8°) for the uncoordinated deprotonated carboxylate moiety is much diminished. The intra-ligand torsion angel about the biphenyl linkage is ∼43°, which enforces a ∼57.5° dihedral angle between the two carboxylate planes. All of these intraligand conformations optimize the supramolecular hydrogen bonding interactions within the 3-D crystal structure of 1 (vide infra).
The strongest of these intermolecular contacts is the charge-separated ionic-character N3–H3N⋯O3 hydrogen bonding interaction11 (N3⋯O3 distance = 2.540(4) Å) between the protonated pendant pyridyl ring of the Hdpa ligand in one complex and an oxygen atom of a negatively charged uncoordinated BPDC carboxylate in a neighboring molecule. The N–H bond distance (which was allowed to freely refine) was longer than usual, at 1.02(4) Å, due to the attraction of the negatively charged carboxylate oxygen. While this strong intermolecular interaction in 1 is best construed as a N+–H⋯O– electrostatic-type attraction, some quasi-covalent N–H⋯O character cannot be ruled out.11Hydrogen bonding has been termed “incipient proton transfer”;12 such an interaction is clearly prevalent in this case.
Through this strong hydrogen bonding interaction, each individual [Co(Hdpa)2(BPDC)2] coordination complex conjoins to four others to construct a (4,4) rhomboid grid pattern oriented parallel to the ab crystal plane. The Co⋯Co distance between directly hydrogen bonded Hdpa and BPDC ligands, delineating the outline of the grid motif, is 24.34 Å. The grids are pinched along the b axis, with Co⋯Co⋯Co angles of 61.5° and 118.5°. Extremely large apertures within the grid, defined by through-space Co⋯Co distances of 41.84 Å and 24.89 Å, allow the parallel interpenetration of four other identical hydrogen bonded (4,4) layers. Therefore 1 manifests five-fold interpenetration of hydrogen bonded rhomboid grid layer motifs, as seen in Fig. 2. Each grid is anchored to its nearest neighbor by weak C–H⋯O interactions between one side of the pendant pyridyl ring of the Hdpa ligands in one grid and ligated BPDC carboxylate oxygen atoms in another grid (C6⋯O1 distance = 3.188(6) Å; C7⋯O2 distance = 3.124(6) Å). Inter-grid supramolecular connectivity is also promoted by π–π stacking (centroid-to-centroid distance = 3.910(4) Å) between pendant Hdpa pyridyl rings and the aromatic ring in the BPDC ligand farthest from cobalt. A network perspective of the interpenetrated layer is shown in Fig. 3, where the hydrogen bonded pairs of Hdpa and BPDC ligands are indicated together as solid lines. There is only one currently reported five-fold parallel interpenetrated (4,4) 2-D covalently connected layer framework, within the structure of the coordination polymer [Co(NCS)2(2,5-bis(4-pyridylethynyl)thiophene)].13 While five-fold 2-D grid interpenetration has also been very recently observed in a co-crystal of tetrakis(4-pyridyl)pentaerythritol and 1,8-diiodoperfluorooctane, promoted by N⋯I halogen bonding,14 to the best of our knowledge 1 is the first reported example of this mode of interpenetration induced by “classical” hydrogen bonding mechanisms.
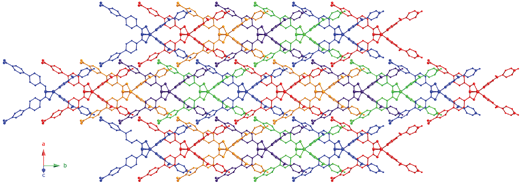 | ||
| Fig. 2 Five-fold interpenetration of hydrogen bonded rhomboid grid motifs in 1. | ||
 | ||
| Fig. 3 Network perspective of the five-fold interpenetrated hydrogen bonded (4,4) layers in 1, with each independent layer drawn in a different color. Each pair of directly hydrogen bonded Hdpa and BPDC ligands is shown as a solid line. | ||
Juxtaposed hydrogen bonded layers in 1 are further aggregated into three dimensions along the c crystal axis via weaker hydrogen bonding patterns mediated by water molecules of crystallization situated in the interlamellar regions, as depicted in Fig. 4. The isolated, uncoordinated water molecules engage in distinct hydrogen bonding interactions with three different [Co(Hdpa)2(BPDC)2] coordination complexes in two adjacent layers. Each water molecule serves as a hydrogen bonding donor both to the uncoordinated BPDC carboxylate oxygen atom O4 in one layer (via H1A, O1W⋯O4 distance = 2.752(4) Å) and to the ligated BPDC carboxylate oxygen atom O2 in another layer (via H1B, O1W⋯O2 distance = 2.752(4) Å). Hydrogen bonding acceptance by the water molecules of crystallization from the Hdpa central amine units (N2–H2N⋯O1W, with an N⋯O distance = 2.750(4) Å) provides the third significant interlayer supramolecular interaction. Taking all direct and through-water molecule classical hydrogen bonding interactions into account, each [Co(Hdpa)2(BPDC)2] neutral complex is conjoined to eight others (four within the layer motif, two within the layer above, two within the layer below). The nearest Co⋯Co contact distances between adjacent layer motifs are 11.203 Å (half the distance of the c unit cell edge length) and 13.791 Å. Full metrical parameters for all classical supramolecular interactions in 1 are given in Table 2.
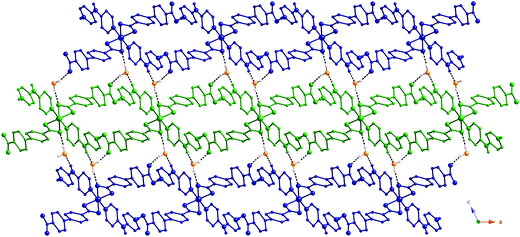 | ||
| Fig. 4 View down b of the interaction of adjacent hydrogen bonded layers through uncoordinated water molecules in 1. Interlayer hydrogen bonding is shown as dashed lines. | ||
| D–H | d(H⋯A)/Å | <DHA/° | d(D⋯A)/Å | Symmetry transformation for A/B |
|---|---|---|---|---|
| 1 | ||||
| O1W–H1A⋯O4 | 1.92(2) | 160(4) | 2.752(4) | x + 1/2, y + 3/2, z |
| O1W–H1B⋯O2 | 1.93(2) | 164(4) | 2.752(4) | –x + 1, –y + 1, –z |
| N2–H2N⋯O1W | 1.89(2) | 167(3) | 2.750(4) | |
| N3–H3A⋯O3 | 1.52(4) | 175(3) | 2.540(4) | x + 1/2, y + 5/2, z |
| 2 | ||||
| O4–H4A⋯N3 | 1.45(3) | 175(3) | 2.606(3) | –x + 3/2, y – 5/2, –z – 1/2 |
Structural description of [Zn(dpa)2(HBPDC)2] (2)
The neutral complex 2 displays an asymmetric unit containing a zinc atom located on a crystallographic two-fold rotation axis, one monodentate pendant dpa ligand, and one singly deprotonated, monodentate 4,4′-biphenyldicarboxylate (HBPDC) ligand. Operation of the crystallographic two-fold rotation axis results in the generation of a single neutral molecular unit of [Zn(dpa)2(HBPDC)2] (Fig. 5). The hydrogen atom attached to oxygen atom O4, part of the unligated carboxylate terminus, was located crystallographically. Protonation at this site is corroborated by the longer C–O bond length relative to that for O3, indicative of greater σ bond character in the C18–O4 bond. Unlike 1, no water molecules of crystallization are present in this case. The tetrahedral [ZnO2N2] coordination environment also stands in contrast to 1. Bond lengths and angles for 2 are standard for four-coordinate Zn coordination complexes (Table 3). The unligated oxygen atom O1 belonging to the bound BPDC carboxylate is 2.82 Å from Zn, well outside the standard range of covalent bonding to this metal.![A complete [Zn(dpa)2(HBPDC)2] coordination complex within 2 with thermal ellipsoids (50% probability) and atom numbering scheme. Hydrogen atoms bound to C are omitted for clarity.](/image/article/2008/CE/b705987f/b705987f-f5.gif) | ||
| Fig. 5 A complete [Zn(dpa)2(HBPDC)2] coordination complex within 2 with thermal ellipsoids (50% probability) and atom numbering scheme. Hydrogen atoms bound to C are omitted for clarity. | ||
| a Symmetry transformations to generate equivalent atoms: #1) –x + 1, y, –z – 1/2. | |||
|---|---|---|---|
| Zn1–O2 | 1.9411(19) | O2–Zn1–O2#1 | 120.16(12) |
| Zn1–O2#1 | 1.9411(19) | O2–Zn1–N1#1 | 116.72(8) |
| Zn1–N1#1 | 2.044(2) | O2#1–Zn1–N1#1 | 101.10(8) |
| Zn1–N1 | 2.044(2) | O2–Zn1–N1 | 101.10(8) |
| O1–C11 | 1.228(3) | O2#1–Zn1–N1 | 116.72(8) |
| O2–C11 | 1.275(3) | N1#1–Zn1–N1 | 99.87(12) |
| O3–C18 | 1.215(3) | ||
| O4–C18 | 1.311(3) |
Individual molecules of 2 subsequently aggregate by strong O–H⋯N hydrogen bonding interactions between the unprotonated pendant dpa termini and protonated BPDC carboxylates in neighboring molecules. The lengthening of the O–H bond (1.16(3) Å) is once again indicative of “incipient proton transfer”.12 Specific data about this interaction is given in Table 2. In order to maximize this interaction, the dpa ligands in 2 exhibit an inter-ring torsion angle of 40.4°, over 54° narrower than 1. In addition, the bound, monodentate carboxylate unit of the HBPDC ligand is twisted by ∼14.8° relative to the benzene ring to which it is attached. Again, this torsion angle is much narrower (by ∼32°) relative to that of the chelating BPDC carboxylate in 1.
The strong hydrogen bonding interaction promotes the formation of five-fold parallel interpenetrated (4,4) rhomboid grid layers virtually identical to those observed in 1, despite the difference in coordination geometry at the metal. The incipient void spaces within the rhomboid grid in 2 deviate only slightly from those in 1, with through-ligand Zn⋯Zn interactions measuring 24.40 Å, through-space Zn⋯Zn distances of 26.15 Å and 41.20 Å and Zn⋯Zn⋯Zn angles of 64.9° and 115.12°. As in 1, the interpenetration of the individual rhomboid grid layers is promoted by weak C–H⋯O interactions (C1⋯O3 distance = 3.160(4) Å) and π–π stacking between dpa and HBPDC moieties in different nets. Layered units in 2 stack primarily by long range crystal packing forces because of the absence of water molecules of crystallization, with a Zn⋯Zn interlayer closest distance of 11.06 Å.
Thermogravimetric analysis
Compound 1 was stable until ∼175 °C, whereupon a slow release of co-crystallized water began to occur. Elimination of all solvent was complete by ∼325 °C, with the 4.0% mass loss exactly matching that predicted for two water molecules of crystallization per coordination complex. Decompositionvia expulsion of both BPDC ligands occurred between ∼325 °C and ∼375 °C (55.6% observed, 52.2% predicted). Combustion of the remaining organic components occurred between ∼410 °C and ∼525 °C with the 9.6% mass remnant roughly consistent with deposition of Co3O4 (8.7% predicted). The TGA trace for complex 1 is depicted in Fig. S1.‡Thermolysis of 1 in air at 180 °C for 24 h resulted in the crumbling of the single crystals into a magenta powder. Powder XRD (Fig. S2‡) revealed the presence of additional peaks in addition to those corresponding to the calculated powder pattern of 1, indicative of either partial decomposition or structural reorganization.The mass of compound 2 remains unchanged until 325 °C whereupon it underwent expulsion of organic moieties. The mass remnant at 700 °C was 11.2% of the original, roughly consistent with possible deposition of zinc oxide (9.1% calcd). Powder XRD indicated that the remnant was amorphous. The TGA trace for compound 2 is depicted in Fig S3.‡Powder XRD of a sample of 2 held at 180 °C for 24 h (Fig S4‡) revealed that the five-fold interpenetrated hydrogen-bonded layer motifs stayed intact.
Conclusions
Employment of long dianionic 4,4′-biphenyldicarboxylate and monoprotonated 4,4′-dipyridylamine ligands has resulted in a neutral cobalt coordination complex with four projecting termini in a pseudo-tetrahedral orientation, with two termini each of opposite charge. This arrangement promotes the aggregation of adjacent molecules through strong charge-separated “ionic” N+–H⋯O–hydrogen bonding into (4,4)-topology rhomboid nets with large apertures which can accommodate the interpenetration of four other identical hydrogen bonded nets. A related zinc complex aggregates into a virtually identical five-fold parallel interpenetrated morphology via strong “covalent” O–H⋯N hydrogen bonding. The subtle differences in the ionic character of the hydrogen bonding are likely caused by the changes in metal coordination, carboxylate binding mode, and local steric environments. Nevertheless, both types of strong hydrogen bonding are of sufficient strength to promote the formation of the interpenetrated supramolecular layers. The five-fold interpenetrated parallel two-dimensional hydrogen bonded networks in crystals of these coordination complexes are the first examples of their type to the best of our knowledge. Continued work exploiting unprotonated and monoprotonated hydrogen-bonding capable 4,4′-dipyridylamine ligands in the construction of novel hydrogen bonded and coordination polymer networks is under way in our laboratory.Experimental
General considerations
CoCl2·6H2O and ZnCl2 (Fisher) and 4,4′-biphenyldicarboxylic acid (Aldrich) were obtained commercially. 4,4′-Dipyridylamine (dpa) was prepared via a published procedure.7Water was deionized above 3 MΩ in-house. Elemental analysis was carried out using a Perkin Elmer 2400 Series II CHNS/O Analyzer. IR spectra were recorded on pure powdered samples with a Perkin Elmer Spectrum One instrument. Thermogravimetric analysis was performed on a TA Instruments TGA 2050 Thermogravimetric Analyzer with a heating rate of 10 °C min–1 up to 600 °C. Powder XRD spectra were acquired on a Rigaku Rotaflex instrument using Θ–2Θ scans.Preparation of {[Co(Hdpa)2(BPDC)2]·2H2O} (1)
CoCl2·6H2O (88 mg, 0.37 mmol), 4,4′-dipyridylamine (127 mg, 0.74 mmol) and 4,4′-biphenyldicarboxylic acid (90 mg, 0.37 mmol) were added to 10 mL of distilled H2O with 1.0 mL of 1.0 M NaOH in a 23 mL Teflon lined Parr acid digestion bomb. The bomb was sealed, heated to 120 °C for 45 h, and then gradually cooled to 23 °C. Magenta rhombs of 1 (55 mg, 32% yield based on H2BPDC) were obtained after filtration, washing with distilled water and acetone, and drying in air. Anal. calc. for C48H40CoN6O101: C, 62.68; H, 4.40; N, 9.14%; Found: C, 63.45; H, 3.90; N, 8.89%. IR (KBr, cm–1): 3403 w, 1691 m, 1591 s, 1508 s, 1434 w, 1381 m, 1330 m, 1306 w, 1208 w, 1171 w, 1098 w, 1018 m, 1005 m, 899 w, 879 w, 846 m, 824 m, 793 w, 763 s, 714 m, 703 m, 676 w.Preparation of [Zn(dpa)2(HBPDC)2] (2)
ZnCl2 (50 mg, 0.37 mmol), 4,4′-dipyridylamine (127 mg, 0.74 mmol) and 4,4′-biphenyldicarboxylic acid (180 mg, 0.74 mmol) were added to 10 mL of distilled H2O with 1.0 mL of 1.0 M NaOH in a 23 mL Teflon lined Parr acid digestion bomb. The bomb was sealed, heated to 120 °C for 45 h, and then gradually cooled to 23 °C. Colorless blocks of 2 (241 mg, 73.2% yield based on Zn) were obtained after filtration, washing with distilled water and acetone, and drying in air. Anal. calc. for C48H36N6O8Zn 2: C, 64.76; H, 4.08; N, 9.44%; Found: C, 64.47; H, 4.08; N, 7.71%. While the elemental analysis value for nitrogen is somewhat lower than expected, phase purity was verified by powder XRD. IR (KBr, cm–1): 3292 w, 3179 w, 1600 s, 1523 s, 1448 m, 1431 m, 1349 s, 1209 s, 1059 m, 1027 s, 902 m, 851 w, 820 s, 769 w, 728 w.X-Ray crystallography
A magenta rhomb of 1 (0.52 mm × 0.28 mm × 0.16 mm) and a colorless block of 2 (0.35 mm × 0.25 mm × 0.20 mm) removed from freshly prepared aqueous reaction solutions were subjected to single crystal X-ray diffraction using a Bruker-AXS SMART 1 k CCD instrument at 273(2) K. Reflection data were acquired using graphite-monochromated Mo-Kα radiation (λ = 0.71073 Å). The data were integrated viaSAINT.15 Lorentz and polarization effect and empirical absorption corrections were applied with SADABS.16 The structures were solved using direct methods and refined on F2 using SHELXTL.17 All non-hydrogen atoms were refined anisotropically. Hydrogen atoms bound to carbon atoms were placed in calculated positions and refined isotropically with a riding model. The hydrogen atoms bound to the central nitrogen atoms of the dpa moieties in both 1 and 2 and the water molecule of crystallization in 1 were found via Fourier difference maps, restrained at fixed positions, and then refined isotropically with thermal parameters 1.2 times the average Uij values of the atoms to which they are bound. The hydrogen atoms involved in the crucial strong hydrogen bonding interactions were located via Fourier difference maps and refined isotropically with thermal parameters 1.2 times the average Uij values of the atoms to which they are bound; however their bond distances were allowed to refine freely. Relevant crystallographic data for 1 and 2 are listed in Table 4. Supramolecular contact information was calculated using PLATON software.18| a R 1 = ∑||Fo| – |Fc||/∑|Fo|. b wR 2 = ∑{[w(Fo2 – Fc2)2]/∑[wFo2]2}1/2. | ||
|---|---|---|
| Empirical formula | C48H40CoN6O10 | C48H36N6O8Zn |
| Formula weight | 919.80 | 890.20 |
| Collection T/K | 293(2) | 293(2) |
| Crystal system | Monoclinic | Monoclinic |
| Space group | C2/c | C2/c |
| a/Å | 24.89(2) | 26.150(7) |
| b/Å | 8.368(8) | 8.241(2) |
| c/Å | 22.23(2) | 21.331(6) |
| β/° | 115.566(12) | 119.933(4) |
| V/Å3 | 4177(6) | 3983.8(18) |
| Z | 4 | 4 |
| D calc/g cm–3 | 1.459 | 1.484 |
| µ/mm–1 | 0.482 | 0.684 |
| Min./max. T | 0.773/0.926 | 0.725/0.872 |
| hklranges | –33 ≤ h ≤ 33, –11 ≤ k ≤ 11, –29 ≤ l ≤ 29 | –33 ≤ h ≤ 33, –10 ≤ k ≤ 10, –27 ≤ l ≤ 26 |
| Total reflections | 24![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 254 254 |
19863 |
| Unique reflections | 5147 | 4643 |
| R(int) | 0.0955 | 0.0559 |
| Parameters/restraints | 306/4 | 291/1 |
| R 1 (all data)a | 0.1250 | 0.0850 |
| R 1 (I > 2σ(I))b | 0.0650 | 0.0475 |
| wR 2 (all data)a | 0.1389 | 0.1038 |
| wR 2 (I > 2σ(I))b | 0.1175 | 0.0917 |
| Max,min residual/e Å–3 | 0.457, –0.725 | 0.368, –0.414 |
| G.O.F. | 1.068 | 1.020 |
Acknowledgements
The authors gratefully acknowledge Michigan State University for financial support of this work.We thank Dr Rui Huang for elemental analysis and Subha Mallika Krishnan and Max Braverman for experimental assistance. The thermogravimetric analyzer at King's College was purchased with a grant from the Alden Trust. We thank one of the reviewers for very helpful suggestions.
References
- S. R. Batten, CrystEngComm, 2001, 3, 67 RSC.
- V. A. Blatov, L. Carlucci, G. Ciani and D. M. Proserpio, CrystEngComm, 2004, 6, 377 RSC.
- S. R. Batten and R. Robson, Angew. Chem., Int. Ed., 1998, 37, 1460 CrossRef.
- A. M. Beatty, CrystEngComm, 2001, 3, 243 RSC.
- C. B. Aakeröy, A. M. Beatty and D. S. Leinen, Angew. Chem., Int. Ed., 1999, 38, 1815 CrossRef CAS.
- P. Dastidar, Z. Stein, I. Goldberg and C. E. Strouse, Supramol. Chem., 1996, 7, 257 CrossRef CAS.
- P. J. Zapf, R. L. LaDuca, R. S. Rarig, K. M. Johnson and J. Zubieta, Inorg. Chem., 1998, 37, 3411 CrossRef CAS.
- R. L. LaDuca, R. S. Rarig and J. Zubieta, Inorg. Chem., 2001, 40, 607 CrossRef CAS.
- D. B. Cordes, L. R. Hanton and M. D. Spicer, Cryst. Growth Des., 2007, 7, 328 CrossRef CAS.
- M. Kurmoo, C. Estournes, Y. Oka, H. Kumagai and K. Inoue, Inorg. Chem., 2005, 44, 217 CrossRef CAS.
- T. Steiner, Angew. Chem., Int. Ed., 2002, 41, 48 CrossRef CAS.
- H. B. Burgi and J. D. Dunitz, Acc. Chem. Res., 1983, 16, 153 CrossRef.
- S. U. Son, B. Y. Kim, C. H. Choi, S. W. Lee, Y. S. Kim and Y. K. Chung, Chem. Commun., 2003, 2528 RSC.
- P. Metrangelo, F. Meyer, T. Pilati, D. M. Proserpio and G. Resnati, Chem.–Eur. J., 2007, 13, 5765 CrossRef.
- SAINT, Software for Data Extraction and Reduction, Version 6.02, Bruker AXS, Inc, Madison, WI, 2002 Search PubMed.
- SADABS, Software for Empirical Absorption Correction, Version 2.03, Bruker AXS, Inc., Madison, WI, 2002 Search PubMed.
- G. M. Sheldrick, SHELXTL, Program for Crystal Structure Refinement, University of Göttingen, Göttingen, Germany, 1997 Search PubMed.
- A. L. Spek, PLATON, A Multipurpose Crystallographic Tool, Utrecht University, Utrecht, The Netherlands, 1998 Search PubMed.
Footnotes |
| † CCDC reference numbers 639657 and 650999. For crystallographic data in CIF or other electronic format see DOI: 10.1039/b705987f |
| ‡ Electronic supplementary information (ESI) available: TGA traces for 1 and 2; powder XRD of 1 and 2. See DOI: 10.1039/b705987f |
| This journal is © The Royal Society of Chemistry 2008 |