Coordination behaviour and network formation with 4,4′,6,6′-tetracarboxy-2,2′-bipyridine and 4,4′-dicarboxy-2,2′-bipyridine ligands with rare and alkaline earth metals†‡
Niamh R.
Kelly
a,
Sandrine
Goetz
a,
Stuart R.
Batten
b and
Paul E.
Kruger
*ac
aSchool of Chemistry, University of Dublin, Trinity College, Dublin 2, Ireland. E-mail: paul.kruger@tcd.ie
bSchool of Chemistry, Monash University, Clayton, VIC 3800, Australia
cDepartment of Chemistry, College of Science, University of Canterbury, Christchurch, 8020, New Zealand. E-mail: paul.kruger@canterbury.ac.nz
First published on 3rd October 2007
Abstract
Reported herein is the synthesis and structural characterisation of the novel ligand, 4,4′,6,6′-tetracarboxylic acid-2,2′-bipyridine (H4tcbp), along with an investigation of its coordination chemistry. Single crystal X-ray diffraction studies showed H4tcbp crystallises as a dihydrate and that intermolecular hydrogen bonding involving adjacent 4,4′-carboxylic acid groups form a R22(8) dimer motif whereas the 6,6′-carboxylic acid groups hydrogen bond together via the lattice water molecules [R44(12)] to form a 2D grid which is 3-fold interpenetrated and yields a 3D hydrogen bonded network. The reaction of H4tcbp under solvothermal conditions in MeOH with Ln(III) (Ln = Yb, Gd and Sm) lead to the isolation of {[Yb(Me2tcbp)2]·H2O·MeOH·H3O}, 1, and {[Ln(Me2tcbp)2]·2H2O·H3O}, where Ln = Gd, 2, or Sm, 3, and when in the presence of Yb(III) and Cu(II) to the mixed-metal species, {[Yb2(Me2tcbp)2]2Cu(OMe)2(H2O)2]·6MeOH}, 4, which were structurally characterised. Under these synthetic conditions the H4tcbp ligand di-esterified in situ at the remote 4- and 4′-carboxylic acid groups to form the 4,4′-dimethylcarboxy-6,6′-dicarboxy-2,2′-bipyridine ligand (Me2tcbp2–). Although 1 is monomeric, extensive hydrogen bonding between lattice water molecules, hydronium counterions and deprotonated carboxylate oxygen atoms results in 2D bilayer structure. Isostructural 2 and 3 are discrete mononuclear complexes whereas the tri-nuclear 4 forms a 2D sheet through face-to-face π–π interactions between adjacent units. In addition, the structural characterization and thermal analysis of four alkaline earth metal 3D coordination networks are reported, two of which feature the H4tcbp ligand: [Ba2(tcbp)(H2O)2] 5 and [CuSr(tcbp)(H2O)3] 6 (a 5,6-connected net), with the remaining two featuring the related 4,4′-dicarboxy-2,2′-bipyridine (H2dcbp) ligand: {[Ba(dcbp)(H2O)0.4]·0.6H2O} 7 (a bi-nodal 6-connected net) and {[Sr(dcbp)]·H2O} 8 (a uni-nodal 6-connected net).
Introduction
Immense current interest revolves around the synthesis and characterization of inorganic polymeric materials because of their potential applications.1 Coordination polymers and porous metal–organic frameworks (MOFs) in particular have attracted the attention of chemists, physicists and materials scientists alike, due to the creation of nanometer-sized spaces with associated, potentially commercial, applications such as in separation,2 gas storage3 and catalysis.2d,e,4 Whilst the incorporation of lanthanide metal ions in combination with organic linkers in the synthesis of these polymeric materials has recently received considerable interest,5 predominantly, transition metals have been utilized. To date, the inclusion of alkaline earth metals into such polymeric materials remains considerably less well explored.5l Although, undoubtedly, control of rare and alkaline earth metal-based frameworks in the construction of coordination polymers remains a challenge due to the metal ions’ lack of preferred geometrical orientation and coordination number, they do offer the advantage of potentially producing structurally interesting and diverse materials with potentially interesting properties. Also, the oxophilic nature of these metal ions renders them ideal candidates for coordination with oxygen donor ligands. For this reason, we have used these metals in the present study in combination with the following ligands: the novel 4,4′,6,6′-tetracarboxy-2,2′-bipyridine (H4tcbp) and the known 4,4′-dicarboxy-2,2′-bipyridine (H2dcbp), Scheme 1. In contrast to the transition metal ions, the rare and alkaline earth metals have no predisposed coordination constraints and possess larger atomic radii. The synthesis of the new ligand H4tcbp should allow coordination of larger metal ions with higher stability within the ‘expanded’ tetradentate (N2O2) chelating site and in addition to the peripheral carboxylate groups. Indeed, the carboxylate groups are able to coordinate to metal ions in a range of diverse modes and provide the potential for the formation of metal–carboxylate clusters or bridging units, which may help enhance the robustness of the resulting network.6 Furthermore, their negative charge provides charge balance on coordination of cations obviating the need for counter-anions which might otherwise occupy any resultant void space within a prospectively porous material. Another advantage that both the H2dcbp and H4tcbp ligands offer is that they may potentially participate in hydrogen bonding interactions through their bipyridyl, carboxylate or carboxylic acid functionalities, thereby allowing for the combination of strong coordination bonding with flexible hydrogen bonding in the preparation of hybrid networks.7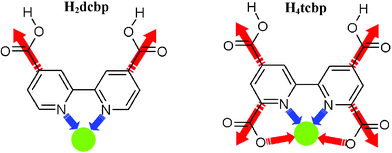 | ||
| Scheme 1 The structures of H2dcbp and H4tcbp showing their potential connectivity through carboxylate and 2,2′-bipyridine coordination sites. | ||
We extend research5c,6,7a,8,9 on H2dcbp-based coordination polymers to now incorporate H4tcbp and the rare and alkaline earth metal ions. We report here the synthesis and characterisation of the new ligand H4tcbp and the solvo-thermal synthesis and structural characterization of a series of complexes featuring H4tcbp and H2dcbp ligands. These complexes range from simple monomeric species through H-bonded networks to 3D coordination polymers and feature an array of multi-faceted carboxylate coordination, details of which are discussed.
Results and discussion
Synthesis and characterisation 4,4′,6,6′-tetracarboxy-2,2′-bipyridine (H4tcbp)
The H4tcbp ligand was prepared via the Na2Cr2O7/H2SO4 mediated oxidation of 4,4′,6,6′-tetramethyl-2,2′-bipyridine (tmbp) following a slight modification to that previously reported for H2dcbp.10tmbp itself has been previously synthesised11 by coupling 2,4-dimethylpyridine over sodium with low yield (2.5%), whereas we used Pd/C mediated coupling to give a far superior 21% yield.12 Micro-analytical and 1H NMR data were consistent with the formation of both tmbp and H4tcbp. IR spectroscopy showed the presence of the carboxylic acid groups of H4tcbp with νas(CO2) and νs(CO2) falling within the range of 1611–1733 cm–1 and 1251–1380 cm–1, respectively. Both H4tcbp and tmbp were isolated as crystalline solids suitable for single crystal diffraction studies from which their structures were determined unambiguously.Crystal structure of H4tcbp·2H2O
The atomic numbering scheme and atom connectivity for H4tcbp·2H2O are shown in Fig. 1(a). The structure was refined in the monoclinic P2/c space group. Crystallographic details are given in Table 2. The two pyridyl rings are twisted with respect to one another about the C5–C5A axis at an angle of approximately 38° from the anti conformation. Additionally, the carboxylic acid groups about C6 and C7 are essentially coplanar with the aromatic ring to which they are attached (ca. 1 and 4° torsion angles, respectively).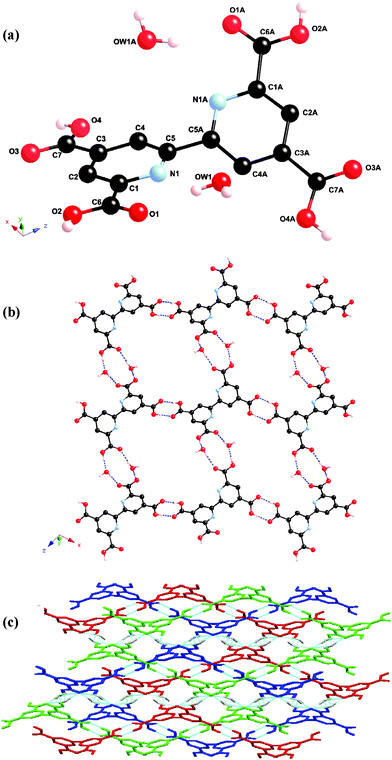 | ||
| Fig. 1 (a) Molecular structure and atomic numbering scheme for H4tcbp·2H2O. Bipyridyl hydrogen atoms omitted for clarity. Symmetry operator: a = –x, y, 0.5 – z. (b) Packing diagram of H4tcbp·2H2O showing 1D hydrogen bonded chains linked to adjacent chains through water molecules to form a wavelike 2D hydrogen bonded sheet. (c) 3-Fold interweaved 3D H4tcbp·2H2O hydrogen bonded network, wherein the 2D sheets hydrogen bond to one another through water molecules. The three networks are presented in blue, red and green. | ||
The overall structure of H4tcbp·2H2O consists of hydrogen bonded chains linking via an R22(8) dimer motif involving the 4- and 4′-carboxylic acid groups. Hydrogen bonding involving the 6- and 6′-carboxylic acid groups and water molecules link these chains to one another through an R44(12) motif to form a wavelike 2D hydrogen bonded grid, Fig. 1(b).13 The ‘aperture’ within the 2D grid is of sufficient size to allow additional 2D grids to pass through it to yield layers of 3-fold 2D → 2D parallel interpenetrating1d,f networks, Fig. 1(c). The inter-weaved networks are then further cross-linked into a single, overall 3D network via further, longer interactions between the water molecules and carboxylic acid groups. A full list of all hydrogen bonding interactions is given in Table 1.
| D–H⋯A | D–H/Å | d(H⋯A)/Å | d(D⋯A)/Å | <(DH⋯A)/° |
|---|---|---|---|---|
| a Symmetry codes: i = x, –y, z – 1/2; ii = –x + 2, –y + 1, –z; iii = –x, –y, –z; iv = x + 1, y, z; v = –x, –y, –z + 1; vi = –x + 1, –y + 1, –z + 1; vii = x, –y + 1, z + 1/2; viii = x + 1/2, –y + 1/2, z + 1/2; ix = x + 1/2, –y + 3/2, z + 1/2; x = –x + 3, –y + 3, –z + 2; xi = x, y – 1, z; xii = –x + 3, –y + 2, –z + 2; xiii = x, y, z – 1; xiv = –x + 2, –y + 2, –z + 1; xv = x – 1, y – 1, z – 1. | ||||
| H4tcbp·2H2O | ||||
| O2–H2⋯O1wi | 0.91 | 1.62 | 2.515(2) | 170 |
| O4–H4⋯O3ii | 0.89 | 1.74 | 2.622(2) | 177 |
| O1w–H1wa⋯O1iii | 0.88 | 1.94 | 2.813(2) | 174 |
| O1w–H1wb⋯O1 | 0.88 | 2.04 | 2.881(2) | 160 |
| 1 | ||||
| O1w–H1w1⋯O15 | 0.84 | 2.01 | 2.850(7) | 175 |
| O1w–H2w1⋯O2iv | 0.84 | 2.12 | 2.870(8) | 148 |
| O18–H18b⋯O10v | 0.85 | 1.88 | 2.730(9) | 174 |
| O18–H18c⋯O1wvi | 0.85 | 2.07 | 2.907(9) | 170 |
| O18–H18d⋯O9 | 0.85 | 2.06 | 2.892(9) | 167 |
| 5 | ||||
| O1w–H1w1⋯O3vii | 0.84 | 1.96 | 2.780(3) | 164 |
| O2w–H1w2⋯O7viii | 0.85 | 1.91 | 2.748(3) | 169 |
| O2w–H2w2⋯O5ix | 0.85 | 2.24 | 2.934(3) | 139 |
| 6 | ||||
| O1w–H1w1⋯O5x | 0.83 | 2.06 | 2.872(7) | 166 |
| O1w–H2w1⋯O6xi | 0.83 | 2.02 | 2.839(7) | 171 |
| O2w–H1w2⋯O1wxii | 0.83 | 2.10 | 2.917(7) | 168 |
| O2w–H2w2⋯O1xiii | 0.83 | 1.95 | 2.709(7) | 150 |
| O3w–H1w3⋯O4xiv | 0.83 | 2.21 | 3.012(7) | 160 |
| O3w–H2w2⋯O7xv | 0.83 | 2.01 | 2.812(7) | 160 |
Synthesis and characterisation of 1–8
The syntheses of the complexes employed solvothermal methods. For the synthesis of 1–3, Ln(CF3SO3)3 (Ln(III) = Yb, Gd, Sm) and H4tcbp·2H2O (1 ∶ 2) were heated under solvothermal conditions in MeOH for the prescribed temperature and time which yielded crystalline products directly after slow cooling. Their infrared spectra are broadly similar possibly attesting to a common structure for each and comparable coordination of the ligand about each metal centre. Microanalytical data were consistent with 1 ∶ 2 metal-to-ligand ratios although solution analyses were frustratingly thwarted by the total lack of solubility of the complexes in common laboratory solvents. It became evident from the crystal structure analysis (see later) that under these synthetic conditions, esterification of the 4- and 4′-carboxylic acid groups had occurred in situ. In an effort to prevent this we then undertook the synthesis in the presence of Cu(II), which in the event was ‘unsuccessful’ as it led to the formation of {[Yb2(Me2tcbp)2]2Cu(OMe)2(H2O)2]·6MeOH}, 4, which was also structurally characterised. When the synthesis was undertaken in neat water no identifiable products could be isolated and only intractable solids were produced.The synthesis of the alkaline earth metal complexes 5–8 followed a typical procedure, where the M(II) salt (acetate or nitrate), H4tcbp or H2dcbp and solvent were reacted solvothermally which also yielded crystalline products directly following slow cooling. In the case of the reaction of H4tcbp with Sr(II) salts we were unable to obtain products of suitable crystal quality for a diffraction study when Sr(II) was employed on its own, however when an equivalent of Cu(OAc)2·2H2O was included, pale blue crystals of the mixed-metal complex 6 were obtained directly.14Infrared spectra of 5–8 showed the presence of carboxylate groups of tcbp4– in the range of 1653–1540 cm–1 and 1422–1229 cm–1 for νas(CO2–) and νs(CO2–), respectively. The carboxylate groups of dcbp2– with νas(CO2–) and νs(CO2–) fall within the ranges of 1732–1538 cm–1 and 1421–1294 cm–1, respectively. Micro-analytical data were consistent with the formation of either a 2 ∶ 1 M(II) ∶ tcbp4– species (5 and 6) or a 1 ∶ 1 M(II) ∶ dcbp2– species (7 and 8). The crystals are indefinitely stable when removed from their mother liquors and insoluble in all common laboratory solvents.
Structures of {[Yb(Me2tcbp)2]·H2O·MeOH·H3O}, 1, and {[Ln(Me2tcbp)2]·2H2O·H3O} (where Ln = Gd, 2, or Sm, 3)
The atomic numbering scheme and atom connectivity for 1 are shown in Fig. 2(a). The structure was refined in the triclinic P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) space group and crystallographic data are shown in Table 2. The Yb(III) metal centre is eight coordinate with two ligands chelating in tetradentate fashion via two bipyridyl nitrogen atoms and the two 6,6′-carboxylate oxygen atoms. Comparison of the two ligands shows that the ligand containing N1 and N2 is essentially planar, whereas the N3 and N4 rings are twisted slightly (∼5°) with respect to each other about their central axis. The ligand mean planes subtend an angle of 85.6° to each other across the Yb(III) centre, and the co-ordination polyhedron around Yb1 may be described as a distorted C2vdodecahedron: the four O-atoms forming an approximate square plane while the N-atoms of each ligand sit above and below this plane, respectively, and are approximately aligned along the two diagonals of the square.15
space group and crystallographic data are shown in Table 2. The Yb(III) metal centre is eight coordinate with two ligands chelating in tetradentate fashion via two bipyridyl nitrogen atoms and the two 6,6′-carboxylate oxygen atoms. Comparison of the two ligands shows that the ligand containing N1 and N2 is essentially planar, whereas the N3 and N4 rings are twisted slightly (∼5°) with respect to each other about their central axis. The ligand mean planes subtend an angle of 85.6° to each other across the Yb(III) centre, and the co-ordination polyhedron around Yb1 may be described as a distorted C2vdodecahedron: the four O-atoms forming an approximate square plane while the N-atoms of each ligand sit above and below this plane, respectively, and are approximately aligned along the two diagonals of the square.15
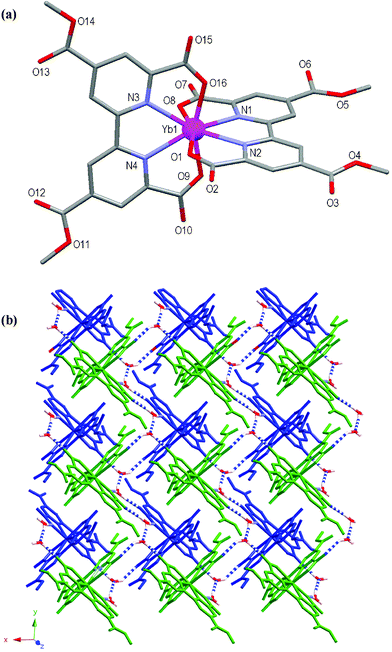 | ||
| Fig. 2 (a) Molecular structure and atomic numbering scheme for 1. Hydrogen atoms and lattice hydronium ion, water and methanol molecules are omitted for clarity. (b) Packing diagram of 1 showing a 2D sheet (green) hydrogen bonding to another 2D sheet (blue) to form a bilayer. Blue and white dashed bonds depict hydrogen bonds. Hydrogen atoms not involved in hydrogen bonding and methanol molecule are omitted for clarity. | ||
The peripheral 4- and 4′-carboxylic acid groups have been (methyl) esterified in situ to form 4,4′-dimethylcarboxy-6,6′-dicarboxy-2,2′-bipyridine (Me2tcbp2–). Disorder is present within two of the methyl ester groups at the oxygen atoms O6 and O13, with O6 disordered over two positions modelled at 55 and 45% occupancy, respectively and O13 disordered over three positions modelled at 24, 38 and 38% occupancy, respectively. Charge balance for the [Yb(Me2tcbp)2]– unit is provided by the presence of a hydronium ion within the crystal lattice.
The overall structure of 1 consists of 2D hydrogen bonded sheets. As shown in Fig. 2(b), both the uncoordinated water molecules and the hydronium cation assist in linking the Yb(III) mononuclear units to one another to form the sheets. The sheets are also supported by offset face-to-face π–π interactions involving the Me2tcbp2– ligands with a 3.38 Å separation at closest contact. Each 2D sheet interacts with one further sheet to form a hydrogen bonded bilayer. A full list of hydrogen bonding interactions is displayed in Table 1.
The structures of the Gd(III) (2) and Sm(III) (3) complexes were also determined and are essentially identical to that of 1 albeit that they crystallise as dihydrates. Crystallographic details and structural diagrams are contained within the ESI.‡
Structure of {Yb2(dmcdcbp)4Cu(OMe)2(H2O)2]·6MeOH}, 4
The atomic numbering scheme and atom connectivity for 4 are shown in Fig. 3. The structure was refined in the P space group with one half of the trinuclear complex related to the other half by inversion symmetry. Crystallographic details are given in Table 2. Akin to complex 1, two ligands coordinate to the Yb(III) centre through their tetradentate chelate sites and each ‘pyridyl’ half is essentially coplanar about its central axis. The ligand mean planes subtend an angle of 88.2° to each other across the Yb(III) centre, and the coordination polyhedron around Yb1 is again best described as a distorted C2vdodecahedron. Furthermore, the peripheral 4- and 4′-carboxylic acid groups have once again been (methyl) esterified to form Me2tcbp2–.
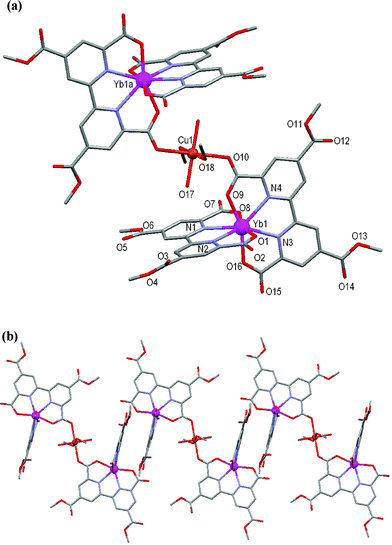 | ||
| Fig. 3 (a) Molecular structure and atomic numbering scheme for 4. Symmetry operator: a = –x + 1, –y, –z. (b) Partial packing diagram showing the formation of chains. Hydrogen atoms and lattice solvent molecules are omitted for clarity. | ||
Two Yb(III) units link to one another through a bridging Cu(II) atom that coordinates to the carboxylate oxygen atom O(10) of each unit, thereby creating a trinuclear species. The Cu(II) lies on the inversion centre. The coordination sphere of the Cu(II) atom is completed by two methanol and two water molecules arranged in a distorted octahedral geometry, with Jahn–Teller elongation evident along the O10–Cu1–O10A axis. Adjacent trinuclear units interact with each other through offset face-to-face π–π interactions involving the Me2tcbp2– ligands with a 3.36 Å separation at closest contact to form a chain, Fig. 3b.
Structure of [Ba2(tcbp)(H2O)2], 5
The atomic numbering scheme and atom connectivity for 5 are shown in Fig. 4(a) and crystallographic details are given in Table 2. The structure was refined in the monoclinic C2/c space group. Two unique Ba(II) environments are present with Ba1 and Ba2 eight and nine coordinate, respectively. The tcbp4– ligand chelates to Ba1 through its tetradentate bipyridyl–carboxylate core (N2O2) with four further oxygen atoms, from four adjacent carboxylate groups, completing its coordination sphere. Two water molecules, O1w and O2w, along with seven carboxylate oxygen atoms from six adjacent tcbp4– ligands, coordinate Ba2. Each of the carboxylate groups are twisted with respect to the pyridyl rings to which they are attached, with those about C11 and C14 twisting by ca. 28° and 1°, respectively, whereas the peripheral carboxylate groups about C12 and C13 twist by ca. 16° and 32°, respectively. Each ligand interacts with eleven Ba(II) metal centres (five Ba1 centres and six Ba2 centres) and likewise each Ba1 and Ba2 metal centre interacts with five and six tcbp4– ligands, respectively. These interactions yield a very complicated 5,6,11-connected 3D polymeric network, Fig. 3(b). The Ba2 bound water molecules participate in hydrogen bonding interactions with the carboxylate oxygen atoms O3 and O5, and the only non-coordinated carboxylate oxygen atom, O7, Fig. 3(b). Details of these hydrogen-bonding interactions within 5 are brought together in Table 1.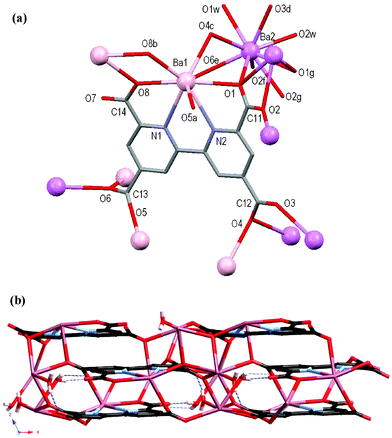 | ||
| Fig. 4 (a) Molecular structure and atomic numbering scheme for 5 with symmetry equivalents of Ba1 (pink) and Ba2 (violet) showing further coordination of the carboxylate oxygen atoms. Hydrogen atoms omitted for clarity. Symmetry operators: a = 0.5 – x, 1.5 – y, –z; b = 0.5 – x, 0.5 – y, –z; c = x, –1 + y, z; d = 1 – x, –1 + y, 0.5 – z; e = 0.5 – x, –0.5 + y, 0.5 – z; f = x, 1 – y, 0.5 + z; g = 1 – x, y, 0.5 – z. (b) Packing diagram of the 3D network of 5 viewed down the crystallographic b-axis. Hydrogen bonds are shown as dashed blue lines. | ||
Structure of [CuSr(tcbp)(H2O)3], 6
The atomic numbering scheme and atom connectivity for 6 are shown in Fig. 5(a) and crystallographic details are given in Table 2. The structure of 6 was refined in the triclinic P space group. Two metal environments, Cu1 and Sr1, are present and are five and eight coordinate, respectively. The tcbp4– ligand coordinates to Cu1 in a tridentate fashion through its bipyridyl–carboxylate core (N2O) using one, rather than two, carboxylate oxygen atoms. The smaller atomic radius of Cu(II) and its associated crystal field effects may account for this coordination mode in comparison with that observed for Ba(II) in 5. A water molecule, O2w, and one carboxylate oxygen atom, O7, from an adjacent tcbp4– ligand complete the coordination sphere about the square pyramidal Cu1. The coordination environment of Sr1 is comprised of two water molecules, O2w and O3w, and six carboxylate oxygen atoms from five different carboxylate groups. The carboxylate groups centred around C11 and C14 twist from the plane of the rings to which they are attached by ∼7° and 47°, respectively, as the former is involved in tridentate coordination to Cu(II) whilst the latter twists to ‘avoid’ coordination to Cu(II). Indeed, the peripheral carboxylate groups centred about C12 and C13 twist from the plane of the rings to which they are attached by ∼26° and 8°, respectively, as the former binds to two Sr1 centres whereas the latter coordinates to only one. The Cu1 and Sr1 metal centres interact with two and five tcbp4– ligands, respectively, and this gives rise to a 3D network, Fig. 5(b). By virtue of the presence of the coordinated water molecules, hydrogen bonding exists between them and the carboxylate oxygen atoms O1, O4, O5, O6 and O7 and also between the water molecules O1w and O2w. Details of these hydrogen-bonding interactions within 6 are brought together in Table 1.
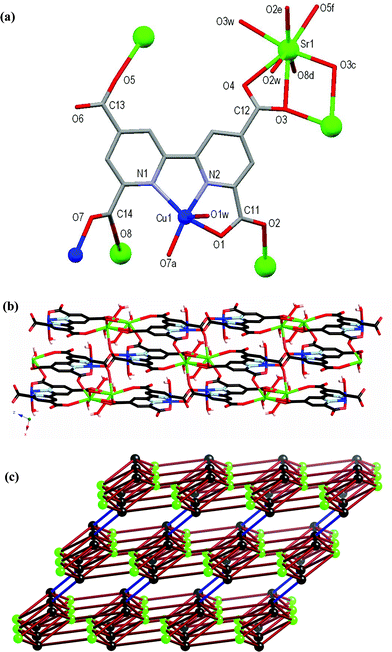 | ||
| Fig. 5 (a) Molecular structure and atomic numbering scheme for 6 with symmetry equivalents of Cu1 and Sr1 included showing further coordination of the carboxylate oxygen atoms. Hydrogen atoms omitted for clarity. Symmetry operators: a = –x, 1 – y, –z; c = 1 – x, 3 – y, 2 – z; d = 1 – x, 2 – y, 1 – z; e = –x, –y, 1 – z; f = 1 – x, 2 – y, 2 – z. (b) Packing diagram of the 3D network of 6 viewed down the crystallographic b-axis. (c) Schematic representation of the underlying network topology. Black spheres represent the tcpb4– ligands (sphere is the midpoint of the C–C bond between pyridyl rings), green spheres represent the Sr atoms, and the ligand–ligand connections via the pairs of Cu atoms are highlighted by the blue bonds. | ||
From a topological point of view, the tcbp4– ligand acts as a six connecting node and the Sr atom acts as a five-connecting node. Each Sr connects to five ligand nodes, while each ligand connects to five Sr atoms and one other ligand via a pair of Cu atoms (one coordinated to the nitrogens of one ligand and an oxygen of the other, while the other, symmetry related Cu shows the same coordination pattern in reverse). The complicated 5,6-connected network formed is shown in Fig. 5(c); the Schläfli symbol is (48.62)(48.67).1d,f
Structure of {[Ba(dcbp)(H2O)0.4]·0.6H2O}, 7
The atomic numbering scheme and atom connectivity for 7 are shown in Fig. 6(a) and crystallographic details are given in Table 2. The structure was refined in the monoclinic P21/c space group. The crystals were found to be non-merohedral twins, as confirmed by the GEMINI16 programme which found two separate twin domains. The Ba(II) centre is ten coordinate with the dcbp2– ligand chelating via its bipyridyl nitrogen atoms, in addition to seven oxygen atoms from five carboxylate groups and one water molecule (refined at 40% occupancy). A disordered water molecule resides within the lattice (refined at 60% occupancy). A ∼10° twist is evident between each ring of the dcbp2– ligand. The dcbp2– ligand interacts with six Ba(II) centres, and likewise each Ba(II) centre interacts with six dcbp2– ligands to generate the neutral 3D coordination polymer. Each of the carboxylate groups are twisted with respect to the rings to which they are attached, with those about C11 and C12 twisting by ca. 4° and 46°, respectively. Each carboxylate group coordinates in a bidentate chelating fashion and also bridges between metal centres, with that about C11 interacting with two and that about C12 with three Ba(II) ions, respectively, Fig. 6(a). The 3D coordination network contains channels that run down the crystallographic c axis, Fig. 6(b). These channels are hydrophilic in nature by virtue of the fact that the carboxylate oxygen atom O2 and coordinated water molecule O1w project into the channel. Indeed, the lattice water molecule O2w resides within this channel and is in close proximity to both O1w and O2.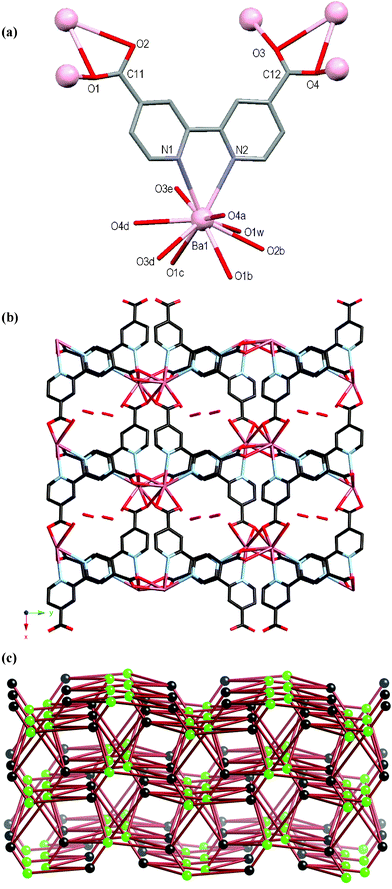 | ||
| Fig. 6 (a) Molecular structure and atomic numbering scheme for 7. Symmetry equivalents of Ba1 are included to show further coordination of the carboxylate oxygen atoms. Hydrogen atoms and lattice water molecule omitted for clarity. Symmetry operators: a = –1 – x, –y, 2 – z; b = 1 + x, y, 1 + z; c = 1 + x, 0.5 – y, 0.5 + z; d = –1 – x, 0.5 + y, 1.5 – z; e = –1 – x, –y, 1 – z. (b) Packing diagram of the 3D network as viewed down the crystallographic c-axis showing the channels with lattice water molecule included. (c) Schematic representation of the complicated underlying binodal six-connected network topology. Black spheres represent the dcbp2– ligands and green spheres represent the Ba atoms. | ||
In the underlying network both the Ba atoms and dcbp2– ligands act as six-connecting nodes, each coordinating to six of the other nodes. The two nodes are topologically different, generating a binodal six-connected network with (49.66)(410.65) topology, Fig. 6(c).
Structure of {[Sr(dcbp)]·H2O}, 8
The atomic numbering scheme and atom connectivity for 8 are shown in Fig. 7(a) and crystallographic details are given in Table 2. The structure was refined in the monoclinic Cc space group. The Sr(II) metal centre is eight coordinate with the dcbp2– ligand chelating through its bipyridyl nitrogen atoms, and six carboxylate oxygen atoms from five adjacent dcbp2– ligands. A ∼3° twist is evident between each pyridyl ring of the dcbp2– ligand. The carboxylate group about C12 coordinates in a syn–syn-bridging fashion to two Sr(II) metal centres, whereas the other about C11 coordinates in a bidentate chelating and bridging mode to three Sr(II) metal centres, Fig. 7(a). Each of the carboxylate groups are twisted with respect to the rings to which they are attached, with those about C11 and C12 twisting by ca. 41° and 19°, respectively. Each Sr(II) metal centre interacts with six dcbp2– ligands and likewise each dcbp2– ligand interacts with six Sr(II) centres to generate the neutral 3D coordination polymer, Fig. 7(b). This network contains channels that run down the crystallographic c axis and within which a disordered water molecule resides, Fig. 7(b). These channels account for 11% of the crystal volume.17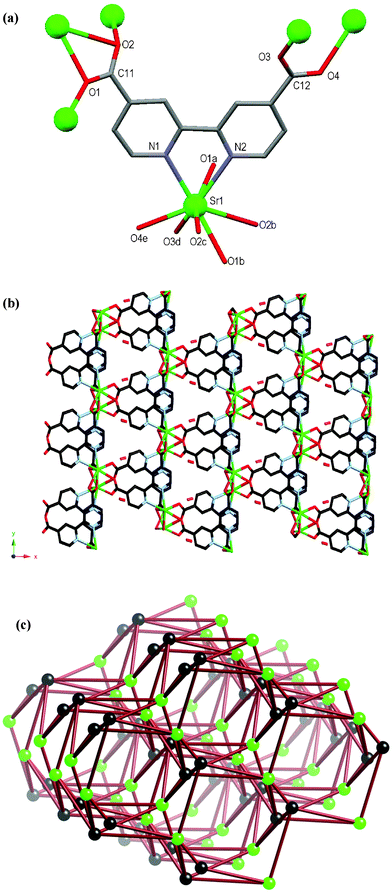 | ||
| Fig. 7 (a) Molecular structure and atomic numbering scheme for 8. Symmetry equivalents of Sr1 are included in order to highlight further coordination of the carboxylate oxygen atoms. Hydrogen atoms and lattice water molecule omitted for clarity. Symmetry operators: a = x, 1 – y, 0.5 + z; b = x, –1 + y, z; c = x, 1 – y, –0.5 + z; d = 0.5 + x, –0.5 + y, z; e = 0.5 + x, –0.5 – y, –0.5 + z. (b) Packing diagram of the 3D network as viewed down the crystallographic c-axis showing the channels with lattice water molecule included. (c) Schematic representation of the underlying six-connected network topology. Black spheres represent the dcbp2– ligands and green spheres represent the Sr atoms. Although the two nodes are represented differently, the idealised net is uninodal. | ||
The polymer can be reduced to a six-connected network by assigning the Sr atoms and dcbp2– ligands as nodes; each connects to six of the others, Fig. 7(c). Although the two nodes are chemically different, in the idealised net all nodes are equivalent, and thus the Schläfli symbol for this uninodal six-connected network is (49.66).
Thermal behaviour of 6, 7 and 8
The thermal behaviour of 6, 7 and 8 was investigated by thermogravimetric analysis (TGA) and the results from this study are displayed in Fig. 8. Insufficiently pure samples of 5 were obtained so TGA was not performed. Analysis of the TGA curve for 6 reveals an initial weight loss of ca. 9.9% between 180 < T < 200 °C consistent with the loss of the three coordinated water molecules. No further weight loss is observed until complex decomposition begins at ca. 325 °C. The TGA trace of 7 shows a gradual weight loss of ca. 4% in the temperature range 110 < T < 235 °C consistent with the loss of the lattice water molecule, followed by a further weight loss of ca. 3% between 235 < T < 470 °C, consistent with loss of the metal-bound water molecule. This is immediately followed by complex decomposition. The TGA trace for 8 displays a gradual weight loss of ca. 5.3% within the temperature range of 100 < T < 230 °C, which is consistent with loss of the lattice water molecule. Complex decomposition begins at ca. 490 °C.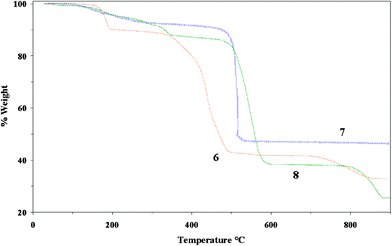 | ||
| Fig. 8 Thermogravimetric curves for 6 (red), 7 (blue) and 8 (green). | ||
The thermal stability of 7 and 8 is greater in comparison with other metal (Cd(II)6 and Mn(II)9a)–dcbp complexes, but are comparable with lanthanide metal (Sm(III))–dcbp coordination polymers (Tdecomp > 470 °C).5c This result infers that the high coordination number of the lanthanide and main group ions in combination with the dcbp ligand has a profound effect on overall thermal stability of the framework.
Conclusions
We have reported the synthesis and structural characterisation of a new ligand 4,4′,6,6′-tetracarboxy-2,2′-bipyridine (H4tcbp) [which includes a new higher yielding route to the 4,4′,6,6′-tetramethyl-2,2′-bipyridine ligand (tmbp)] and shown H4tcbp capable of coordinating to rare earth metal ions and demonstrated that the 2,2′-bipyridyl–polycarboxylate ligands, H2dcbp and H4tcbp, are capable of acting as versatile organic linkers in combination with alkaline earth metals for the generation of 3D coordination polymers. In this regard we have presented the synthesis, structural and thermal analysis of four new Sr(II), Ba(II) and mixed Sr(II)–Cu(II) examples. Utilization of the H2dcbp ligand resulted in the synthesis of 3D coordination polymers when reacted solvothermally with Ba(II) and Sr(II) which display high thermal stability. Furthermore, many various types of multi-faceted carboxylate coordination have been noted within 4–8 and these are summarised in Scheme 2. It is clear that when forming coordination polymers the increase in the number of carboxylate groups about the 2,2′-bipyridyl-scaffold, on moving from H2dcbp to H4tcbp, leads to an increase in the number of metal ions connected about the ligand viz. 6 and 6 vs. 11 and 7, for H2dcbp to H4tcbp, respectively. We are currently extending the use of these ligands to include other metals of the transition metal, main group and lanthanide series in the solvothermal synthesis of 3D coordination networks. We will report upon our recent successes in the near future.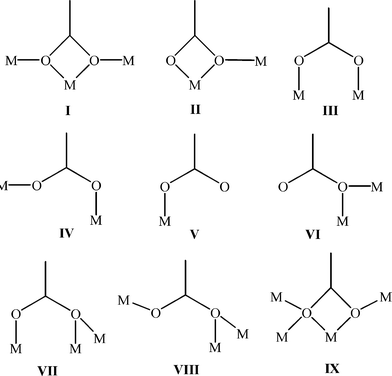 | ||
| Scheme 2 The multi-faceted modes of carboxylate coordination as observed within compounds 4 (IV), 5 (VI, VII, VIII and IX), 6 (II, III, IV and V) 7 (I and II) and 8 (I and III). | ||
Experimental
Materials and methods
Solvents and reagents were purchased from Aldrich and used as received. Water was triply distilled in house prior to use. H2dcbp10 was prepared as detailed previously. Solvothermal syntheses were carried out using a Parr Instrument general-purpose digestion bomb employing a Teflon insert with either a 23 or 45 ml capacity. IR spectra were recorded on a Perkin Elmer Spectrum One FTIR spectrometer using a universal ATR sampling accessory in the 4000–650 cm–1 region. Elemental analyses were performed at the Micro-analytical laboratories, University College Dublin. Thermogravimetric analyses were carried out on a Mettler TC 11 system under a flow of nitrogen at a heating rate of 10 °C min–1 for all measurements.Synthesis of 4,4′,6,6′-tetramethyl-2,2′-bipyridine (tmbp)
2,4-Dimethylpyridine (40 ml) was heated at reflux (160 °C) over 10% Pd/C (5 g) for 3 d. Toluene (40 ml) was added and the mixture heated at reflux for a further 1.5 h. The Pd/C was filtered off whilst hot and washed with hot toluene (2 × 30 ml). The filtrate was evaporated to dryness under vacuum and the yellow solid residue recrystallised from ethyl acetate to produce large colourless crystals. Yield 7.73 g. Mp 144–145 °C. Found: C, 78.9; H, 7.4; N, 13.3%. C14H16N2 requires: C, 79.2; H, 7.6, N, 13.2%. νmax/cm–1: 2920 w, 1598 s, 1566 m, 1443 m, 1399 m, 1371 m, 1277 w, 1185 m, 1038 m, 992 w, 943 m, 898 w, 856 s, 761 m. δH (400 MHz; CDCl3): 8.0 (s, 2H, H3,3′), 7.0 (s, 2H, H5,5′), 2.6 (s, 6H, CH3), 2.4 (s, 6H, CH3). MS (ESI): m/z 213.14 [MH]+.Synthesis of 4,4′,6,6′-tetracarboxy-2,2′-bipyridine, H4tcbp·2H2O
Adapting the method used in the synthesis of 4,4′-dicarboxy-2,2′-bipyridine by employing tmbp in place of 4,4′-dimethyl-2,2′-bipyridine gave H4tcbp in 63% yield. Recrystallisation from HNO3 yielded crystals suitable for a diffraction study. Found: C, 45.6; H, 3.0; N, 7.4%. C14H8N2O8·2H2O requires: C, 45.6; H, 3.3, N, 7.6%. νmax/cm–1: 3114 w, 2427 m, br, 1887 m, br, 1705 s, br, 1604 m, 1562 m, 1459 m, 1364 m, 1281 s, 1265 s, 1239 s, 1140 m, 1070 m, 1011 s, 992 w, 913 m, 865 m, 822 m, 765 s, 721 m, 679 s, 662 m. δH (400 MHz; D2O/NaOD): 8.50 (s, 2H, H3,3′), 8.17 (s, 2H, H5,5′).Synthesis of {[Yb(Me2tcbp)2]·H2O·MeOH·H3O}, 1, and {[Ln(Me2tcbp)2]·2H2O·H3O}, where Ln = Gd (2) or Sm (3)
The synthesis of 1–3 followed essentially the same procedure with appropriate Ln(CF3SO3)3 used in each. The synthesis for 1 is representative: Yb(CF3SO3)3 (0.074 g; 0.12 mmol) and H4tcbp·2H2O (0.088 g; 0.24 mmol) were placed in a 45 ml Teflon®-lined acid digestion bomb in MeOH (12 ml). The contents within the bomb were stirred for 10 min, sealed and heated to 150 °C and held at that temperature for 3 d before being slowly cooled (3 °C h–1) to room temperature. Colourless needle crystals suitable for a diffraction study were isolated by filtration, washed with MeOH and air-dried. Yield 0.047 g. Found: C, 41.9; H, 3.5; N, 6.1%. YbC33H29N4O19 requires: C, 41.3; H, 3.0, N, 5.8%. νmax/cm–1: 3435 w, br, 3070 m, br, 1733 m, 1643 s, 1611 s, 1567 m, 1460 m, 1440 m, 1424 m, 1396 m, 1328 m, 1311 w, 1270 s, 1251 s, 1200 m, 1152 m, 1021 m, 980 m, 923 w, 900 w, 833 w, 830 w, 813 w, 789 w, 769 m, 733 s, 708 m, 693 s.Synthesis of {Yb2(dmcdcbp)4Cu(OMe)2(H2O)2]·6MeOH} 4
Yb(CF3SO3)3 (0.074 g; 0.12 mmol), Cu(OAc)2·2H2O (0.048 g; 0.24 mmol) and H4tcbp·2H2O (0.088 g; 0.24 mmol) were placed in a 45 ml Teflon®-lined acid digestion bomb in MeOH (12 ml). The bomb was stirred for 10 min, sealed and heated to 140 °C for 3 d and before being slowly cooled (3 °C h–1) to room temperature. A mixture of prism-like crystals amongst a white powder was produced. The crystals were separated from the powder by hand under the microscope. Elemental analyses were not performed as insufficient ‘clean’ sample could be separated from the powder. νmax/cm–1: 3271 w, br, 2952 w, br, 1611 m, 1553 m, 1424 w, 1380 s, 1234 w, 1090 w, 1075 w, 1029 w, 944 w, 914 w, 873 w, 845 w, 796 w, 780 m, 728 m, 701 m, 667 w.Synthesis of [Ba2(tcbp)(H2O)2], 5
Ba(NO3)2 (0.104 g, 0.4 mmol) and H4tcbp (0.133 g, 0.4 mmol) were placed in a 45 ml Teflon®-lined digestion bomb with 4 ml water and 4 ml ethanol. The bomb was sealed and heated to 200 °C for 7 d before slowly cooling to room temperature (3 °C h–1). Colourless needle crystals, associated with an inseparable cream powder (most likely H4tcbp as indicated by IR and elemental analyses), were obtained directly. Satisfactory elemental analysis could not be obtained as sufficient ‘clean’ crystals could not be isolated. νmax/cm–1: 3341 m, br, 1653 m, 1596 m, 1571 m, 1540 s, 1422 m, 1396 m, 1376 m, 1293 w, 1229 m, 1140 w, 1114 w, 1032 m, 1002 m, 905 m, 857 w, 798 m, 779 m, 687.Synthesis of [CuSr(tcbp)(H2O)3], 6
Sr(OAc)2 (0.021 g, 0.1 mmol), Cu(OAc)2·H2O (0.02 g, 0.1 mmol) and H4tcbp (0.033 g, 0.1 mmol) were placed in a 23 ml Teflon®-lined digestion bomb with 4 ml water and 4 ml methanol. The bomb was sealed and heated to 160 °C for 4 d before slowly cooling to room temperature (3 °C h–1). Pale green needle crystals were obtained directly. Yield 0.038 g. Found: C, 31.7; H, 1.8; N, 5.0%. C14H10N2O11SrCu requires: C, 31.5; H, 1.9; N, 5.3%. νmax/cm–1: 3402 m, br, 1650 m, 1603 s, 1569 m, 1552 m, 1408 m, 1383 m, 1350, 1309 w, 1286 m, 1240 w, 1201 w, 1103 w, 1054 w, 1003 w, 921 m, 836 m, 796 w, 782 m, 740 s, 723 s, 697 s.Synthesis of {[Ba(dcbp)(H2O)0.4]·0.6H2O}, 7
Ba(NO3)2 (0.104 g, 0.4 mmol) and H2dcbp (0.098 g, 0.4 mmol) were placed in a 45 ml Teflon®-lined digestion bomb with 4 ml water and 4 ml ethanol. The bomb was sealed and heated to 200 °C for 36 h before slowly cooling to room temperature (5 °C h–1). Small colourless plate crystals were obtained directly. Yield 0.12 g. Found: C, 34.8; H, 2.2; N, 6.6%. C12H8N2O5Ba·H2O requires: C, 34.8; H, 2.4; N, 6.8%. νmax/cm–1: 3336 w, br, 1596 m, 1576 m, 1540 m, 1421 m, 1396 m, 1376 m, 1229 w, 1032 w, 1003 w, 857 w, 798 vw, 774 m, 731 w, 688 s, 662 w.Synthesis of {[Sr(dcbp)]·H2O}, 8
Sr(OAc)2 (0.021 g, 0.1 mmol) and H2dcbp (0.024 g, 0.1 mmol) were placed in a 23 ml Teflon®-lined digestion bomb with 4 ml water and 4 ml methanol. The bomb was sealed and heated to 160 °C for 4 d before slowly cooling to room temperature (5 °C h–1). Small pale yellow plate crystals were obtained directly. Yield 0.02 g. Found: C, 41.6; H, 2.4; N, 8.0%. C12H8N2O5Sr requires: C, 41.4; H, 2.3; N, 8.1%. νmax/cm–1: 2451 w, br, 1732 m, br, 1708 m, 1573 w, 1538 m, 1428 w, 1397 m, 1365 m, 1294 m, 1228 w, 1144 w, 1071 w, 1042 w, 1006 w, 910 w, 869 w, 800 w, 779 s, 765 m, 695 m, 687 s.Crystallographic measurements
Single crystal data and experimental details for H4tcbp·2H2O and 1, 4–8 are summarised in Table 2, whilst those for tmbp, 2 and 3 are contained within Table S1.‡ Single crystal analyses were performed at 153 K with a Bruker SMART APEX CCD diffractometer using graphite mono-chromated Mo-Kα radiation (λ = 0.71073 Å). A full sphere of data was obtained for each using the omega scan method. Data were collected, processed and corrected for Lorentz and polarization effects using SMART18 and SAINT-NT19 software. Absorption corrections were applied using SADABS.20 The structures were solved using direct methods and refined by full matrix least squares against F2 using the SHELXTL21 programme package. Data for the non-merohedral twinned crystals (7) were indexed using the GEMINI16 programme. The twinned data was processed using the twin output of SAINT (version 6.45) and corrected for absorption using TWINABS.22 The twinned structure 7 was solved as for single crystals using HKLF4 data for a single domain containing overlap and successfully refined on HKLF5 data with the required number of BASF parameters within the SHELXTL programme package. All non-hydrogen atoms were refined anisotropically. Aromatic hydrogen atoms were assigned to calculated positions with isotropic thermal parameters fixed at 1.2 times that of the attached carbon atom. Disorder in 1 is present at the carboxyl-oxygen atoms O6 and O13; O6 being disordered over two positions modeled at 55% and 45% occupancy, respectively with O13 being disordered over three positions modeled at 24%, 38% and 38% occupancy, respectively. Disorder is also present in 2 at the carboxyl-oxygen atom O3 and was modeled over two positions with occupancy of 50% at each position. Water hydrogen atoms were located where possible (5 and 6) from difference maps and refined with O–H distances restrained to 0.84 Å, and isotropic thermal parameters fixed at 1.5 times that of the respective oxygen atom. No significant reflections were found beyond 48° for 5 so data beyond this threshold were not collected. The lattice water molecule in 7 is disordered over two positions refined at 30% occupancy each and the coordinated water molecule was refined at 40% occupancy. The lattice water molecule in 8 is also disordered over two positions refined at 60% and 40% occupancy, respectively. Hydrogen atoms bonded to the disordered water molecules could not be modelled. To ascertain sample homogeneity 6–10 single crystals from each batch were randomly selected and separately indexed on the diffractometer. In each case the cell parameters were consistent with those found for the single crystals used in the full data collection and ensure phase purity.| Compound | H4tcbp·2H2O | 1 | 4 | 5 | 6 | 7 | 8 |
|---|---|---|---|---|---|---|---|
| a R 1 =∑||Fo| – |Fc||/∑|Fo|, wR2 = [∑w(Fo2 – Fc2)2/∑w(Fo2)2]1/2. | |||||||
| Chemical formula | C14N2H12O10 | YbC33H29N4O19 | Yb2CuC70H74N8O43 | Ba2C14H8N2O10 | CuSrC14H10N2O11 | BaC12H8N2O5 | SrC12H8N2O5 |
| Formula weight | 368.26 | 958.64 | 2124.99 | 638.90 | 533.40 | 395.53 | 347.82 |
| Crystal system | Monoclinic | Triclinic | Triclinic | Monoclinic | Triclinic | Monoclinic | Monoclinic |
| Space group | P2/c |
P |
P |
C2/c |
P |
P21/c | Cc |
| µ (MoKα)/mm–1 | 0.143 | 2.763 | 2.728 | 5.034 | 7.054 | 3.120 | 4.546 |
| a/Å | 9.065(1) | 10.2706(9) | 10.4424(6) | 27.590(1) | 9.2075(6) | 9.696(1) | 17.284(2) |
| b/Å | 7.164(1) | 10.8551(9) | 12.9331(7) | 9.7108(4) | 9.6199(6) | 19.241(2) | 9.9314(8) |
| c/Å | 11.730(2) | 16.487(1) | 15.4313(9) | 13.2117(5) | 10.2189(7) | 7.2146(7) | 7.0022(6) |
| α/° | 90 | 99.169(2) | 79.426(1) | 90 | 107.787(1) | 90 | 90 |
| β/° | 102.472(3) | 103.656(2) | 87.329(2) | 117.092(1) | 91.857(1) | 107.827(3) | 97.462(2) |
| γ/° | 90 | 94.272(2) | 74.204(1) | 90 | 115.389(1) | 90 | 90 |
| V/Å3 | 743.2(2) | 1751.2(3) | 1971.3(2) | 3151.3(2) | 764.46(9) | 1281.3(2) | 1191.7(2) |
| Z | 2 | 2 | 1 | 8 | 2 | 4 | 4 |
| D c/g cm–3 | 1.644 | 1.818 | 1.790 | 2.693 | 2.317 | 2.050 | 1.939 |
| T/K | 153(2) | 153(2) | 153(2) | 153(2) | 153(2) | 153(2) | 153(2) |
| 2θmax | 50.00 | 48.00 | 50.00 | 59.96 | 60.94 | 52.00 | 49.96 |
| Min/Max trans. factor | 0.7193/1.0000 | 0.7903/1.0000 | 0.7718/1.0000 | 0.7531/1.000 | 0.5935/1.000 | 0.7684/1.000 | 0.7061/1.000 |
| R int | 0.0350 | 0.0488 | 0.0529 | 0.0255 | 0.0244 | 0.000 | 0.031 |
| R 1, wR2 [I > 2σ(I)]a | 0.0430, 0.1099 | 0.0453, 0.0909 | 0.0436, 0.0974 | 0.0277, 0.0547 | 0.0336, 0.0769 | 0.0694, 0.1825 | 0.0263, 0.0577 |
| R 1, wR2 (all data) | 0.0659, 0.1221 | 0.0616, 0.0958 | 0.0635, 0.1052 | 0.0255, 0.0588 | 0.0445, 0.0908 | 0.0746, 0.1864 | 0.0287, 0.0588 |
| Reflections: collected | 5487 | 12476 | 15903 | 16970 | 8484 | 4782 | 4546 |
| unique | 1314 | 5501 | 6935 | 4491 | 4256 | 4782 | 2075 |
| observed | 937 | 4586 | 5519 | 4201 | 3564 | 4292 | 1977 |
Acknowledgements
This publication has emanated from research conducted with the financial support of Science Foundation Ireland, which is gratefully acknowledged. We also thank Mr Trevor Woods, School of Physics (TCD), for assistance with TGA studies.References
- (a) C. Janiak, Dalton Trans., 2003, 2781 RSC; (b) R. Robson, J. Chem. Soc., Dalton Trans., 2000, 3735 RSC; (c) B. Moulton and M.J. Zaworotko, Chem. Rev., 2001, 101, 1629 CrossRef CAS; (d) S.R. Batten and R. Robson, Angew. Chem., Int. Ed., 1998, 37, 1460 CrossRef; (e) B. J. Holliday and C. A. Mirkin, Angew. Chem., Int. Ed., 2001, 40, 2022 CrossRef CAS; (f) S. R. Batten, CrystEngComm, 2001, 3, 67 RSC; (g) M. D. Hollingsworth, Science, 2002, 295, 2410 CAS; (h) G. R. Desiraju, J. Mol. Struct., 2003, 656, 5 CrossRef CAS; (i) S. Kitagawa, R. Kitaura and S.-I. Noro, Angew. Chem., Int. Ed., 2004, 43, 2334 CrossRef CAS; (j) L. Brammer, Chem. Soc. Rev., 2004, 33, 476 RSC; (k) B. Conerney, P. Jensen, P. E. Kruger, B. Moubaraki and K. S. Murray, CrystEngComm, 2003, 5, 454 RSC; (l) P. E. Kruger, H. Grove, M. Julve, F. Lloret, K. W. Tornroos and J. Sletten, Inorg. Chim. Acta, 2001, 325, 115 CrossRef CAS; (m) G. J. Halder, C. J. Kepert, B. Moubaraki, K. S. Murray and J. D. Cashion, Science, 2002, 298, 1762 CrossRef CAS.
- (a) O. M. Yaghi, C. E. Davis, G. Li and H. Li, J. Am. Chem. Soc., 1997, 119, 2861 CrossRef CAS; (b) R. Kitaura, K. Fujimoto, S. Noro, M. Kondo and S. Kitagawa, Angew. Chem., 2002, 114, 141 CrossRef; (c) B. Chen, C. Liang, J. Yang, D. S. Contreras, Y. L. Clancy, E. B. Lobkovsky, O. M. Yaghi and S. Dai, Angew. Chem., Int. Ed., 2006, 45, 1390 CrossRef CAS; (d) B. Kesanli and W. B. Lin, Coord. Chem. Rev., 2003, 246, 305 CrossRef CAS; (e) J. S. Seo, D. Whang, H. Lee, S. I. Jun, J. Oh, Y. J. Jeon and K. Kim, Nature, 2000, 404, 982 CrossRef CAS; (f) R. Custelcean, T. J. Haverlock and B. A. Moyer, Inorg. Chem., 2006, 45, 6446 CrossRef CAS.
- (a) M. O'Keeffe, Chem. Eng. News, 2005, 83, 42; (b) A. R. Millward and O. M. Yaghi, J. Am. Chem. Soc., 2005, 127, 17998 CrossRef CAS; (c) J. L. C. Roswell and O. M. Yaghi, Angew. Chem., Int. Ed., 2005, 44, 4670 CrossRef CAS; (d) N. L. Rosi, J. Eckert, M. Eddaoudi, D. T. Vodak, J. Kim, M. O'Keeffe and O. M. Yaghi, Science, 2003, 300, 1127 CrossRef CAS; (e) M. Eddaoudi, J. Kim, N. Rosi, D. T. Vodak, J. Wachter, M. O'Keeffe and O. M. Yaghi, Science, 2002, 295, 469 CrossRef; (f) B. Chen, N. W. Ockwig, A. R. Millward, D. S. Contreras and O. M. Yaghi, Angew. Chem., Int. Ed., 2005, 44, 4745 CrossRef CAS; (g) S. Kitagawa, Nature, 2006, 441, 584 CrossRef CAS; (h) D. N. Dybtsev, H. Chun and K. Kim, Angew. Chem., Int. Ed., 2004, 43, 5033 CrossRef CAS; (i) D. N. Dybtsev, H. Chun, S. H. Yoon, D. Kim and K. Kim, J. Am. Chem. Soc., 2004, 126, 32 CrossRef CAS; (j) L. Pan, M. B. Sander, X. Huang, J. Li, M. Smith, E. Bittner, B. Bockrath and J. K. Johnson, J. Am. Chem. Soc., 2004, 126, 1308 CrossRef CAS; (k) Y. Kubota, M. Takata, R. Matsuda, R. Kitaura, S. Kitagawa, K. Kato, M. Sakata and T. C. Kobayashi, Angew. Chem., Int. Ed., 2005, 44, 920 CrossRef CAS; (l) L. Pan, B. Parker, X. Huang, D. H. Olson, J. Y. Lee and J. Li, J. Am. Chem. Soc., 2006, 128, 4180 CrossRef CAS; (m) L. Pan, K. M. Adams, H. E. Hernandez, X. Wang, C. Zheng, Y. Hattori and K. Kaneko, J. Am. Chem. Soc., 2003, 125, 3062 CrossRef CAS; (n) E. J. Cussen, J. B. Claridge, M. J. Rosseinsky and C. J. Kepert, J. Am. Chem. Soc., 2002, 124, 9574 CrossRef CAS; (o) D. Bradshaw, T. J. Prior, E. J. Cussen, J. B. Claridge and M. J. Rosseinsky, J. Am. Chem. Soc., 2004, 126, 6106 CrossRef CAS; (p) H. Chun, D. N. Dybtsev, H. Kim and K. Kim, Chem.–Eur. J., 2005, 11, 3521 CrossRef CAS; (q) Y. Li and R. T. Yang, J. Am. Chem. Soc., 2006, 128, 8136 CrossRef CAS.
- (a) D. Bradshaw, J. B. Claridge, E. J. Cussen, T. J. Prior and M. J. Rosseinsky, Acc. Chem. Res., 2005, 38, 273 CrossRef CAS; (b) M. Fujita, Y. J. Kwon, S. Washizu and K. Ogura, J. Am. Chem. Soc., 1994, 116, 1151 CrossRef CAS; (c) S.-H. Cho, B. Ma, S. T. Nguyen, J. T. Hupp and T. E. Albrecht-Schmitt, Chem. Commun., 2006, 2563 RSC; (d) C.-D. Wu, A. Hu, L. Zhang and W.B. Lin, J. Am. Chem. Soc., 2005, 127, 8940 CrossRef CAS.
- (a) T. M. Reineke, M. Eddaoudi, M. Fehr, D. Kelley and O. M. Yaghi, J. Am. Chem. Soc., 1999, 121, 1651 CrossRef CAS; (b) H.-L. Gao, L. Yi, B. Ding, H.-S. Wang, P. Cheng, D.-Z. Liao and S.-P. Yan, Inorg. Chem., 2006, 45, 481 CrossRef CAS; (c) J.-Y. Wu, T.-T. Yeh, Y.-S. Wen, J. Twu and K.-L. Lu, Cryst. Growth Des., 2006, 6, 467 CrossRef CAS; (d) H.-L. Gao, L. Yi, X.-Q. Zhao, P. Cheng, D.Z. Liao and S.-P. Yan, Inorg. Chem., 2006, 45, 5980 CrossRef CAS; (e) R. Cao, D. Sun, Y. Liang, M. Hong, K. Tatsumi and Q. Shi, Inorg. Chem., 2002, 41, 2087 CrossRef CAS; (f) S. Varughese and V.R. Pedireddi, Chem. Commun., 2005, 1824 RSC; (g) B. D. Chandler, D. T. Cramb and G. K. H. Shimizu, J. Am. Chem. Soc., 2006, 128, 10403 CrossRef CAS; (h) L. Pan, E. B. Woodlock, X. Wang and C. Zheng, Inorg. Chem., 2000, 39, 4174 CrossRef; (i) O. Guillou, C. Daiguebonne, M. Camara and N. Kerbellec, Inorg. Chem., 2006, 45, 8468 CrossRef CAS; (j) T. Devic, C. Serre, N. Audebrand, J. Marrot and G. Férey, J. Am. Chem. Soc., 2005, 127, 12788 CrossRef CAS; (k) C. L. Cahill, D. T. de Lill and M. Frisch, CrystEngComm, 2007, 9, 15 RSC; (l) L. Pan, T. Frydel, M. B. Sander, X. Huang and J. Li, Inorg. Chem., 2001, 40, 1271 CrossRef CAS; (m) J. P. Leonard, P. Jensen, T. McCabe, J. E. O'Brien, R. D. Peacock, P. E. Kruger and T. Gunnlaugsson, J. Am. Chem. Soc., 2007, 129, 10986 CrossRef CAS.
- Y.-H. Lie, Y.-L. Lu, H.-C. Wu, J.-C. Wang and K.-L. Lu, Inorg. Chem., 2002, 41, 2592 CrossRef CAS.
- (a) E. Tynan, P. Jensen, P. E. Kruger, A. C. Lees and M. Nieuwenhuyzen, Dalton Trans., 2003, 1223 RSC; (b) C. J. Matthews, M. R. J. Elsegood, G. Bernardinelli, W. Clegg and A. F. Williams, Dalton Trans., 2004, 492 RSC; (c) R. P. Doyle, P. E. Kruger, M. Julve, F. Lloret and M. Nieuwenhuyzen, CrystEngComm, 2002, 4, 13 RSC; (d) C. B. Aakeroy and A. M. Beatty, Aust. J. Chem., 2001, 54, 409 CrossRef CAS; (e) C. B. Aakeroy, A. M. Beatty and D. S. Leinen, Angew. Chem., Int. Ed., 1999, 38, 1815 CrossRef CAS.
- (a) E. Tynan, P. Jensen, N. R. Kelly, P. E. Kruger, A. C. Lees, B. Moubaraki and K. S. Murray, Dalton Trans., 2004, 3440 RSC; (b) E. Tynan, P. Jensen, P. E. Kruger and A. C. Lees, Chem. Commun., 2004, 776 RSC; (c) E. Tynan, P. Jensen, A. C. Lees, B. Moubaraki, K. S. Murray and P. E. Kruger, CrystEngComm, 2005, 7, 90 RSC; (d) P. E. Kruger, R. P. Doyle, M. Julve, F. Lloret and M. Nieuwenhuyzen, Inorg. Chem., 2001, 40, 1726 CrossRef.
- (a) T. Schareina, C. Schick and R. Kempe, Z. Anorg. Allg. Chem., 2001, 627, 131 CrossRef CAS; (b) T. Schareina, C. Schick, B. F. Abrahams and R. Kempe, Z. Anorg. Allg. Chem., 2001, 627, 1711 CrossRef CAS.
- A. R. Oki and R. J. Morgan, Synth. Commun., 1995, 25, 4093 CrossRef CAS.
- R. H. Linnell, J. Org. Chem., 1957, 22, 1691 CrossRef CAS.
- L. Della-Ciana, W. J. Dressick and A. von Zelewsky, J. Heterocycl. Chem., 1990, 27, 163 Search PubMed.
- M. C. Etter, Acc. Chem. Res., 1990, 23, 120 CrossRef CAS.
- We have often noted that crystallisation is assisted when mixed-metal salts are used when ‘neat’ metal salts fail: N. R. Kelly, Synthesis and Structural Analysis of Mononuclear to Polymeric 2,2′–Bipyridyl Lanthanide, Main Group and Transition Metal Complexes, Ph.D. Thesis, University of Dublin, 2007.
- J. C. G. Bunzli, L. J. Charboniere and R. F. Ziessel, J. Chem. Soc., Dalton Trans., 2000, 1917 RSC.
- GEMINI, Autoindexing Program for Twinned Crystals, Version 1.2 Release 05/2000, Bruker-AXS Inc., Madison, WI, 1999 Search PubMed.
- (a) A. L. Spek, PLATON – A Multipurpose Crystallographic Tool Utrecht University, Utrecht, The Netherlands, 2005 (v1.08) Search PubMed.
- Bruker SMART, Version 5.629, Bruker-AXS Inc., Madison, WI, 1997–2003 Search PubMed.
- Bruker SAINT-NT, Version 6.45, Bruker-AXS Inc., Madison, WI, 1997–2003 Search PubMed.
- SADABS, Program within Bruker SAINT-NT, Version 6.45, Bruker-AXS Inc., Madison, WI, 1997–2003 Search PubMed.
- G. M. Sheldrick, SHELXTL, Version 5.1, Bruker-AXS Inc., Madison, WI, 1998 Search PubMed.
- TWINABS, Version Beta Test, Bruker-AXS Inc., Madison, WI, 2003 Search PubMed.
Footnotes |
| † CCDC reference numbers 648001–648004 and 655787–655789. For crystallographic data in CIF or other electronic format see DOI: 10.1039/b711469a |
| ‡ Electronic supplementary information (ESI) available: Single crystal data for tmbp, 2 and 3; structural description, molecular structure and atomic numbering scheme for tmbp, 2 and 3. See DOI: 10.1039/b711469a |
| This journal is © The Royal Society of Chemistry 2008 |