DOI:
10.1039/B709938J
(Paper)
CrystEngComm, 2008,
10, 54-67
Evaluation of the interchangeability of C–H and C–F groups: insights from crystal packing in a series of isomeric fluorinated benzanilides†‡
Received
29th June 2007
, Accepted 17th August 2007
First published on 6th September 2007
Abstract
A series of mono- and difluorinated benzanilides have been synthesized and characterized to unravel the importance of interactions involving organic fluorine. The effect of fluorine substitution on the molecular conformation and the solid-state organization of the molecules in the crystalline lattice have been discussed in terms of changes in molecular conformation and the supramolecular aggregation. It is noteworthy that the meta- and para-isomers (mono and difluorinated) are isostructural, with the ortho-isomers possessing minor conformational variations. Furthermore, the molecular conformation as obtained by theoretical DFT calculations is different from the solid-state conformation highlighting the importance of co-operative features brought by N–H⋯O and C–H⋯O hydrogen bonds, along with weak intermolecular interactions involving organic fluorine and aromatic C–H⋯π contacts in the solid state.
Introduction
Non-covalent forces play an important role in processes like folding and self-assembly.1 In recent years, chemists have been engaged in the development of foldamers2 and artificial molecules that utilize non-covalent forces to modulate folding or helical architectures. In this regard the strong directional characteristics associated with hydrogen bonds like O–H⋯O,3 N–H⋯O4,5 and N–H⋯N6,7 are highly effective in the assembly of supramolecular structures. Recently there has been a lot of interest invoked in fluorine containing compounds in the field of medicine and agrochemicals.8 Although it has been well established that the fluoride ion acts as a very strong proton acceptor9,10 it has been reported that covalently bonded fluorine is a very weak intermolecular hydrogen-bond acceptor.11,12 Intermolecular interactions in fluoro-aromatic molecules such as C–H⋯F, C–F⋯π and C–F⋯F–C contacts have been the focus of recent structural, photophysical, topochemical, polymorphism and DSC studies.13–17 Amongst intermolecular interactions involving fluorine, C–H⋯F bonds have directional characteristics and are stabilized by electrostatic forces,13 C–F⋯π interaction14 is considered as destabilizing and F⋯F interactions are ubiquitous16 and recent studies in polymorphism16b unravel the importance of these weak but important interactions. Most of the previously reported studies on such C–H⋯F–C hydrogen bonds have been limited to a selection of compounds where potential strong hydrogen bond donors and acceptors are absent.18 However it has been reported that weak intermolecular interactions involving halogens are also observed in the presence of strong hydrogen bonds like O–H⋯O and N–H⋯O.19 In addition to steric and electrostatic factors, two other effects could determine the changes involved depending on the type of fluorine substitution pattern. The first is its electron withdrawing effect, which increases the acidity of close hydrogen atoms thus enhancing their capacity to participate in hydrogen bonding, and the second is the ability of the fluorine atom to participate in weak interactions. The latter aspect may be of particular relevance, and although it is documented that weak fluorine based interactions are easily overridden by the stronger heteroatom based intermolecular interactions, cases have been reported in which the crystal structure is controlled by weak interactions of the type C–H⋯F–C.20 Furthermore, comparison of the structures of several of the fluorinated cyclopropanes with those of the non-fluorinated counterparts revealed that close intermolecular contacts of fluorine substituents with hydrogen are not solely due to crystal packing effects but are caused by weak X–H⋯F–C hydrogen bridges.21 Recently it has been observed that intra-molecular N–H⋯F hydrogen bonding could be used to promote the stability of hydrazide-based quadruple hydrogen bonded hetero-dimers.22 Recent studies highlight that rationally designed crescent and helical conformations of aromatic amides utilizing intra-molecular N–H⋯F hydrogen bonding might provide a novel approach to the formation of rigid and well-established conformations.23 All these studies no doubt improve the understanding of crystal packing in fluorine containing molecules, but the fact that such studies have been carried out on diverse molecular skeletons makes a proper analysis of various intermolecular interactions somewhat tedious.
Furthermore, fluorinated organic compounds are widely used in the synthesis of pharmaceuticals and agrochemicals. Due to their favourable chemical and biological properties such as solubility and bioavailability, they enhance the lipophilicity and thus increase the rate of cell penetration and transport of a drug to an active site.24 In general, the introduction of a fluorine atom in a molecule does not result in a considerable change in the size or the shape of the compound. Due to the minimal conformational effects, the fluorinated molecule generally also fits in the active site of the receptor and the fluorine atom is thus a suitable isosteric substitution of hydrogen.25 The C–F bond is stronger than the original C–H bond and fluorinated compounds are more resistant to metabolic degradation. The importance of fluorinated aromatic amino acids such as 3-fluorotyrosine (3-FTyr) and 5- and 6-fluorotryptophan are amongst few unnatural amino acids that can be incorporated into proteins by microorganisms.26 An aromatic C–F bond is longer and the direction of the bond dipole is opposite to that of the C–H bond. Also fluorine is a weak hydrogen bond acceptor. It is reported that the change in conformation of 3-FTyr accompanying fluorine substitution is not yet understood and the ability of fluorine to participate in the formation of O–H and N–H hydrogen bonds is significant.27
With this background we reasoned that if such fluorine based interactions are ideally examined in a family of crystal structures by gradually increasing the extent of fluorination in molecules, then their structural diversity and interplay amongst the variety of intermolecular interactions can be analyzed on an invariant molecular scaffold. In this regard we have synthesized and characterized a small library of mono- and difluorinated isomeric benzanilides to evaluate the effect that fluorine substitution has on the supramolecular packing of molecules and its role in organizing molecules in the solid state. This class of compounds has been used actively as drugs. Recent patents highlight the unique role of such substituted benzanilides, which have been used as CCR5 receptor ligands, anti-inflammatory agents and antiviral agents.28 Furthermore these find application in the study of voltage dependent potassium channel openers.29
Experimental: synthesis and crystal growth
The starting materials [fluoro benzoyl chlorides (o, m, p), fluoroanilines (o, m, p), dimethylaminopyridine (DMAP)] were all purchased from Sigma Aldrich (Analytical Grade). The amines were further purified by distillation while the benzoyl chlorides were used directly from the bottle. The solvents used for crystallization were of Analytical Grade. Dichloromethane used for the reaction was dried using calcium hydride and stored over molecular sieves.
The fluorinated amines and corresponding fluorinated benzoyl chlorides were taken in a round bottomed flask (Scheme S1‡), in the presence of DMAP and dry DCM and cooled to 0 °C in an ice bath (Table S1‡). The reaction was allowed to take place for a period of 10–12 h and the completion of the reaction was monitored using TLC. At the end of the reaction, the compound was isolated by solvent-extraction procedure using dichloromethane, dried using sodium sulfate and finally purified by column chromatography using silica by eluting with EtOAc (ethyl acetate)/hexane. IR (Table S2‡) and melting point measurements techniques were used (Table S3‡) for characterization of the synthesized compounds.
Single crystals of desired size and quality were grown by slow evaporation at 0 and 25 °C by dissolving the substance in different solvent combinations like DCM/hexane, EtOAc/hexane and acetone/hexane, respectively. In most of the cases the quality of crystals obtained were not good. Morphologies ranging from thin plates to fibrous needles were obtained in a majority of cases. It is of interest to note that in the article,23 the researchers made repeated attempts to crystallize some of the difluorinated benzanilides (sample code in our study: DC7, DC9, DC13, DC15) for studying the N–H⋯F hydrogen bonding but were unsuccessful. In our studies, we could crystallise DC7 and DC15. Further we also observed concomitant polymorphism in DC9.16b
Single crystal X-ray diffraction data were collected on a Bruker AXS SMART APEX CCD diffractometer using graphite monochromated Mo Kα (λ = 0.7107 Å) radiation at 290(2) K and the intensities were measured using a ω scan with a scan-width of 0.3°. A total of 606 frames per set were collected in different settings of φ (φ = 0, 90 and 180° if the system is monoclinic; φ = 0, 90, 180 and 270° if it is triclinic) keeping the sample-to-detector distance of 6.03 cm. Crystal data were reduced by SAINTPLUS30 and an empirical absorption correction was applied using the package SADABS30 available in the Bruker software package. All the crystal structures were solved by direct methods using SIR9231 and refined by the full matrix least squares method using SHELXL9732 present in the program suite WinGX (Version 1.63.04a).33 ORTEP diagrams of all compounds were generated using ORTEP3234 and packing diagrams were generated using CAMERON35 and Mercury software. Geometrical calculations were done using PARST36 and PLATON.37
DC1: R1 = F; R2 = R3 = R1′ = R2′ = R3′ = H; DC2: R2 = F; R1 = R3 = R1′ = R2′ = R3′ = H; DC3: R3 = F; R1 = R2 = R1′ = R2′ = R3′ = H; *DC4: R1′ = F; R1 = R2 = R3 = R2′ = R3′ = H; DC5: R1 = R2 = R3 = R1′ = R3′ = H; R2′ = F; DC6: R1 = R2 = R3 = R1′ = R2′ = H; R3′ = F; DC7: R1 = R1′ = F; R2 = R3 = R2′ = R3′ = H; *DC8: R1′ = R2 = F; R1 = R3 = R2′ = R3′ = H;#DC9: R3 = R1′ = F; R1 = R2 = R2′ = R3′ = H; DC10: R1 = R2′ = F; R2 = R3 = R1′ = R3′ = H; #DC11: R1 = R3 = R1′ = R3′ = H; R2 = R2′ = F; DC12: R3 = R2′ = F; R1′ = R1 = R2 = R3′ = H; *DC13: R1 = R3′ = F; R2 = R3 = R1′ = R2′ = H; *DC14 : R2 = R3′ = F; R1 = R3 = R1′ = R2′ = H;DC15: R3 = R3′ = F; R1 = R2 = R1′ = R2′ = H;
Results
Table 1 lists the relevant crystallographic and refinement data. Relevant dihedral angles obtained from experiment and from DFT calculations at the B3LYP/6-31G** level are given in Table 2. DC 1–6 is mono-fluorinated (isomeric) and DC 7–15 are di-fluorinated (isomeric) molecules. The symbol * (Scheme 1) indicates that these compounds do not yield crystals suitable for single crystal X-ray diffraction and # indicates that dimorphs were obtained and their structures have been analyzed and reported elsewhere.16b,c All crystal structures have been analyzed by considering the distances for H⋯O, H⋯F, H⋯C (sp2) to be greater than the van der Waals limit and an angular cut-off greater than 110° has been adopted. Table 3 lists all relevant intra- and intermolecular interactions in all structures.
 |
| | Scheme 1 Chemical scheme of the compounds studied. | |
Table 1 Crystallographic and refinement data
|
DATA
|
DC1 |
DC2 |
DC3 |
|
Mixed: Amino hydrogen located and refined isotropically, remaining atoms fixed geometrically. Refined all: All the hydrogen atoms were located and refined isotropically.
|
| Crystal size/mm |
0.22 × 0.11 × 0.07 |
0.36 × 0.41 × 0.09 |
0.25 × 0.19 × 0.05 |
|
Solvent
|
EtOAc/Hexane |
EtOAc/Hexane |
EtOAc/Hexane |
| Formula |
C13 H10 N1 O1 F1 |
C13 H10 N1 O1 F1 |
C13 H10 N1 O1 F1 |
| Formula weight/g mol–1 |
215.2 |
215.2 |
215.2 |
| Temperature/K |
290(2) |
290(2) |
290(2) |
| Wavelength/Å |
0.7107 |
0.7107 |
0.7107 |
| Crystal system |
Monoclinic |
Monoclinic |
Triclinic |
| Space group |
P21 |
P21/c |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
|
a/Å |
5.421(2) |
8.036(2) |
5.349(3) |
|
b/Å |
6.258(2) |
5.360(9) |
7.599(4) |
|
c/Å |
15.534(4) |
23.640(5) |
12.945(6) |
|
α/° |
90 |
90 |
92.124(8) |
|
β/° |
97.454(5) |
90.074(4) |
101.179(7) |
|
γ/° |
90 |
90 |
90.162(8) |
| Volume/Å3 |
522.5(2) |
1018.2(4) |
515.8(5) |
|
Z
|
2 |
4 |
2 |
| Density/g cm–3 |
1.37 |
1.40 |
1.39 |
|
µ/mm–1 |
0.099 |
0.102 |
0.101 |
|
F(000)
|
224 |
443.9 |
224 |
|
θ
min,max
|
2.6, 25.0 |
1.7, 27.2 |
1.6, 20.8 |
| hmin,max, kmin,max, lmin,max |
(–6,6), (–7,7), (–18,18) |
(–9,9), (–6,6), (–28,28) |
(–5,5), (–7,7), (–12,12) |
| Treatment of hydrogensa |
Mixed |
Mixed |
Refined all |
|
No of reflns measured |
3606 |
6666 |
3381 |
|
No of unique reflns |
1783 |
1761 |
1082 |
|
No of parameters |
149 |
168 |
185 |
|
R_all, R_obs |
0.127, 0.063 |
0.061, 0.071 |
0.047, 0.032 |
|
wR
2_all, wR2_obs |
0.111, 0.093 |
0.142, 0.136 |
0.112, 0.082 |
| Δρmin,max/eÅ–3 |
–0.11, 0.12 |
–0.16, 0.24 |
–0.16, 0.16 |
| GoF |
1.001 |
1.132 |
1.222 |
|
CRYSTAL
DATA
|
DC5 |
DC6 |
DC7 |
| Crystal size |
0.40 × 0.30 × 0.20 |
0.20 × 0.18 × 0.08 |
0.58 × 0.53 × 0.13 |
|
Solvent
|
EtOAc/Hexane |
EtOAc/Hexane |
EtOAc/Hexane |
| Formula |
C13 H10 N1 O1 F1 |
C13 H10 N1 O1 F1 |
C13 H9 N1 O1 F2 |
| Formula weight/gm mol–1 |
215.2 |
215.2 |
233.2 |
| Temperature/K |
290(2) |
290(2) |
290(2) |
| Wavelength/Å |
0.7107 |
0.7107 |
0.7107 |
| Crystal system |
Monoclinic |
Triclinic |
Orthorhombic |
| Space group |
P21/c |
P |
P
212121 |
|
a/Å |
8.069(2) |
5.369(1) |
5.186(9) |
|
b/Å |
5.391(1) |
7.862(2) |
5.867(2) |
|
c/Å |
23.238(6) |
12.892(2) |
35.127(9) |
|
α/° |
90 |
107.47(3) |
90 |
|
β/° |
90.17(4) |
99.49(3) |
90 |
|
γ/° |
90 |
90.71(3) |
90 |
| Volume/Å3 |
1011.0(4) |
511.24(6) |
1068.9(5) |
|
Z
|
4 |
2 |
4 |
| Density/g cm–3 |
1.41 |
1.40 |
1.45 |
|
µ/mm–1 |
0.103 |
0.101 |
0.116 |
|
F(000)
|
447.9 |
224 |
479.9 |
|
θ
min,max
|
1.8, 27.3 |
2.7, 27.4 |
1.2, 25.0 |
| hmin,max, kmin,max, lmin,max |
(–10,9), (–6,6), (–28,28) |
(–6,6), (–8,9), (–15,11) |
(–6,6), (–7,7), (–42,44) |
| Treatment of hydrogensa |
Refined all |
Refined all |
Mixed |
|
No of reflns measured |
7141 |
3399 |
7182 |
|
No of parameters |
185 |
185 |
158 |
|
R_all, R_obs |
0.065, 0.052 |
0.071,0.063 |
0.089, 0.076 |
|
wR
2_all, wR2_obs |
0.129, 0.121 |
0.120, 0.116 |
0.135, 0.130 |
| Δρmin,max/eÅ–3 |
–0.18, 0.22 |
–0.18, 0.19 |
–0.24, 0.35 |
| GoF |
1.022 |
1.244 |
1.090 |
|
CRYSTAL DATA |
DC10 |
DC12 |
DC15 |
| Formula |
C13 H9 N1 O1 F2 |
C13 H9 N1 O1 F2 |
C13 H9 N1 O1 F2 |
| Size |
0.57 × 0.38 × 0.08 |
0.45 × 0.43 × 0.05 |
0.53 × 0.34 × 0.09 |
|
Solvent
|
EtOAc/Hexane |
EtOAc/Hexane |
EtOAc/Hexane |
| Formula weight/gm mol–1 |
233.2 |
233.2 |
233.2 |
| Temperature/K |
290(2) |
290(2) |
290(2) |
| Radiation |
Mo Kα |
Mo Kα |
Mo Kα |
| Wavelength/Å |
0.7107 |
0.7107 |
0.7107 |
| Crystal system |
Orthorhombic |
Monoclinic |
Triclinic |
| Space group |
Pca21 |
P21/c |
P |
|
a/Å |
8.244(2) |
12.235(2) |
5.460(2) |
|
b/Å |
5.203(1) |
10.879(9) |
7.693(3) |
|
c/Å |
24.660(5) |
7.865(9) |
13.038(5) |
|
α/° |
90 |
90 |
106.758(6) |
|
β/° |
90 |
90.232(2) |
100.371(6) |
|
γ/° |
90 |
90 |
90.016(6) |
| Volume/Å3 |
1057.8(4) |
1046.8(2) |
515.0(3) |
|
Z
|
4 |
4 |
2 |
| Density/g cm–3 |
1.46 |
1.48 |
1.50 |
|
µ/mm–1 |
0.117 |
0.119 |
0.120 |
|
F(000)
|
479.9 |
479.9 |
240.0 |
|
θ
min,max
|
1.6, 25.0 |
1.7, 26.4 |
1.7, 25.4 |
| hmin,max, kmin,max, lmin,max |
(–29,28), (–6,6), (–9,9) |
(–15,15), (–12,13), (–9,9) |
(–6,6), (–9,9), (–15,15) |
| Treatment of hydrogensa |
Mixed |
Refined all |
Refined all |
|
No of reflns measured |
6748 |
7786 |
3767 |
|
No. unique reflns |
1796 |
2051 |
1862 |
|
No of parameters |
155 |
190 |
190 |
|
R_all, R_obs |
0.126, 0.085 |
0.068, 0.050 |
0.079, 0.061 |
|
wR
2_all, wR2_obs |
0.149, 0.136 |
0.100,0.093 |
0.173, 0.162 |
| Δρmin,max/eÅ–3 |
–0.24, 0.24 |
–0.23, 0.19 |
–0.24, 0.45 |
| GoF |
1.148 |
1.149 |
1.059 |
Table 2 Dihedral angles formed by the least squares planes. Plane 1: C1/C6, Plane 2: C13/N1/O1, Plane 3: C7/C12
| Angle |
Planes 1 & 2 |
Planes 1 & 3 |
Planes 2 & 3 |
|
The values in italics are obtained from theoretical B3LYP/6-31G** calculations.
|
| DC1/° |
28.3(2) |
5.2(2) |
23.0(3) |
| |
179.98
a
|
179.98
|
0.04
|
| DC2/° |
30.5(2) |
62.0(1) |
31.9(2) |
| |
26.12
|
178.98
|
4.46
|
| DC3/° |
29.5(1) |
62.2(1) |
32.9(1) |
| |
25.65
|
178.77
|
5.09
|
| DC5/° |
31.0(1) |
62.2(1) |
31.6(1) |
| |
25.85
|
178.91
|
3.83
|
| DC6/° |
32.4(1) |
61.3(1) |
29.2(1) |
| |
26.18
|
179.17
|
4.69
|
| DC7/° |
17.5(4) |
1.0(2) |
16.6(4) |
| |
179.99
|
179.89
|
0.011
|
| DC10/° |
27.3(3) |
1.5(2) |
27.0(3) |
| |
179.98
|
179.98
|
179.98
|
| DC12/° |
30.9(1) |
59.6(1) |
29.4(1) |
| |
23.10
|
178.52
|
3.2
|
| DC15/° |
28.3(1) |
58.0(1) |
30.1(1) |
| |
25.50
|
178.94
|
4.95
|
Table 3 Intra- and intermolecular interactions in DC1: Cg1 and Cg2 refer to the center of gravity of the rings formed by C1–C6 and C7–C12
| D–H⋯A |
r(D–H)/Å |
r(D⋯A)/Å |
r(⋯A)/Å |
∠D–H⋯A/° |
Symmetry operator |
![[D with combining low line]](https://www.rsc.org/images/entities/char_0044_0332.gif) ![[C with combining low line]](https://www.rsc.org/images/entities/char_0043_0332.gif) ![[1 with combining low line]](https://www.rsc.org/images/entities/char_0031_0332.gif)
|
| N1–H1N⋯F1 |
0.91(4) |
2.772(4) |
2.17(4) |
123(3) |
— |
| C8–H8⋯O1 |
0.93 |
2.867(5) |
2.34 |
116 |
— |
| N1–H1N⋯O1 |
0.91(4) |
3.254(4) |
2.41(4) |
153(4) |
x – 1, y, z |
| C5–H5⋯F1 |
0.93 |
3.379(5) |
2.59 |
143 |
–x, y – 1/2, –z + 1 |
| C4–H4⋯Cg1 |
0.93 |
3.749(5) |
2.99 |
141 |
– x + 1, y – 1/2, –z +1 |
| C10–H10⋯Cg2 |
0.93 |
3.759(5) |
2.98 |
143 |
–x, y + 1/2, –z |
![[2 with combining low line]](https://www.rsc.org/images/entities/char_0032_0332.gif)
|
| C8–H8⋯O1 |
0.93 |
2.925(3) |
2.46 |
111 |
— |
| N1–H1N⋯O1 |
0.88(4) |
3.176(4) |
2.36(4) |
156(3) |
x, y + 1, z |
| C12–H12⋯O1 |
0.93 |
3.405(4) |
2.75 |
129 |
x, y + 1, z |
| C4–H4⋯F1 |
0.93 |
3.408(4) |
2.62 |
142 |
–x, y + 1/2, –z + 3/2 |
| C8–H8⋯Cg1 |
0.93 |
3.616(5) |
2.95 |
130 |
–x + 1, –y, –z |
| C5–H5⋯Cg2 |
0.93 |
3.636(4) |
2.90 |
137 |
–x + 1, –y + 1, –z |
| C2–H2⋯Cg2 |
0.93 |
3.634(4) |
2.92 |
133 |
–x, –y, –z |
![[3 with combining low line]](https://www.rsc.org/images/entities/char_0033_0332.gif)
|
| N1–H1N⋯O1 |
0.83(3) |
3.165(4) |
2.39(3) |
155(3) |
— |
| C8–H8⋯O1 |
0.93(3) |
2.925(3) |
2.46(2) |
111(2) |
— |
| C12–H12⋯O1 |
0.94(3) |
3.393(4) |
2.71(3) |
131(2) |
x – 1, y, z |
| C11–H11⋯F1 |
0.97(3) |
3.344(3) |
2.62(3) |
132(2) |
x – 1, y, z – 1 |
| C8–H8⋯Cg1 |
0.93(3) |
3.567(4) |
2.88(3) |
131(2) |
–x + 1, –y + 1, –z |
| C12–H12⋯Cg1 |
0.93(3) |
3.672(4) |
3.00(2) |
130(2) |
–x + 2, –y, –z |
| C3–H3⋯Cg2 |
0.98(3) |
3.626(4) |
2.96(3) |
127(2) |
–x + 1, –y, –z |
| C5–H5⋯Cg2 |
0.96(3) |
3.568(4) |
2.81(2) |
138(2) |
–x + 2, –y + 1, –z |
![[5 with combining low line]](https://www.rsc.org/images/entities/char_0035_0332.gif)
|
| C8–H8⋯O1 |
0.95(2) |
2.897(2) |
2.45(2) |
110(1) |
— |
| N1–H1N⋯O1 |
0.88(3) |
3.178(2) |
2.37(3) |
153(2) |
x, y + 1, z |
| C12–H12⋯O1 |
0.91(2) |
3.289(3) |
2.64(2) |
129(2) |
x, y + 1, z |
| C3–H3⋯F1 |
1.02(2) |
3.806(2) |
2.79(2) |
172(1) |
x, –y + 1/2, z + 1/2 |
| C10–H10⋯F1 |
0.96(2) |
3.443(2) |
2.63(2) |
143(2) |
–x + 1, y + 1/2, –z + 1/2 |
| C8–H8⋯Cg1 |
0.95(2) |
3.655(3) |
2.92(2) |
135(2) |
–x, –y + 2, –z |
| C11–H11⋯Cg1 |
0.96(2) |
3.646(3) |
2.91(2) |
134(1) |
–x + 1 , –y + 1, –z |
| C2–H2⋯Cg2 |
0.94(2) |
3.633(3) |
2.92(2) |
135(2) |
–x + 1, –y + 2, –z |
| C5–H5⋯Cg2 |
1.00(1) |
3.720(3) |
2.99(2) |
132(2) |
–x, –y + 1, –z |
![[6 with combining low line]](https://www.rsc.org/images/entities/char_0036_0332.gif)
|
| C8–H8⋯O1 |
0.93(3) |
2.894(3) |
2.43(3) |
110(2) |
— |
| N1–H1N⋯O1 |
0.86(3) |
3.165(3) |
2.37(3) |
154(3) |
x – 1, y, z |
| C12–H12⋯O1 |
0.93(3) |
3.298(4) |
2.64(3) |
129(2) |
x – 1, y, z |
| C5–H5⋯F1 |
0.96(3) |
3.439(4) |
2.76(4) |
129(3) |
x, y , z – 1 |
| C8–H8⋯Cg1 |
0.95(4) |
3.593(4) |
2.88(4) |
133(3) |
–x + 1, –y + 1, –z + 1 |
| C11–H11⋯Cg1 |
0.96(4) |
3.605(4) |
2.94(3) |
127(3) |
–x, –y, –z + 1 |
| C2–H2⋯Cg2 |
0.93(3) |
3.580(3) |
2.90(3) |
131(2) |
–x + 1, –y, –z + 1 |
| C5–H5⋯Cg2 |
0.94(4) |
3.580(4) |
2.88(3) |
132(3) |
–x, –y + 1, –z + 1 |
![[7 with combining low line]](https://www.rsc.org/images/entities/char_0037_0332.gif)
|
| N1–H1N⋯F2 |
0.80(6) |
2.613(8) |
2.14(6) |
118(5) |
— |
| N1–H1N⋯F1 |
0.80(6) |
2.746(9) |
2.23(6) |
123(6) |
— |
| C8–H8⋯O1 |
0.93 |
2.836(8) |
2.33 |
114 |
— |
| N1–H1N⋯O1 |
0.80(6) |
3.314(7) |
2.70(6) |
135(6) |
x – 1, y, z |
| C5–H5⋯O1 |
0.93 |
3.545(7) |
2.66 |
160 |
x – 1, y + 1, z |
| C8–H8⋯F1 |
0.93 |
3.309(7) |
2.52 |
143 |
x + 1, y – 1, z |
| C3–H3⋯Cg1 |
0.93 |
3.879(6) |
3.12 |
140 |
x + 1/2, –y + 5/2, –z |
| C10–H10⋯Cg2 |
0.93 |
3.954(7) |
3.14 |
148 |
–x + 1, y – 1/2, –z + 1/2 |
| C11–H11⋯F2 |
0.93 |
3.463(7) |
2.72 |
138 |
–x, y – 1/2, –z + 1/2 |
![[0 with combining low line]](https://www.rsc.org/images/entities/char_0030_0332.gif)
|
| N1–H1N⋯F1 |
1.03(1) |
2.745(6) |
2.10(1) |
118(1) |
— |
| C8–H8⋯O1 |
0.93 |
2.853(7) |
2.37 |
112 |
— |
| N1–H1N⋯O1 |
1.03(1) |
3.058(6) |
2.20 |
139 |
x, y – 1, z |
| C4–H4⋯F2 |
0.93 |
3.412(7) |
2.67 |
137 |
x – 1/2, –y + 1, z |
| C11–H11⋯F2 |
0.93 |
3.715(9) |
2.79 |
171 |
–x + 1/2, y – 1, z + 1/2 |
| C12–H12⋯Cg1 |
0.93 |
3.897(8) |
3.12 |
142 |
–x, –y, z + 1/2 |
| C3–H3⋯Cg2 |
0.93 |
3.870(7) |
3.09 |
142 |
–x, –y + 1, z – 1/2 |
|
|
| C8–H8⋯O1 |
0.91(2) |
2.920(2) |
2.45(2) |
112(1) |
— |
| N1–H1N⋯O1 |
0.82(2) |
3.311(2) |
2.51(2) |
165(2) |
–x + 1, y – 1/2, –z + 1/2 |
| C6–H6⋯O1 |
0.972) |
3.449(2) |
2.74(2) |
130(2) |
–x + 1, y – 1/2, –z + 1/2 |
| C10–H10⋯F1 |
0.95(2) |
3.199(3) |
2.62(2) |
120(1) |
x + 1, –y + 3/2, z – 1/2 |
| C11–H11⋯F1 |
0.95(2) |
3.224(2) |
2.62(2) |
122(2) |
x + 1, –y + 3/2, z – 1/2 |
| C3–H3⋯F2 |
0.96(2) |
3.594(2) |
2.71(2) |
154(2) |
x – 1, y, z |
| C5–H5⋯Cg1 |
0.97(2) |
3.644(3) |
2.85(2) |
139(2) |
x, –y + 1/2, z – 1/2 |
| C2–H2⋯Cg2 |
0.94(2) |
3.495(3) |
2.80(2) |
132(1) |
–x + 1, –y + 1, –z + 1 |
| C11–H11⋯Cg2 |
0.95(2) |
3.613(3) |
2.97(2) |
126(2) |
x, –y + 1/2, z + 1/2 |
|
|
| C8–H8⋯O1 |
0.92(3) |
2.898(3) |
2.41(3) |
113(2) |
— |
| N1–H1N⋯O1 |
0.92(4) |
3.250(3) |
2.46(4) |
143(3) |
x – 1, y, z |
| C12–H12⋯O1 |
1.00(1) |
3.340(4) |
2.62(3) |
130(2) |
x – 1, y, z |
| C11–H11⋯F1 |
0.99(3) |
3.520(3) |
2.82(3) |
128(2) |
x – 1, y, z – 1 |
| C3–H3⋯F2 |
0.95(3) |
3.358(4) |
2.79(4) |
120(3) |
x + 1, y, z + 1 |
| C8–H8⋯Cg1 |
0.92(3) |
3.549(4) |
2.89(3) |
130(2) |
–x + 1 , –y + 1, –z |
| C11–H11⋯Cg1 |
1.00(3) |
3.583(4) |
2.86(3) |
131(2) |
–x, –y + 2, –z |
| C2–H2⋯Cg2 |
1.02(4) |
3.539(4) |
2.76(3) |
134(2) |
–x + 1, –y + 2, –z |
| C5–H5⋯Cg2 |
0.97(3) |
3.540(4) |
2.84(3) |
130(2) |
–x, –y + 1, –z |
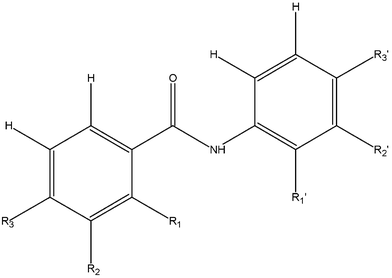
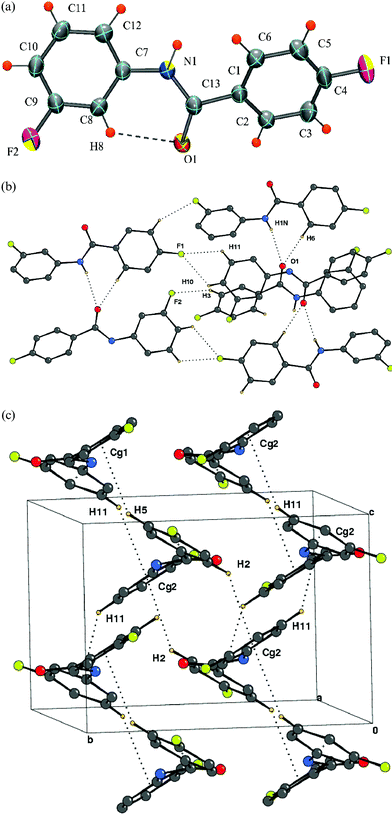
The molecule [Fig. 1(a)] crystallizes in a non-centrosymmetric monoclinic space group, P21 with Z = 2. Intramolecular N–H⋯F and C–H⋯O hydrogen bonds, involving H1N and H8, stabilize the non-planar conformation of DC1 [Table 2]. An intermolecular N–H⋯O hydrogen bond (involving H1N) forms molecular chains along the crystallographic a-axis with weak C–H⋯F interactions involving the acidic hydrogen H5 along the crystallographic screw b axis [Fig. 1(b), Table 3]. C–H⋯π intermolecular interactions, (involving H4 and H10) hold the molecules together, generating a “molecular staircase” [Fig. 1 (c)].
 |
| | Fig. 1 (a): ORTEP of DC1 drawn with 50% ellipsoidal probability. Dotted lines indicate intra-molecular hydrogen bonds. Fig. 1b: Packing of molecules (DC1) via strong N–H⋯O hydrogen bonds and C–H⋯F chains down the ac-plane. Fig. 1c: Staircase arrangement due to the involvement of C–H⋯π interactions (hydrogens H4 and H10) in DC1. | |
Structure of 3-fluoro-N-phenyl benzamide [DC2]
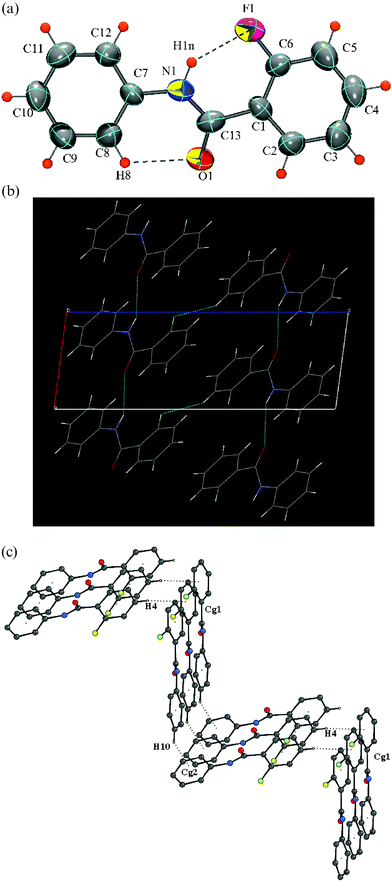
The molecule displays an intramolecular C–H⋯O hydrogen bond while crystallizing in a monoclinic space groupP21/c with Z = 4. [Fig. 2a]. The molecule is distinctly non-planar in the crystal structure as compared to the gas-phase conformation (Table 2) demonstrating the significance of intermolecular interactions. The fluorine atom is disordered, resulting in two conformations, the major conformer having an occupancy of 0.78(2). Strong N–H⋯O hydrogen bonds (involving H1N) generate molecular chains along the crystallographic b-axis, assisted by C–H⋯O interactions (involving H12). Molecules in adjacent layers are held by C–H⋯F interactions, involving H4 and the fluorine F1, forming molecular chains along the crystallographic screw b-axis, respectively [Fig. 2b]. Further stability is imparted by C–H⋯π interactions, resulting in the formation of dimers involving the hydrogens H2, H5, as shown in Fig. 2c and H8 as in Fig. 2d [Table 3].
 |
| | Fig. 2 (a): ORTEP of DC2 drawn with 50% ellipsoidal probability. Dotted line indicates an intramolecular hydrogen bond (only the major conformer is shown). Fig. 2b: Packing diagram (DC2) via strong N–H⋯O hydrogen bonds, and C–H⋯F chains down the bc-plane. Fig. 2c: Formation of molecular dimers via C–H⋯π interaction in DC2 (hydrogens H2 and H5). Molecules at (′) and (″) have the symmetry code (–x, –y, –z) and (–x + 1, –y + 1, –z) respectively. Cg1 and Cg2 are the centers of gravity of the phenyl rings C1/C6 and C7/C12. Fig. 2d: Formation of molecular dimers via C–H⋯π interaction in DC2. Molecules at (x, y, z) and (–x + 1, –y, –z) have been shown. | |
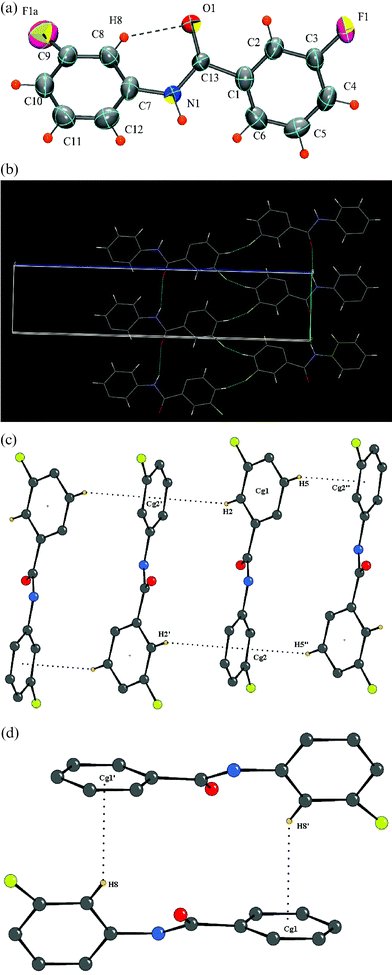
 |
| | Fig. 3 (a): ORTEP of DC3 drawn with 50% ellipsoidal probability. Dotted line indicates an intramolecular hydrogen bond. Fig. 3b: Packing of molecules (DC3) via N–H⋯O, C–H⋯O and C–H⋯F interactions in the crystal lattice down the ac-plane. Fig. 3c: Formation of N–H⋯O, C–H⋯O, C–H⋯π (hydrogens H5 and H8) and C–H⋯F interactions in DC3. Molecules at (′), (″), (*) and (′′′) have the symmetry codes (x – 1, y, z), (x – 1, y, z – 1), (–x + 2, –y + 2, –z) and (–x + 1, –y + 1, –z). Fig. 3d: Formation of C–H⋯π interaction (hydrogens H3 and H12) in DC3. Molecules at (′) and (″) have the symmetry codes (–x + 2, –y, –z) and (–x + 1, –y, –z). | |
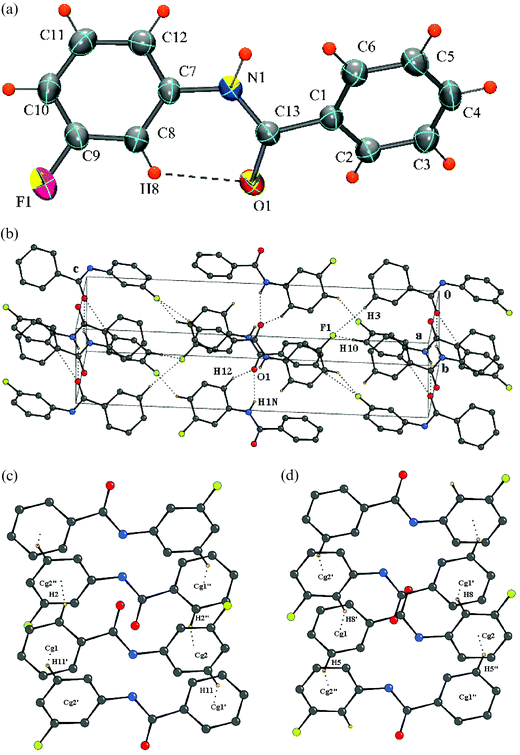 |
| | Fig. 4 (a) ORTEP of DC5 drawn with 50% ellipsoidal probability. Dotted lines indicate an intramolecular hydrogen bond. Fig. 4b: Packing of molecules (DC5) via N–H⋯O, C–H⋯O and C–H⋯F interactions in the crystal lattice. Fig. 4c: Formation of C–H⋯π (hydrogens H2 and H11) interactions in DC5. Molecules at (′) and (″) have the symmetry codes (–x + 1, –y + 1, –z) and (–x + 1, –y + 2, –z). Fig. 4d: Formation of C–H⋯π interactions(hydrogens H5 and H8) in DC5. Molecules at (′) and (″) have the symmetry codes (–x, –y + 2, –z) and (–x, –y + 1, –z). | |
Structure of N-(4-fluorophenyl)benzamide [DC6]
The crystal structure is centrosymmetric, space groupP with Z = 2. Fig. 5a represents the molecular conformation along with the C–H⋯O intramolecular hydrogen bond. The crystal structure displays a sheet-like motif where intermolecular N–H⋯O, C–H⋯O and C–H⋯F interactions become operative [Fig. 5b]. Molecular C–H⋯π dimers involving the acidic hydrogen atoms H2, H5, H8, H11 impart an additional strength to the assembly of molecules [Fig. 5c–d, Table 3].
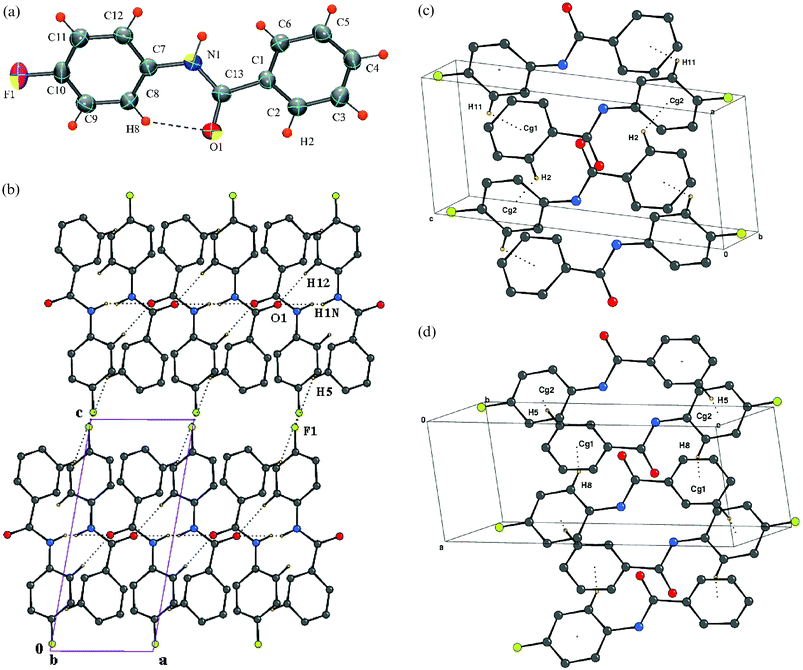 |
| | Fig. 5 (a): ORTEP of DC6 drawn with 50% ellipsoidal probability. Dotted line indicates an intramolecular hydrogen bond. Fig. 5b: Packing of molecules (DC6) via N–H⋯O, C–H⋯O and C–H⋯F interactions in the crystal lattice down the ac-plane. Fig. 5c: Molecular dimers in DC6 involving hydrogens H2 and H11. Fig. 5d: Molecular dimers in DC6 involving hydrogens H5 and H8. | |
Structure of 2-fluoro-N-(2-fluorophenyl)benzamide [DC7]
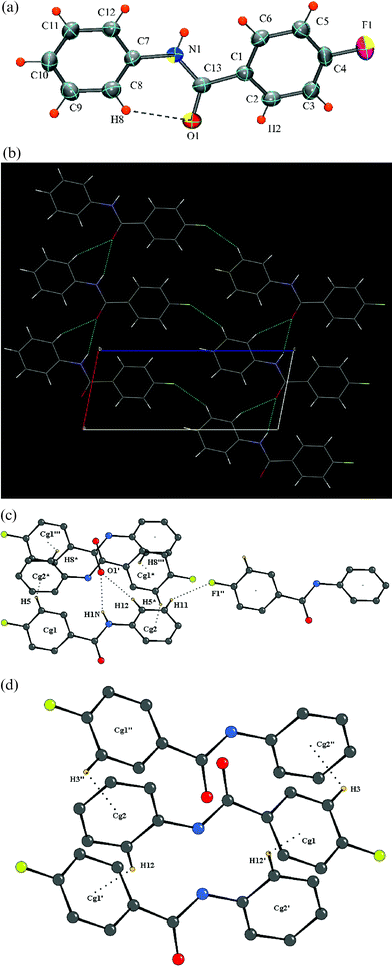
The presence of two fluorine atom substitutions, one each in the ortho position results in a non-centrosymmetric orthorhombic space groupP212121 with Z = 4. Intramolecular bifurcated N–H⋯F hydrogen bonding, involving the hydrogen atoms H1N and fluorine atoms F1 and F2, locks the molecular conformation and is further stabilized by an intramolecular C–H⋯O hydrogen bond, characteristic of these benzanilides [Fig. 6a]. The molecules are packed with the involvement of N–H⋯O hydrogen bonds, generating molecular chains along the a-axis, further stabilized by C–H⋯O hydrogen bonds. C–H⋯F interactions involving fluorine atoms, F1 with hydrogen H8, F2 with hydrogen H11, [Fig. 6b–c] generate molecular chains along the screw b-axis. C–H⋯π interactions, involving the hydrogen atoms H3 and H10 provide an additional stability to the packing of the molecules along the screw a- and b-axis, respectively [Fig. 6c, Table 3].
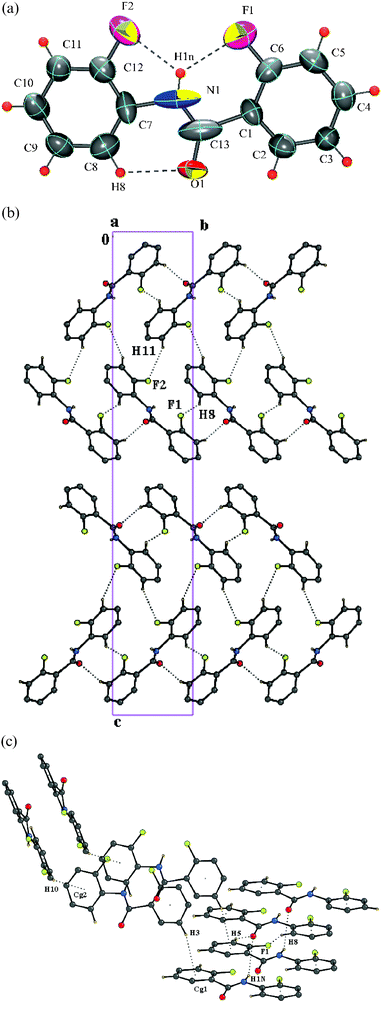 |
| | Fig. 6 (a): ORTEP of DC7 drawn with 50% ellipsoidal probability. Dotted lines indicate intramolecular hydrogen bonds. Fig. 6b: Packing of molecules via N–H⋯O interactions in DC7 down the ac-plane. Fig. 6c: Regions of N–H⋯O, C–H⋯O, C–H⋯F and C–H⋯π interactions in DC7. | |
Structure of 2-fluoro-N-(3-fluorophenyl)benzamide [DC10]
The molecule crystallizes in the non-centrosymmetric orthorhombic space groupPca21 with Z = 4. An intramolecular N–H⋯F hydrogen bond, involving fluorine atom F1 and hydrogen atom H1N, stabilizes the molecular conformation along with an intramolecular C–H⋯O hydrogen bond [Fig. 7a]. It is noteworthy that atom F1 is not involved further in the formation of intermolecular interactions. N–H⋯O hydrogen bonded chains run along the crystallographic b-axis, while C–H⋯F interactions hold the molecules along the a- and c-axis, respectively. In addition, C–H⋯π interactions hold the molecules along the screw c-axis (involving the hydrogens H3 and H12 alternately) leading to the formation of a well defined three-dimensional structure [Fig. 7c, Table 3].
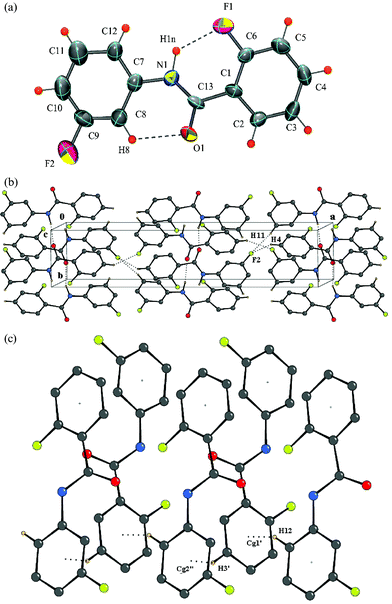 |
| | Fig. 7 (a): ORTEP of DC10 drawn with 50% ellipsoidal probability. Dotted lines indicate intramolecular hydrogen bonds. Fig. 7b: Packing of molecules in DC10via N–H⋯O, C–H⋯O, C–H⋯F and C–H⋯π interactions in the lattice. Fig. 7c: C–H⋯π (involving H3 and H12) interactions in DC10. Cg2 is the center of gravity of the phenyl ring C7/C12. | |
Structure of 4-fluoro-N-(3-fluorophenyl)benzamide [DC12]
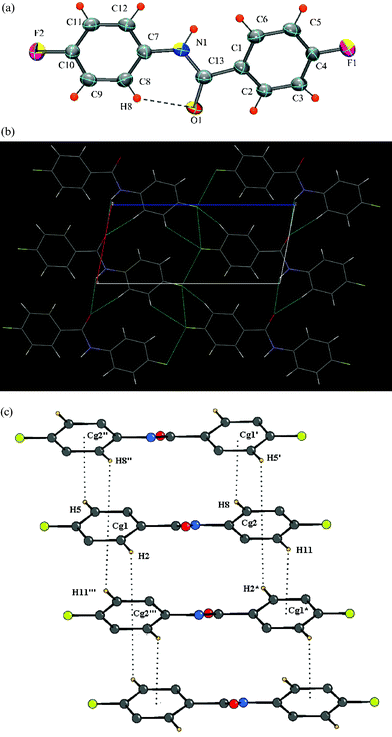
The molecule crystallizes in the centrosymmetric monoclinic space groupP21/c with Z = 4, with the characteristic intramolecular C–H⋯O hydrogen bond [Fig. 8a]. In addition to N–H⋯O hydrogen bonded chains, involving H1N, C–H⋯O interaction, involving H6, along the crystallographic b-axis, stabilizes the crystal structure. C–H⋯F interactions involving hydrogen atoms H11, H10 and fluorine F1, generate molecular chains along the c-axis. Also, the fluorine atom F2 and the hydrogen atom H3 are involved in the formation of chains along the a-axis [Fig. 8b]. Finally, C–H⋯π interactions involving H5 and H11 hold the molecules along the crystallographic c-axis leading to the formation of C–H⋯π dimers [Fig. 8c, Table 3].
 |
| | Fig. 8 (a): ORTEP of DC12 drawn with 50% ellipsoidal probability. Dotted lines indicate an intramolecular hydrogen bond. Fig. 8b: Packing of molecules in DC12via N–H⋯O and C–H⋯F interactions.Fig. 8c: C–H⋯π intermolecular interactions (involving hydrogens H2, H5 and H11) in DC12. | |
Structure of 4-fluoro-N-(4-fluorophenyl)benzamide [DC15]
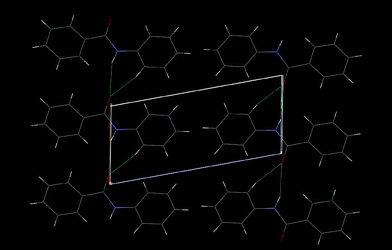
The structure depicts [Fig. 9a] the space groupP with Z = 2 with the presence of a typical intramolecular C–H⋯O hydrogen bond. It is noteworthy that even though this compound is isostructural with both DC3 and DC6 [Fig. 9b] the intermolecular interactions in the packing are entirely different. Familiar N–H⋯O hydrogen bonds, C–H⋯O interactions and weak C–H⋯F interactions, involving fluorine F1 and F2 with hydrogen H3 and H11, respectively, form molecular chains along the crystallographic c-axis, [Fig. 9b] generating a sheet-like structure. The fluorine atoms F1 and F2 in such heterodimeric motifs form an F⋯F contact [d = 2.953(3) Å, symmetry code: x + 1, y, z + 1]. A further atom F1 forms an additional F⋯F contact with the fluorine atom F2 of the adjacent layers, the distance being 2.928(3) Å (x, y, z + 1), thus providing an additional stability. The role of fluorine interaction in dictating packing modes is well demonstrated in this example. Once again, the C–H⋯π interactions forming molecular dimers involving H2, H8, H5 and H11 contribute to the packing in the lattice. [Fig. 9c, Table 3].
 |
| | Fig. 9 (a): ORTEP of DC15 drawn with 50% ellipsoidal probability. Dotted line indicates intramolecular hydrogen bond. Fig. 9b: Packing of molecules via involvement of N–H⋯O, C–H⋯O, C–H⋯F and F⋯F interactions in DC15 viewed down the ac-plane. Fig. 9c: Formation of C–H⋯π interactions (involving hydrogens H2, H5, H8 and H11) in DC15. Molecules at (′), (″), (‴) and (*) have the symmetry codes (–x + 1, –y + 1, –z), (–x, –y + 1, –z), (–x, –y + 2, –z), (–x + 1, –y + 2, –z). | |
Discussion
Detailed crystallographic studies on mono- and difluorinated benzanilides allows for the understanding of intermolecular contacts, which stabilize the packing of molecules in the crystal lattice. The control and pre-organization of molecules is typically by strong hydrogen bonds of the type N–H⋯O, which are further stabilized by C–H⋯O interactions forming linear molecular chains. This layer of molecules is held with the adjacent layers by the involvement of weak intermolecular C–H⋯F, C–F⋯F–C intermolecular interactions operating across the layers, generating a sheet-like structure. Successive layers are held by the presence of weak C–H⋯π interactions, which provide further stability to the crystal packing. It is of interest to compare the similarities and differences, which occur in the molecular conformation and crystal packing on substitution of a fluorine atom on the benzanilide ring. On substitution of the fluorine atom on the three different ring positions ortho-, meta- and para- on either side of the amido group, in the series of substituted monofluorinated benzanilides, some interesting similarities in the supramolecular features are observed. Due to the delocalization of the lone pair of electrons on the nitrogen atom over the carbonyl group, the nitrogen atom becomes positively charged. Spreading of this positive charge over the adjacent phenyl ring leads to ortho and para hydrogens acquiring sufficient acidic character to participate in the H-bonding. The effect of the fluorine substitution is to enhance the acidity of the neighbouring hydrogen atoms leading to increase in H-bond participation.
The parent structure, benzanilide, PhNHCOPh, exists in two polymorphic forms, a triclinic P form38 and a monoclinic C2/c form.39 The molecular conformation of the triclinic form is almost the same as that of DC10 whereas the monoclinic form has conformational characteristics similar to those of DC2/3/5/6/12/15. Also, the triclinic form has lattice parameters and packing characteristics close to those of DC3/6/15 (Fig. 10). The monoclinic form (Fig. 11) is isostructural with the plate form of DC1116c,d (Fig. 11).
 |
| | Fig. 10 Packing of molecules down the ac-plane in the triclinic form of unsubstituted benzanilide (only the major conformer shown), which has an identical characteristics with DC3/6/15. | |
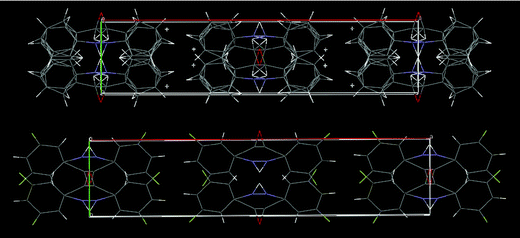 |
| | Fig. 11 Packing of molecules down the ab-plane in the monoclinic form of benzanilide (both conformers shown), which is similar to the plate form of DC11. | |
An analysis of the Cambridge Structural Database (CSD, version 5.26, November 2006)40 was performed to evaluate the changes in the molecular conformation and crystal packing due to the presence of different substituents (other than fluorine or in combination with fluorine) on the phenyl rings of benzanilide. This CSD search41 resulted in 303 hits, containing various substituents. On careful examination of the crystal structures of all these compounds it was observed that the following REFCODES41 have either similar lattice parameters or conformational features with those of the fluorinated benzanilides. The lattice parameters a, b, and c in monofluorinated benzanilides DC2, DC5 and in difluorinated benzanilideDC10 are similar, suggesting isostructurality (Table 2). DC2, DC5 crystallise in a centrosymmetric monoclinic space groupP21/c whereas DC10 crystallizes in a non-centrosymmetric orthorhombic space groupPca21. Further, the molecular conformations of DC2 and DC5 are similar while DC10 shows deviations w.r.t the dihedral angle between the two-phenyl rings (62° in DC2/DC5 and 1.5° in DC10). Likewise, DC3, DC6 (fluorine in the para-position on either phenyl ring) and DC15 (fluorine atoms F1 and F2 in the para-position on either phenyl ring) crystallize in the triclinic space groupP depicting isostructurality. Similarly, DC3, DC6 (monofluorinated) and DC15 (difluorinated) have similar conformations. It must be noted, however, that the packing features generated in the presence of the fluorine atom in all isostructural compounds are different and generate motifs of different dimensionalities. This provides strong evidence for the presence of C–H⋯F and in the case of DC15, F⋯F interactions and the accompanying directional preferences.
DFT calculations at the B3LYP/6-31G** were performed with crystallographic coordinates as a starting set to obtain the optimised geometry of an isolated molecule. It is observed that in DC1, which is a monofluorinated benzanilide, and in DC7 and DC10, both difluorinated benzanilides, the gas phase conformation is planar while in the remaining derivatives the conformation angles are significantly out of plane (Table 2). In DC1, DC7 and DC10 intramolecular N–H⋯F42 and C–H⋯O interactions tend to lock the entire molecule in a planar configuration. Deviations from planarity observed in these three cases in the crystal structures (Table 2) hence point out to the significance of intermolecular interactions, which dictate the overall packing of molecules in the lattice. The formation of the “trimeric motif” consisting of N–H⋯O and C–H⋯F interactions (in DC2 and DC5) is unique and is also present in the crystal structure of 3-fluoro-N-(4-methylphenyl) benzamide.19 The occurrence of N–H⋯O hydrogen bonds in isostructural compounds DC2, DC5 and DC10 is specific in generating chains along the crystallographic b’-axis while the chains are along the a-axis in isostructural compounds DC3, DC6 and DC15. DC916b and DC1116c,d exists in two polymorphic modifications. DC9 crystallizes in two non-centrosymmetric modifications (namely, needles: Pca21, plates: P21) whereas DC11 crystallizes as plates (C2/c) and needles (P21). It is noteworthy that the needle form of DC9 and the plate form of DC11 are also isostructural with DC2/5/10. Also, the plate form of DC9 and the needle form in DC11 have identical lattice parameters (a = 5.06, b = 5.35, c = 19.3) and display identical packing characteristics with DC1 and DC7. It is noteworthy that both fluorine atoms in DC9 are ordered whereas in DC11 these are disordered. Although the molecular conformation in both forms (DC9 and DC11) is similar, the supramolecular features associated are different. Apart from the presence of strong hydrogen bonds, it is the presence of weak, yet directional interactions involving fluorine that results in molecules adopting different packing modes in the crystal lattice.
All crystal structures display N–H⋯O, C–H⋯O, C–H⋯π and C–H⋯F intermolecular interactions in the crystalline lattice. F⋯F contacts in DC15 are not observed across the crystallographic center of symmetry. Such contacts are ubiquitous and recent charge density studies,43 using high resolution X-ray data, have established unequivocally the existence of such weak contacts. Also, a quantitative estimation based on topological analysis has established that the C–H⋯F interaction has weak hydrogen bonding characteristics.
Conclusion
The introduction of fluorine atom/atoms on the phenyl ring involves an isosteric replacement of the hydrogen atom. The change in the position of the fluorine atom from ortho and meta to a para position generates altered packing modes in the crystal lattice in these isomeric molecules. On replacement of the C–H group by C–F group the only significant change observed is the variation in the crystal system and space group (from centrosymmetric to noncentrosymmetric) as was observed in the case of DC2/DC5/DC10. However, no such change is observed in DC3/DC6/DC15 with all compounds crystallizing in triclinic centrosymmetric P. The influence of interactions involving fluorine is demonstrated in the altered packing modes in the otherwise isostructural compounds, a striking example being the C–H⋯F and F⋯F contacts in DC15. Furthermore, the C–H⋯O and C–H⋯π intermolecular interactions play an important role in providing stability to the overall packing of molecules. It is noteworthy that the CSD search revealed that the dependence of the nature of molecular conformation and packing features is sensitive to the type and electronic features associated with the substituents on the phenyl rings. However, there are a few exceptions (11 hits out of 303), which are insensitive to the features associated with the substituent.
Acknowledgements
We thank DST, India for data collection on the CCD facility at IISc, Bangalore. DC thanks Dr A. R. Choudhury for discussions and suggestions, CSIR, India for SRF, Mr Saikat Sen for extensive help during the synthesis and Prof Goverdhan Mehta for allowing me to use his laboratory facilities. DC thanks Mr Padaikanta for DSC measurements, Mr Girish Kunte for DTA measurements, Ms Shampa Ghosh for SHG measurements and Mr Jarali for IR measurements, Mr Rajamani for theoretical calculations.
References
- D. S. Lawrence, T. Jiang and M. Levett, Chem. Rev., 1995, 95, 2229 CrossRef CAS.
-
(a) D. Seebach and J. L. Matthews, Chem. Commun., 1997, 2015 RSC;
(b) S. H. Gellman, Acc. Chem. Res., 1998, 31, 173 CrossRef CAS;
(c) K. D. Stigers, M. J. Soth and J. S. Nowick, Cur. Opin. Chem. Biol., 1999, 3, 714 Search PubMed;
(d) D. J. Hill, M. J. Mio, R. B. Prince, T. S. Hughes and J. S. Moore, Chem. Rev., 2001, 101, 3893 CrossRef CAS;
(e) M. S. Cubberley and B. L. Iverson, Cur. Opin. Chem. Biol., 2001, 5, 650 Search PubMed;
(f) A. R. Sanford and B. Gong, Curr. Org. Chem., 2003, 7, 1649 CrossRef CAS;
(g) C. Schmuck, Angew. Chem., 2003, 115, 2552 CrossRef.
- T. Steiner, Angew. Chem., Int. Ed., 2002, 41, 48 CrossRef CAS.
-
(a) P. S. Corbin, S. C. Zimmerman, P. A. Thiessen, N. A. Hawryluk and J. Murray, J. Am. Chem. Soc., 2001, 123, 10475 CrossRef CAS;
(b) K. Gademann, A. Hane, M. Rueping, B. Jaun and D. Seebach, Angew. Chem., 2003, 115, 1573 (
Angew. Chem., Int. Ed.
, 2003
, 42
, 1534
) CrossRef;
(c) J. S. Nowick and J. O. Brower, J. Am. Chem. Soc., 2003, 125, 876 CrossRef CAS;
(d) J. S. Park, H. S. Lee, J. R. Lai, B. M. Kim and S. H. Gellman, J. Am. Chem. Soc., 2003, 125, 8539 CrossRef CAS.
-
(a) B. Gong, H. Zeng, J. Zhu, L. Yuan, Y. Han, S. Cheng, M. Furukawa, R. D. Parra, A. Y. Kovalevsky, J. L. Mills, E. Skrzypczak-Jankun, S. Martinovic, R. D. Smith, C. Zheng, T. Szyperski and X. C. Zeng, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 11583 CrossRef CAS;
(b) J.-L. Hou, X.-B. Shao, G.-J. Chen, Y.-X. Zhou, X.-K. Jiang and Z.-T. Li, J. Am. Chem. Soc., 2004, 126, 12386 CrossRef CAS;
(c) D. Kanamori, T. Okamura, H. Yamamoto and N. Ueyama, Angew. Chem., 2005, 117, 995 (
Angew. Chem., Int. Ed.
, 2005
, 44
, 969
) CrossRef.
-
(a) Y. Hamuro, S. J. Geib and A. D. Hamilton, J. Am. Chem. Soc., 1996, 118, 7529 CrossRef CAS;
(b) E. Kolomiets, V. Berl, I. Odriozola, A.-M. Stadler, N. Kyritsakas and J. -M. Lehn, Chem. Commun., 2003, 2868 RSC;
(c) B. Huang, M. A. Prantil, T. L. Gustafson and J. R. Parquette, J. Am. Chem. Soc., 2003, 125, 14518 CrossRef CAS;
(d) H. Jiang, J.-M. Léger and I. Huc, J. Am. Chem. Soc., 2003, 125, 3448 CrossRef CAS;
(e) J. Garric, J. -M. Léger and I. Huc, Angew. Chem., Int. Ed., 2005, 117, 1990 (
Angew. Chem., Int. Ed.
, 2005
, 44
, 1954
) CrossRef.
- G. R. Desiraju, Chem. Commun., 1997, 1475 RSC.
-
(a) H. J. Bohm, D. Banner, S. Bendels, M. Kansy, B. Kuhn, K. Muller, U. O. Sander and M. Stahl, ChemBioChem, 2004, 5, 637 CrossRef;
(b) P. Jeschke, ChemBioChem, 2004, 5, 570 CrossRef CAS;
(c) F. Leroux, ChemBioChem, 2004, 5, 644 CrossRef CAS.
-
(a) S. A. Harrell and D. H. McDaniel, J. Am. Chem. Soc., 1964, 86, 4497 CrossRef CAS;
(b) S. J. Coles, J. G. Frey, P. A. Gale, M. B. Hursthouse, M. E. Light, K. Navakhun and G. L. Thomas, Chem. Commun., 2003, 568 RSC.
-
(a)
L. Pauling, The Nature of the Chemical Bond, Cornell University Press, Ithaca, NY, 3rd edn, 1960, p. 464 Search PubMed;
(b) B. Dietrich, J.-M. Lehn, J. Guilhem and C. Pascard, Tetrahedron Lett., 1989, 30, 4125 CrossRef CAS.
- J. A. K. Howard, V. J. Hoy, D. O' Hagan and G. T. Smith, Tetrahedron, 1996, 52, 12613 CrossRef CAS.
-
(a) J. D. Dunitz and R. Taylor, Chem.–Eur. J., 1997, 3, 89 CrossRef CAS;
(b) J. D. Dunitz, ChemBioChem, 2004, 5, 614 CrossRef CAS.
-
(a) G. W. Coates, L. A. R. Dunn, M. Henling, J. W. Ziller, E. B. Lobkovsky and R. H. Grubbs, J. Am. Chem. Soc., 1998, 120, 3641 CrossRef CAS;
(b) M. L. Renak, G. P. Bartholomew, S. Wang, P. J. Ricatto, R. J. Lachicotte and G. C. Bazan, J. Am. Chem. Soc., 1999, 121, 7787 CrossRef CAS;
(c) C. Dai, P. Nguyen, T. B. Marder, A. J. Scott, W. Clegg and C. Viney, Chem. Commun., 1999, 2493 RSC;
(d) W. J. Feast, P. W. Lövenich, H. Puschmann and C. Taliani, Chem. Commun., 2001, 505 RSC;
(e) J. C. Collings, K. P. Roscoe, R. L. Thomas, A. S. Batsanov, L. M. Stimson, J. A. K. Howard and T. B. Marder, New J. Chem., 2001, 25, 1410 RSC.
-
(a) V. R. Thalladi, H.-C. Weiss, D. Bläser, R. Boese, A. Nangia and G. R. Desiraju, J. Am. Chem. Soc., 1998, 120, 8702 CrossRef CAS;
(b) H. Lee, C. B. Knobler and M. F. Hawthorne, Chem. Commun., 2000, 2485 RSC.
-
(a) N. Hayashi, T. Mori and K. Matsumoto, Chem. Commun., 1998, 1905 RSC;
(b) I. S. Neretin, K. A. Lyssenko, M. Y. Antipin, Y. L. Slovokhotov, O. V. Boltalina, P. A. Troshin, A. Y. Lukonin, L. N. Sidorov and R. Taylor, Angew. Chem., Int. Ed., 2000, 39, 3273 CrossRef CAS.
-
(a) D. Chopra, K. Nagarajan and T. N. Guru Row, Cryst. Growth Des., 2005, 5, 1035 CrossRef CAS;
(b) D. Chopra and T. N. Guru Row, Cryst. Growth Des., 2005, 5, 1679 CrossRef CAS;
(c) D. Chopra and T. N. Guru Row, Cryst. Growth Des., 2007 Search PubMed (unpublished results);
(d) Deepak Chopra, PhD Thesis, Indian Institute of Science, Bangalore, 2007 Search PubMed.
-
(a) J. Kowalik, D. vanDerveer, C. Clower and L. M. Tolbert, Chem. Commun., 1999, 2007 RSC;
(b) V. R. Thalladi, H.-C. Weiss, R. Boese, A. Nangia and G. R. Desiraju, Acta Crystallogr., Sect. B, 1999, 55, 1005 CrossRef;
(c) O. J. Dautel and M. Fourmigué, J. Org. Chem., 2000, 65, 6479 CrossRef CAS.
- A. R. Choudhury and T. N. Guru Row, Cryst. Growth Des., 2004, 4, 47 CrossRef.
- D. Chopra and T. N. Guru Row, J. Mol. Struct., 2005, 733, 133 CrossRef CAS.
- A. R. Choudhury and T. N. Guru Row, CrystEngComm, 2006, 6, 265 RSC.
- R. Frohlich, T. C. Rosen, O. G. J. Meyer, K. Rissanen and G. Haufe, J. Mol. Struct., 2006, 50, 787.
- X. Zhao, X.-Z. Wang, X.-K. Jiang, Y.-Q. Chen, Z.-T. Li and G.-J. Chen, J. Am. Chem. Soc., 2003, 125, 15128 CrossRef CAS.
- C. Li, S. F. Ren, J. L. Hou, H. P. Yi, S. Z. Zhu, X. K. Jiang and Z. T. Li, Angew. Chem., Int. Ed., 2005, 44, 5725 CrossRef CAS.
-
(a) C. Minks, R. Huber, L. Moroder and N. Budisa, Biochemistry, 1999, 38, 10649 CrossRef CAS;
(b) E. Neil and G. March, Chem. Biol., 2000, 7, R153 CrossRef CAS;
(c) C. Minks, R. Huber, L. Moroder and N. Budisa, Anal. Biochem., 2000, 284, 29 CrossRef CAS.
-
(a) R. H. Rich and P. A. Barlett, J. Org. Chem., 1996, 61, 3916 CrossRef CAS;
(b) J. K. Sutherland, W. J. Watkins, J. P. Bailey, A. K. Chapman and G. M. Davies, J. Chem. Soc., Chem. Commun., 1989, 1386 RSC;
(c) R. Brettle, R. Cross, M. Fredrickson, E. Haslam, F. S. Macbeath and G. M. Davies, Bioorg. Med. Chem. Lett., 1996, 6, 1275 CrossRef CAS;
(d) F. J. Weigert and A. Shenvi, J. Fluor. Chem., 1994, 66, 19 Search PubMed.
- J. H. Bae, P. P. Pal, L. Moroder, R. Huber and N. Budisa, ChemBioChem, 2004, 5, 720 CrossRef CAS.
- G. Xiao, J. F. Parsons, R. N. Armstrong and G. L. Gilliland, J. Am. Chem. Soc., 1997, 119, 9325 CrossRef CAS.
-
W. E. Bondiwell and J. A. Chan, US Patent 6515027..
-
A. O. Wickenden, M. F. Gross and G. A. M. Smith, US Patent 6989398.
- Bruker (2004). SMART, SAINT, SADABS, XPREP, SHELXTL. Bruker AXS Inc., Madison, Wisconsin, USA.
- A. Altomare, G. Cascarano, C. Giacovazzo and A. Guagliardi, J. Appl. Crystallogr., 1993, 26, 343 CrossRef.
-
G. M. Sheldrick, SHELXL97, University of Göttingen, Germany. 1997.
- L. J. Farrugia, WinGX, J. Appl. Crystallogr., 1999, 32, 837 CrossRef.
- L. J. Farrugia, J. Appl. Crystallogr., 1997, 30, 565 CrossRef CAS.
-
D. M. Watkin, L. Pearce and C. K. Prout, CAMERON - A Molecular Graphics Package. Chemical Crystallography Laboratory, University of Oxford, UK. 1993.
- M. Nardelli, J. Appl. Crystallogr., 1995, 28, 569.
- A. L. Spek, Acta Crystallogr., Sect. A, 1990, 46, C34.
- K. F. Bowes, C. Glidewell, J. N. Low, J. M. S. Skakle and J. K. Wardell, Acta Crystallogr., Sect. C, 2003, 59, o1.
- S. Kashino, K. Ito and M. Haisa, Bull. Chem. Soc. Jpn., 1979, 52, 365 CAS.
-
(a) F. H. Allen, The Cambridge Structural Database: A quarter of a million crystal structures and rising., Acta Crystallogr., Sect. B, 2002, 58, 380 CrossRef;
(b) F. H. Allen and W. D. S. Motherwell, Acta Crystallogr., Sect. B, 2002, 58, 407 CrossRef.
- The CSD searches were performed with the following restrictions: 3D-coordinates determined, R-factor less than 0.10, no polymeric species, no ions and only organics. The obtained REFCODES are ACALAR (having a nitro group connected to a benzoyl group), ACALEV (having two methyl substituents on amino phenyl and one nitro group on benzoyl moiety), BUTDID (having a nitro group connected to amino phenyl moiety), BUTDOJ (methoxy group connected to amino phenyl moiety), JODTEB (two chlorine atoms on aminophenyl and one methoxy group on benzoyl group), PADTIX (amino group), QUKVUM (pentafluoro substituents on the aminophenyl moiety), SAMBOY (chloro on benzoyl group and hydroxy on aminophenyl moiety), SAMCEP (fluoro on benzoyl group and hydroxy on aminophenyl moiety), SAMBUE (chloro on benzoyl group and methyl on aminophenyl group), SAMCAL (fluoro on benzoyl group and methyl on aminophenyl group), SAQKEA (hydroxy connected to aminophenyl group) and VECCEL (carboxylic group on benzoyl moiety and methyl on an aminophenyl group). These molecules have either identical packing or similar conformational features with those of the fluorinated benzanilides.
- P. Hobza, Int. J. Quantum Chem., 2002, 90, 1071 CrossRef CAS.
- D. Chopra, T. S. Cameron, J. D. Ferrera and T. N. Guru Row, J. Phys. Chem. A, 2006, 110, 10465 CrossRef CAS.
Footnotes |
| † CCDC reference numbers 285374–285381. For crystallographic data in CIF or other electronic format see DOI: 10.1039/b709938j |
| ‡ Electronic supplementary information (ESI) available: Synthetic procedure (Scheme S1); amounts of reactants taken for the synthesis of benzanilides (Table S1); description of various measuring techniques applied; important vibrational spectroscopic assignments (Table S2); melting point of the prepared compounds (Table S3). See DOI: 10.1039/b709938j |
|
| This journal is © The Royal Society of Chemistry 2008 |
Click here to see how this site uses Cookies. View our privacy policy here.