Hydrogen bonding patterns in a series of 1-arylcycloalkanecarboxamides†
Andreas
Lemmerer
and
Joseph P.
Michael
*
Molecular Sciences Institute, School of Chemistry, University of the Witwatersrand, PO Wits 2050, Johannesburg, South Africa. E-mail: joseph.michael@wits.ac.za; Fax: +27 11 717 6749; Tel: +27 11 717 6753
First published on 13th September 2007
Abstract
Single-crystal structures of five 1-arylcycloalkanecarboxamides, viz.1-phenylcyclopentanecarboxamide (1), 1-phenylcyclohexanecarboxamide (2), 1-(2-fluorophenyl)cyclohexanecarboxamide (3), 1-(2-chlorophenyl)cyclohexanecarboxamide (4) and 1-(2-bromophenyl)-cyclohexanecarboxamide (5), are reported. The primary hydrogen-bonded motif consists of centrosymmetric or non-centrosymmetric R22(8) dimers between the carboxamide functional groups. In compound 1, the dimers are further linked by hydrogen bonds to form infinite two-dimensional sheets, while in 4 and 5 additional hydrogen bonding with molecules not involved in dimer formation links the dimers into infinite chains. The cycloalkane rings in the five compounds adopt noticeably different conformations. As a consequence of these various effects, none of the five compounds is isostructural.
Introduction
The rapidly growing discipline of crystal engineering is predicated on molecular recognition between discrete molecules, which permits them to assemble in highly ordered supramolecular arrays by capitalising on various intermolecular interactions.1,2 Dominant among such molecular interactions is hydrogen bonding, which is frequently the over-riding factor in the design of supramolecular assemblages.3 As a constituent of ‘supramolecular synthons’2—the building blocks from which solid-state superstructures are generated—the amide functional group has few equals for the versatility with which it can engage in hydrogen bonding to create a diverse range of packing arrangements.4,5 Notable is the effect that subtle variations in molecular structure or in the substituents can have on the packing of homomeric hydrogen-bonded amide aggregates.6 Changes in packing motifs resulting from small variations in molecular structure or substituents have also been observed with other compounds capable of homomeric hydrogen bonding (for example, carboxylic acids,7oximes8 and secondary enaminones9), as well as in many heteromeric hydrogen-bonded structures (for example, bisphenol–amine co-crystals).10Our interest in the title compounds originated from synthetic investigations first performed in the 1980s, when we prepared several 1-(2-halophenyl)cyclohexanecarboxamides as part of a model study to identify suitable precursors for the construction of oxindoles bearing spirocyclic rings at C-3.11,12 These spiro-oxindoles in turn served as models for developing methodology aimed at the total synthesis of the complex spiro-oxindole alkaloid gelsemine.13 Related 1-phenylcycloalkanecarboxamides have been tested for neuroprotective activity.14 In this article we report a range of distinctly different hydrogen bonding patterns in five representatives of the title compound, 1–5 (Scheme 1), as revealed by single-crystal X-ray diffraction studies.
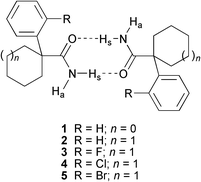 | ||
| Scheme 1 Structures of compounds 1–5, showing the primary hydrogen-bonded dimeric motif. | ||
Results and discussion
Crystallographic descriptions of the structures of 1-arylcycloalkanecarboxamides 1–5
Table 1 provides crystallographic details for compounds 1–5. The atomic numbering scheme for all five compounds is given in Fig. 1, which also shows the contents of the asymmetric unit in each case. The distances and angles within the five compounds reported are generally as expected.15 In all five structures, hydrogen bonds (summarised in Table 2) play a major part in controlling the supramolecular assembly of the molecules. In describing the hydrogen-bonding patterns in the five carboxamide structures reported in this study, we shall use unitary (N1) and binary (N2) graph set (GS) analysis.16 All structures have simple N–H⋯O hydrogen bonds; and, as expected, the primary hydrogen-bonded motif between molecules (Scheme 1) is a cyclic dimer [Synthon 1, R22(8)]. In distinguishing between the two H atoms on the primary amide group, we shall follow the procedure of Desiraju5 in labelling the N–H atom syn to the carbonyl group (i.e., the N–H atom involved in the formation of the cyclic dimer) as ‘Hs’ and the anti-orientated H atom as ‘Ha’. More interestingly, three of the five structures show additional hydrogen bonding as described below.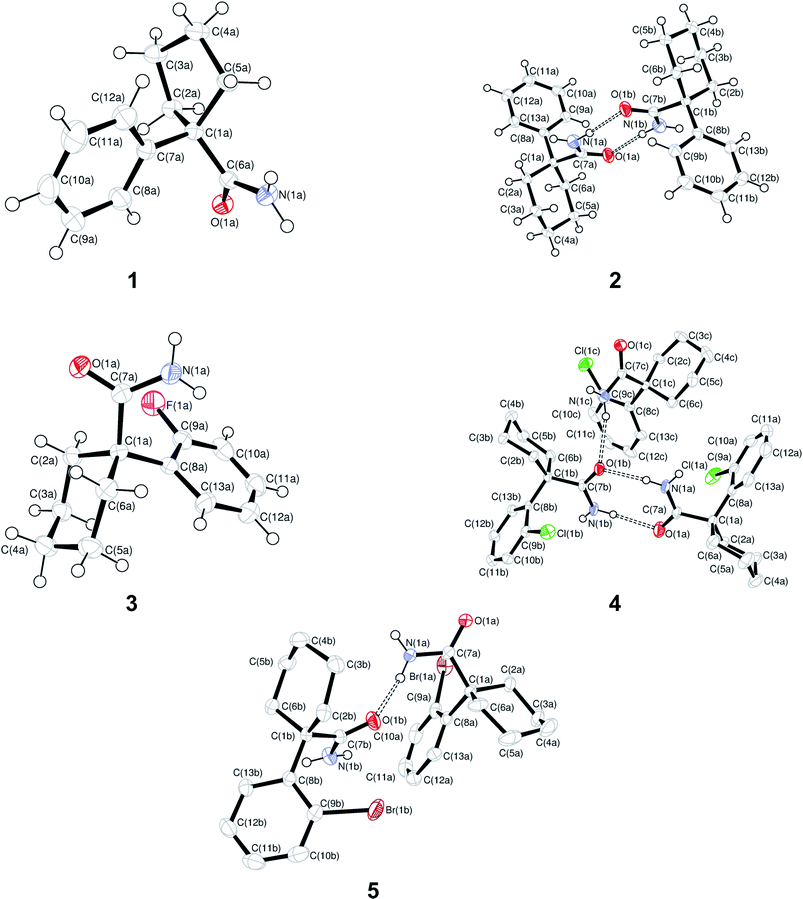 | ||
| Fig. 1 The contents of the asymmetric unit for structures 1–5, showing the atomic numbering scheme. Displacement ellipsoids are shown at the 50% probability level. H atoms not involved in hydrogen bonding interactions are omitted in 4 and 5 for clarity. Atoms labelled with small letters a, b or c refer to molecules A, B and C in the asymmetric unit. | ||
| 1 | 2 | 3 | 4 | 5 | |
|---|---|---|---|---|---|
| Formula | C12H15NO | C13H17NO | C13H16FNO | C13H16ClNO | C13H16BrNO |
| M r | 189.25 | 203.28 | 221.27 | 237.72 | 282.18 |
| Temperature/K | 173 | 173 | 173 | 173 | 173 |
| Crystal size/mm | 0.30 × 0.20 × 0.10 | 0.40 × 0.36 × 0.14 | 0.40 × 0.32 × 0.08 | 0.56 × 0.26 × 0.20 | 0.38 × 0.32 × 0.16 |
| Crystal system | Monoclinic | Orthorhombic | Monoclinic | Monoclinic | Triclinic |
| Space group | P21/c | Pca21 | P21/c | P21/c |
P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) |
| a/Å | 14.140(2) | 13.8232(19) | 11.792(5) | 13.314(2) | 8.9257(13) |
| b/Å | 7.3843(11) | 6.1326(8) | 8.646(4) | 9.3311(13) | 10.8387(16) |
| c/Å | 10.2066(15) | 26.176(4) | 12.170(5) | 29.104(4) | 13.7825(19) |
| α/° | 90 | 90 | 90 | 90 | 69.877(10) |
| β/° | 109.291(10) | 90 | 111.510(7) | 90.109(10) | 87.223(11) |
| γ/° | 90 | 90 | 90 | 90 | 82.095(11) |
| V/Å3 | 1005.8(3) | 2219.0(5) | 1154.4(8) | 3615.6(9) | 1240.1(3) |
| Z | 4 | 8 | 4 | 12 | 4 |
| D calc/g cm–3 | 1.250 | 1.217 | 1.273 | 1.310 | 1.511 |
| µ(Mo-Kα)/mm–1 | 0.079 | 0.077 | 0.091 | 0.295 | 3.294 |
| θ range/° | 1.53 to 28.00 | 1.56 to 28.00 | 1.86 to 28.00 | 1.40 to 28.00 | 2.02 to 28.00 |
| No. unique data | 2430 | 2731 | 2791 | 8724 | 5986 |
| No. data with I ≥ 2σ(I) | 1963 | 2282 | 2003 | 6264 | 4856 |
| R 1 | 0.0459 | 0.0434 | 0.0436 | 0.0471 | 0.0319 |
| wR 2 (all data) | 0.1184 | 0.1126 | 0.1190 | 0.1576 | 0.0958 |
| Absolute parameter17 | — | –10(10) | — | — | — |
| D–H⋯A | D–H/Å | H⋯A/Å | D⋯A/Å | <(D–H⋯A)/° | Symmetry transformations |
|---|---|---|---|---|---|
| 1 | |||||
| N(1A)–H(1As)⋯O(1A) | 0.91(2) | 2.15(2) | 3.047(1) | 169(1) | –x + 1, –y + 2, –z + 1 |
| N(1A)–H(1Aa)⋯O(1A) | 0.89(2) | 2.23(2) | 3.104(1) | 165(1) | x, –y + 3/2, z – 1/2 |
| 2 | |||||
| N(1A)–H(1As)⋯O(1B) | 0.85(4) | 2.09(4) | 2.933(3) | 172(4) | — |
| N(1B)–H(1Bs)⋯O(1A) | 0.99(5) | 1.95(4) | 2.921(3) | 169(4) | — |
| 3 | |||||
| N(1A)–H(1As)⋯O(1A) | 0.87(2) | 1.99(2) | 2.860(2) | 174(2) | –x + 1, –y + 1, –z |
| 4 | |||||
| N(1A)–H(1As)⋯O(1B) | 0.85(3) | 2.03(3) | 2.881(2) | 176(2) | — |
| N(1B)–H(1Bs)⋯O(1A) | 0.88(3) | 2.13(3) | 2.999(2) | 172(2) | — |
| N(1B)–H(1Ba)⋯O(1C) | 0.83(3) | 2.13(3) | 2.903(2) | 157(2) | x, y + 1, z |
| N(1C)–H(1Ca)⋯O(1B) | 0.87(3) | 2.22(3) | 3.043(2) | 159(2) | — |
| 5 | |||||
| N(1A)–H(1As)⋯O(1A) | 0.88(3) | 2.11(3) | 2.981(2) | 174(2) | –x + 1, –y, –z + 1 |
| N(1A)–H(1Aa)⋯O(1B) | 0.86(3) | 2.08(3) | 2.874(2) | 153(2) | — |
| N(1B)–H(1Ba)⋯O(1A) | 0.84(3) | 2.21(3) | 2.959(3) | 149(3) | x + 1, y, z |
The structure of 1-phenylcyclopentanecarboxamide (1), which has one molecule (labelled A) in the asymmetric unit, contains two unique hydrogen bonds. The first, N(1A)–H(1As)⋯O(1A), forms the expected R22(8) dimers that point alternately in the [–110] and [110] directions (Fig. 2a); while the second, N(1A)–H(1Aa) ⋯O(1A), results in the formation of a C(4) chain along the c-axis. The unitary GS notation is N1 = C(4)R22(8), and the binary GS notation is N2 = C12(4)C22(8). The combination of the two types of hydrogen bonds creates a larger ring, a hexamer with GS notation R46(16). The net result is the formation of a 2-D network of alternating R22(8) and R46(16) rings parallel to the bc-plane (Fig. 2b). This 2-D network has a wave-like pattern in which adjacent R22(8) dimers along the b-axis are related by a centre of inversion (Fig. 2c). As a consequence of the inversion centre, both N(1A)–H(1As)⋯O(1A) hydrogen bond distances are equal [d(H⋯O) = 2.15(2) Å].
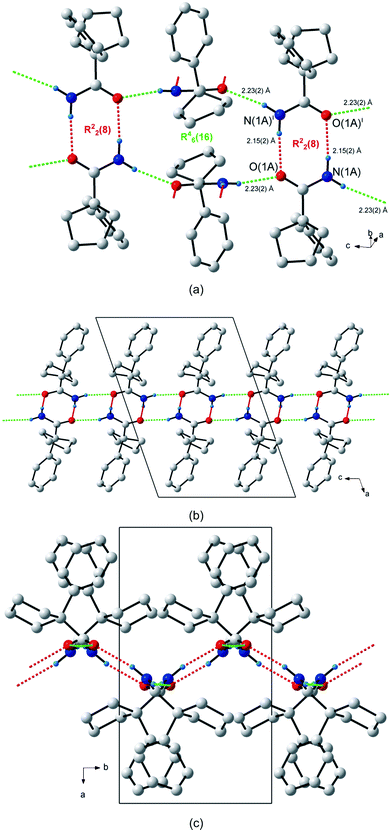 | ||
| Fig. 2 Hydrogen bonding in 1-phenylcyclopentanecarboxamide (1). (a) The two unique hydrogen bonds, shown as dashed red and green lines, forming the R22(8) centrosymmetric hydrogen-bonded dimer and the R46(16) ring. (b) The 2-D hydrogen-bonded pattern formed by the R22(8) and R46(16) rings. (c) Side-on view of the wave-like hydrogen bonded pattern. Atoms marked with a superscript (i) are at the symmetry position (1 – x, 2 – y, 1 – z). H atoms not involved in hydrogen bonding interactions are omitted for clarity. | ||
The structure of 1-phenylcyclohexanecarboxamide (2) has two molecules (labelled A and B) in the asymmetric unit. The two molecules are related by a pseudo inversion centre and the unitary GS notation is N1 = DD. As a consequence, the N2 = R22(8) dimers, connected by N(1A)–H(1As)⋯O(1B) and N(1B)–H(1Bs)⋯O(1A) hydrogen bonds, are non-centrosymmetric. These stack along the direction of the a-axis and point alternately in the [–101] and [101] directions (Fig. 3a). The H⋯O bond distances for N(1A)–H(1As)⋯O(1A) and N(1B)–H(1Bs)⋯O(1B) differ by 0.19 Å. The two anti-orientated H atoms, H(1Aa) on molecule A and H(1Ba) on molecule B, do not participate in hydrogen bonding.
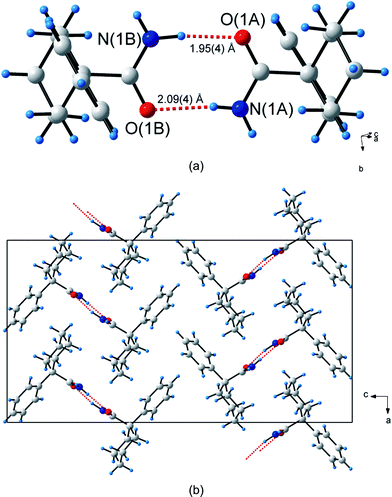 | ||
| Fig. 3 Hydrogen bonding in 1-phenylcyclohexanecarboxamide (2). (a) The two unique hydrogen bonds, shown as dashed red lines, forming the R22(8) non-centrosymmetric hydrogen bonded dimer. (b) Packing diagram of 2. | ||
The asymmetric unit in the crystal structure of 1-(2-fluorophenyl)cyclohexanecarboxamide (3) contains only one molecule. The centrosymmetric R22(8) dimers are joined by N(1A)–H(1As)⋯O(1A) hydrogen bonds (Fig. 4a). These dimers stack in a parallel head-to-head fashion along the c-axis; and they themselves point approximately along the [101] direction (Fig. 4b). Atom H(1Aa) does not participate in any hydrogen bonding interactions. The unitary GS motif is N1 = R22(8). In contrast to the halogen-containing analogues 4 and 5 (vide infra), structure 3 has no C–H⋯π interactions.
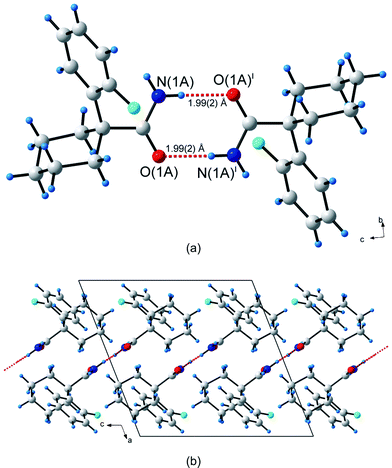 | ||
| Fig. 4 Hydrogen bonding in 1-(2-fluorophenyl)cyclohexanecarboxamide (3). (a) The single unique hydrogen bonds, shown as dashed red lines, forming the R22(8) centrosymmetric hydrogen bonded dimer. (b) Packing diagram of 3. Atoms marked with a superscript (i) are at the symmetry position (1 – x, 1 – y, –z). | ||
The comparatively simple crystal structure of 3 contrasts markedly with the complexity shown by that of 1-(2-chlorophenyl)cyclohexanecarboxamide (4), which has no fewer than three molecules (labelled A, B and C) in its asymmetric unit (Z′ = 3)—the most among all of the compounds reported in this study. Molecules A and B form non-centrosymmetric R22(8) dimers that result from the N(1A)–H(1As)⋯O(1B) and N(1B)–H(1Bs)⋯O(1A) hydrogen bonds (Fig. 5a). These dimers extend along the crystallographic a-direction. Adjacent dimers are linked by molecule C to form an infinite 1-D chain, GS notation C22(8), along the b-axis (Fig. 5b and 5c). The hydrogen bonds creating the chain are N(1B)–H(1Ba)⋯O(1C) and N(1C)–H(1Ca)⋯O(1B). The unitary GS motif is N1 = DDDD and the binary GS pattern is N2 = C22(8)R22(8). Hydrogen atoms H(1Aa) on molecule A and H(1Cs) on molecule C are not involved in any hydrogen bonded interactions. Additionally, there is a weak C–H⋯π interaction present between molecules B and C, with atom C(6B) interacting with the centroid Cg3 of the aromatic group C(8C)–C(13C) through atom H(6B1) [d(C–H⋯Cg3 = 2.76 Å; ∠(C–H⋯Cg3) = 141°; cf.Fig. 5a]. However, there appear to be no significant C–H⋯Cl interactions.
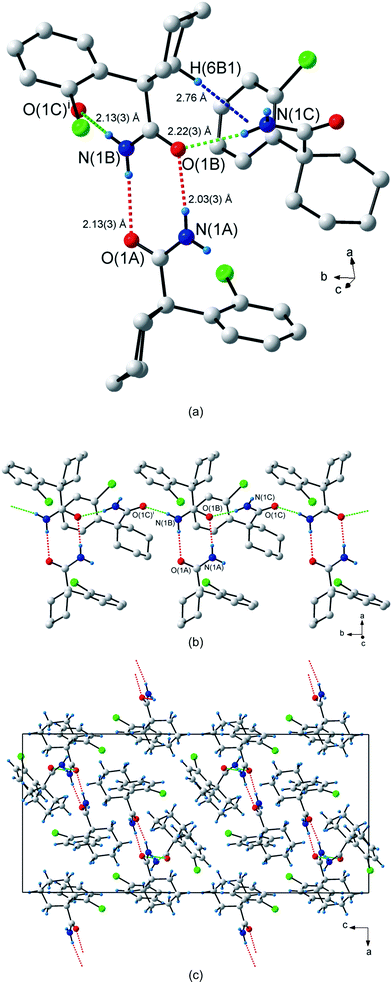 | ||
| Fig. 5 Hydrogen bonding in 1-(2-chlorophenyl)cyclohexanecarboxamide (4). (a) The three types of hydrogen bonds observed in 4. The non-centrosymmetric R22(8) dimer hydrogen bonds are shown as dashed red lines, the hydrogen bonds in the C22(8) chain are shown as dashed green lines and the C–H⋯π hydrogen bond as dashed blue lines. (b) The dimers connected to form an infinite 1-D chain along the b-axis. (c) Packing diagram shown down the b-axis. Atoms marked with a superscript (i) are at the symmetry position (x, 1 + y, z). H atoms not involved in hydrogen bonding interactions are omitted for clarity in (a) and (b). | ||
The crystal structure of 1-(2-bromophenyl)cyclohexanecarboxamide (5) has two molecules (labelled A and B) in the asymmetric unit. Two molecules of A form centrosymmetric R22(8) dimers pointing along the c-axis (Fig. 6a). Neighbouring dimers are linked by molecule B to form a C22(8) chain of hydrogen bonds. The same hexamer with GS notation R46(16) is formed as in (1). The unitary and binary GS notations are N1 = DDR22(8) and N2 = C22(8) respectively. Together, the R22(8) dimer and R46(16) hexamer form an infinite hydrogen bonded ribbon along the a-axis (Fig. 6b and 6c). Atom H(1Bs) plays no part in the hydrogen bonding interactions, i.e. the C12(4) chain observed in 1 is absent and hence no 2-D network is created. However, there is a weak C–H⋯π interaction present between molecules A and B, with atom C(6A) interacting through atom H(6A1) with centroid Cg2 of the aromatic group C(8B)–C(13B) [d(C–H⋯Cg2) = 2.88 Å; ∠(C–H⋯Cg2) = 142°; cf.Fig. 6a]. There is no apparent involvement of the bromine atoms in hydrogen bonding.
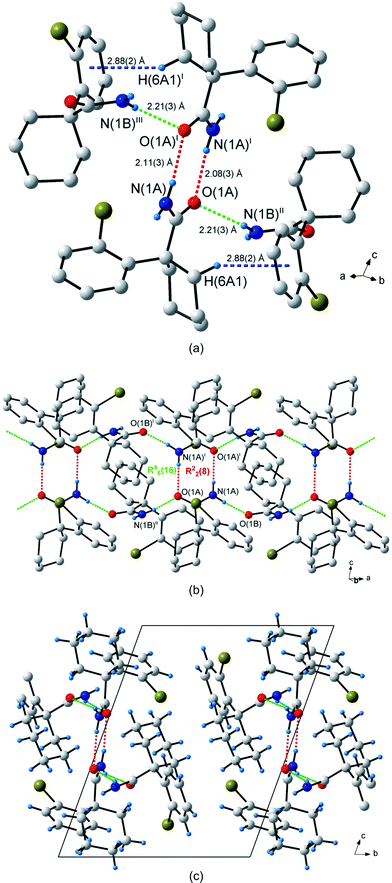 | ||
| Fig. 6 Hydrogen bonding in 1-(2-bromophenyl)cyclohexanecarboxamide (5). (a) The three types of hydrogen bonds observed in 5. The centrosymmetric R22(8) dimer hydrogen bonds are shown as dashed red lines, the hydrogen bonds in the C22(8) chain are shown as dashed green lines and the C–H⋯π hydrogen bond as dashed blue lines. (b) The R22(8) and R46(16) rings connected to form an infinite ribbon pattern along the b-axis. (c) Packing diagram shown down the a-axis. Atoms marked with superscripts (i), (ii) and (iii) are at the symmetry positions (1 – x, –y, 1– z), (–1 + x, y, z) and (2 – x, –y, 1 – z) respectively. H atoms not involved in hydrogen bonding interactions are omitted for clarity in (a) and (b). | ||
Comparison of structures 1–5
The packing and hydrogen-bonding modes in compounds 1–5 are summarised in Table 3. Despite the structural similarity between 1-phenylcyclopentanecarboxamide (1) and 1-phenylcyclohexanecarboxamide (2), the difference in the packing of the two molecules is striking. This difference can be ascribed to the conformation that each cycloalkane ring adopts. In 1, the cyclopentane ring is approximately a half-chair, with the phenyl group in the axial position and the carboxamide in the more exposed equatorial position. This allows neighbouring carboxamide groups to approach each other closely, thereby facilitating not only the formation of the cyclic dimer (the primary supramolecular synthon) but also further interactions that result in the formation of the 2-D hydrogen-bonded network. By contrast, although the cyclohexane ring in 2 adopts a classic chair conformation, the orientations of the carboxamide and phenyl substituents are reversed, and they lie in axial and equatorial positions, respectively. The result is that the dimers are H-shaped (Fig. 3b), with the hydrogen-bonded ring forming the cross-bar. This shape sterically prevents neighbouring dimers from approaching each other and engaging in additional interactions [N(1A)–H(1Aa)⋯O(1A): D⋯A = 3.94(1) Å; H⋯A = 3.08 Å; N(1B)–H(1Ba)⋯O(1B): D⋯A = 3.94(1) Å; H⋯A = 3.09 Å]. Surprisingly, the only closely related structure found in a search of the Cambridge Structural Database18 (Ver. 5.28; November 2006 release) is for cyclopentanecarboxamide itself (reference code BARFEF).19 This compound possesses the same hydrogen bonding pattern as 1; and although the cyclopentane ring adopts a disordered envelope conformation, the amide substituent is effectively equatorial.| 1 | 2 | 3 | 4 | 5 | |
|---|---|---|---|---|---|
| Hydrogen bonding pattern | Centrosymmetric dimers connected to form 2-D network | Non-centrosymmetric discrete dimer | Centrosymmetric discrete dimer | Non-centrosymmetric dimers connected to form 1-D chain | Centrosymmetric dimers connected to form 1-D ribbon |
| Graph set notation | N 1 = C(4)R22(8) | N 1 = DD | N 1 = R22(8) | N 1 = DDDD | N 1 = DDR22(8) |
| N 2 = C12(4)C22(8) | N 2 = R22(8) | N 2 = C22(8)R22(8) | N 2 = C22(8) | ||
| Number of molecules in the asymmetric unit (Z′) | 1 | 2 | 1 | 3 | 2 |
| Position of carboxamide | Equatorial (A) | Axial (A and B) | Equatorial (A) | Equatorial (A and B); axial (C) | Equatorial (A); axial (B) |
| Position of aryl ring | Axial (A) | Equatorial (A and B) | Axial (A) | Axial (A and B); equatorial (C) | Axial (A); equatorial (B) |
| Torsion angle between carboxamide and aryl ring/° | C(7A)–C(1A)–C(6A)–O(1A) = 97.52(12) | C(8A)–C(1A)–C(7A)–O(1A) = –93.4(3); C(8B)–C(1B)–C(7B)–O(1B) = 91.7(3) | C(8A)–C(1A)–C(7A)–O(1A) = 155.82(12) | C(8A)–C(1A)–C(7A)–O(1A) = 148.2(2); C(8B)–C(1B)–C(7B)–O(1B) = –147.13(18); C(8C)–C(1C)–C(7C)–O(1C) = –134.91(19) | C(8A)–C(1A)–C(7A)–O(1A) = –143.24(18); C(8B)–C(1B)–C(7B)–O(1B) = –128.48(19) |
| Packing efficiency25 (%) | 70.7 | 69.7 | 68.4 | 68.5 | 68.0 |
Compounds 2 and 3 both form isolated R22(8) dimers. However, while the dimers in 2 are arranged in a non-centrosymmetric fashion, those in 3 are centrosymmetric. This is reflected in the asymmetric unit in 2 being twice that of 3. Most interestingly, the replacement of an ortho-hydrogen atom in 2 by a fluorine atom in 3 results in a surprising conformational inversion of the cyclohexane ring: the orientations of the carboxamide and aryl rings have switched to equatorial and axial, respectively, making the molecular disposition more like 1 than like 2. Notwithstanding the more spacious arrangement of the dimeric supramolecular synthon that results in the equatorial location of the carboxamide groups, there are no additional hydrogen-bonding interactions between the dimers. In addition, the dihedral angle between the carboxamide and aryl substituents changes substantially upon halogenation of the latter, as one might expect from the greater degree of steric interference; for 1 and 2 the angle is a little greater than ±90°, while for 3–5 it is around ±145° for the molecules involved in dimer formation (Table 3).
The structures of the chloro and bromo analogues 4 and 5 are even more unexpected, as they now contain extra hydrogen-bonded interactions. As described above, the non-centrosymmetric R22(8) dimers in 4 (molecules A and B) are linked through a third, independent, molecule (C) to create a chain-type motif (Fig. 5b); and in 5, the centrosymmetric dimers (molecules A) are linked by non-dimeric molecules (B) to give a larger hexameric hydrogen-bonded motif (Fig. 6b) that associates further to form ribbons. Once again, the changes appear to be related to the conformations adopted by the cyclohexane rings. In all three of the halogen-containing compounds, the molecules that form the dimers have their carboxamide functional groups in equatorial positions. However, the cyclohexane ring is again inverted in the non-dimeric molecules in 4 and 5. In the chloro compound 4, the molecule that links the dimers into chains (molecule C) has an axial carboxamide functional group; and the same holds for the molecule in the bromo compound 5 (molecule B) that links the dimers into hexameric units. The torsion angle between the carboxamide and aryl substituents deviates correspondingly between the molecules that form dimers and the molecules that link the dimers (Table 3). In both 4 and 5, the aryl ring of the non-dimeric linking molecules further rigidifies the crystal structure by C–H⋯π association with an axial hydrogen atom adjacent to the substituted carbon atom on the cyclohexane ring; but in none of the halogenated compounds is the halogen atom involved in hydrogen bonding.
Conclusion
All five carboxamide derivatives reported in this article form hydrogen bonded dimers in the solid state with both centrosymmetric and non-centrosymmetric R22(8) motifs observed. The packing of these dimers varies in the crystal structure and is related at least in part to variation in the conformation of the cycloalkane ring, with consequences for the relative axial and equatorial orientations of the carboxamide and aryl substituents. However, it is also possible that the dimer observed in all five compounds is such a strong motif that it may be the primary unit for packing, in which case the conformational differences observed for the cycloalkane rings may be the consequence, rather than the cause, of the supramolecular arrangements. The extent to which the nature of the ortho-substituent in 2–5 influences the ring conformations and the packing modes is no doubt also significant, but a more substantial investigation would be required before one could attempt to predict outcomes. Furthermore, the effect of cycloalkane ring size on hydrogen-bonding patterns, while clearly important, needs to be explored further; it would be particularly interesting to probe the consequences of incorporating large-ring systems with greater conformational flexibility and variability. Finally, the likelihood of our having crystallised out specific polymorphs of compounds 1–5 cannot be discounted, and there remain exciting possibilities for preparing polymorphs that might reveal conformational variations in the molecular structure.Experimental
Materials
Compounds 3–5 were prepared by hydrolysis of the corresponding nitriles with a mixture of acetic acid and concentrated sulfuric acid at 50 °C, as described previously.12 Compounds 1 and 2, prepared by the same method in this work, have previously been obtained by treatment of the corresponding acid chlorides with ammonia.20 Physical and spectroscopic properties of the five compounds agreed with those reported in the earlier publications. For the X-ray diffraction studies, crystals of compounds 1 and 2 were grown from benzene–hexane, 3 from acetone, and 4 and 5 from chloroform–hexane.X-Ray crystallography
Intensity data were collected on a Bruker SMART 1 K CCD area detector diffractometer with graphite monochromated Mo-Kα radiation (50 kV, 30 mA) and performed at T = 173 K. The collection method involved ω-scans of width 0.3°. Data reduction was carried out using the program SAINT+, version 6.0221 and face indexed absorption corrections were made using the program XPREP.21The crystal structure was solved by direct methods using SHELXS-97.22 Non-hydrogen atoms were first refined isotropically followed by anisotropic refinement by full matrix least-squares calculations based on F2 using SHELXL-97.22Hydrogen atoms were first located in the difference map and all CH hydrogen atoms were then positioned geometrically and allowed to ride on their respective parent atoms. NH hydrogen atom positions were allowed to refine freely and their isotropic thermal parameters were assigned as 1.2 times those of their parent atoms. Diagrams and publication material were generated using WinGx,23 ORTEP,24 PLATON25 and DIAMOND.26 Graph set notations were assigned using the program RPLUTO.27 Further crystallographic data are summarised in Table 1.
Acknowledgements
This work was supported by grants from the National Research Foundation, Pretoria (GUN 2053652) and the University of the Witwatersrand. We thank the University for providing the infrastructure required to do this work. We are also grateful to Dr M. A. Fernandes (University of the Witwatersrand) and Dr M. Rademeyer (University of KwaZulu–Natal) for their valuable comments.References
- See, among others, the following reviews: D. Braga, L. Brammer and N. R. Champness, CrystEngComm, 2005, 7, 1–19 Search PubMed; C. B. Aakeröy and D. J. Salmon, CrystEngComm, 2005, 7, 439–448 RSC; C. V. K. Sharma, Cryst. Growth Des., 2002, 2, 465–474 RSC; G. R. Desiraju, Curr. Opin. Solid State Mater. Sci., 1997, 2, 451–454 CrossRef CAS; C. B. Aakeröy, Acta Crystallogr., Sect. B, 1997, 53, 569–586 CrossRef CAS; G. R. Desiraju, in Comprehensive Supramolecular Chemistry, ed. D. D. MacNicol, F. Toda and R. Bishop, Pergamon Press, Oxford, 1996, vol. 6, ch. 1, pp. 1–22 and references cited therein CrossRef.
- G. R. Desiraju, Angew. Chem., Int. Ed. Engl., 1995, 34, 2311–2327 CrossRef CAS.
- See, among others, the following reviews: C. B. Aakeröy and A. M. Beatty, Aust. J. Chem., 2001, 54, 409–421 Search PubMed; G. R. Desiraju, Acc. Chem. Res., 1996, 29, 441–449 CrossRef CAS; C. B. Aakeröy and K. R. Seddon, Chem. Soc. Rev., 1993, 22, 397–407 CrossRef CAS; M. C. Etter, J. Phys. Chem., 1991, 95, 4601–4610 RSC and references cited therein.
- L. Leiserowitz and A. T. Hagler, Proc. R. Soc. London, Ser. A, 1983, 388, 133–175 CrossRef CAS.
- S. S. Kuduva, D. Bläser, R. Boese and G. R. Desiraju, J. Org. Chem., 2001, 66, 1621–1626 CrossRef CAS and numerous references cited therein.
- For a recent example, see: N. Boechat, L. C. Maciel, A. C. Pinto, S. M. S. V. Wardell, J. M. S. Skakle and R. A. Howie, Acta Crystallogr., Sect. C, 2005, 61, o270–o275 Search PubMed.
- D. Das, R. K. R. Jetti, R. Boese and G. R. Desiraju, Cryst. Growth Des., 2003, 3, 675–681 CrossRef CAS; R. Centore and A. Tuzi, Cryst. Eng., 2003, 6, 87–97 CrossRef CAS.
- E. A. Bruton, L. Brammer, F. C. Pigge, C. B. Aakeröy and D. S. Leinen, New J. Chem., 2003, 27, 1084–1094 RSC.
- V. Bertolasi, P. Gilli, V. Ferretti and G. Gilli, Acta Crystallogr., Sect. B, 1998, 54, 50–65 CrossRef.
- P. I. Coupar, C. Glidewell and G. Ferguson, Acta Crystallogr., Sect. B, 1997, 53, 521–533 CrossRef.
- I. Fleming, M. A. Loreto, J. P. Michael and I. H. M. Wallace, Tetrahedron Lett., 1982, 23, 2053–2056 CrossRef CAS.
- I. Fleming, M. A. Loreto, J. P. Michael and I. H. M. Wallace, J. Chem. Soc., Perkin Trans. 1, 1986, 349–359 RSC.
- C. Clarke, I. Fleming, J. M. D. Fortunak, P. T. Gallagher, M. C. Honan, A. Mann, C. O. Nübling, P. R. Raithby and J. J. Wolff, Tetrahedron, 1988, 44, 3931–3944 CrossRef.
- I. Roufos, S. J. Hays, D. J. Dooley, R. D. Schwarz, G. W. Campbell and A. W. Probert, Jr., J. Med. Chem., 1994, 37, 268–274 CrossRef CAS; S. N. Calderon, S. Izenwasser, B. Heller, J. S. Gutkind, M. V. Mattson, T.-P. Su and A. H. Newman, J. Med. Chem., 1994, 37, 2285–2291 CrossRef CAS.
- F. H. Allen, O. Kennard, D. G. Watson, L. Brenner, A. G. Orpen and R. Taylor, J. Chem. Soc., Perkin Trans. 2, 1987, S1–S19 RSC.
- J. Bernstein, R. E. Davis, L. Shimoni and N.-L. Chang, Angew. Chem., Int. Ed. Engl., 1995, 34, 1555–1573 CrossRef CAS; M. C. Etter, J. C. MacDonald and J. Bernstein, Acta Crystallogr., Sect. B, 1990, 46, 256–262 CrossRef; M. C. Etter, Acc. Chem. Res., 1990, 23, 120–126 CrossRef CAS.
- H. D. Flack, Acta Crystallogr., Sect. A, 1983, 39, 876–881 CrossRef.
- F. H. Allen, Acta Crystallogr., Sect. B, 2002, 58, 380–388 CrossRef.
- H. J. Winter, H. J. Hecht and H. Bradaczek, Acta Crystallogr., Sect. B, 1981, 37, 2183–2185 CrossRef.
- A. Kalir and Z. Pelah, Isr. J. Chem., 1967, 5, 223–229 CAS.
- Bruker, SAINT+. Version 6.02 (includes XPREP and SADABS), Bruker AXS Inc., Madison, Wisconsin, USA, 1999 Search PubMed.
- G. M. Sheldrick, SHELX, release 97–2 (includes SHELXS and SHELXL), University of Göttingen, Germany, 1997 Search PubMed.
- L. J. Farrugia, WinGX, J. Appl. Crystallogr., 1999, 32, 837–838 CrossRef.
- L. J. Farrugia, J. Appl. Crystallogr., 1997, 30, 565 CrossRef CAS.
- A. L. Spek, J. Appl. Crystallogr., 2003, 36, 7–13 CrossRef CAS.
- K. Brandenburg, Diamond. Version 2.1e., Crystal Impact GbR, Bonn, Germany, 1999 Search PubMed.
- RPLUTO, Cambridge Crystallographic Data Centre, Cambridge, UK, 2004–2007, http://www.ccdc.cam.ac.uk/free_services/rpluto/. Search PubMed.
Footnote |
| † CCDC reference numbers 649368–649372. For crystallographic data in CIF or other electronic format see DOI: 10.1039/b708333e |
| This journal is © The Royal Society of Chemistry 2008 |