A microwave assisted intramolecular-furan-Diels–Alder approach to 4-substituted indoles†
Filip
Petronijevic
,
Cody
Timmons‡
,
Anthony
Cuzzupe§
and
Peter
Wipf
*
Department of Chemistry, University of Pittsburgh, Pittsburgh, Pennsylvania, 15260, USA. E-mail: pwipf@pitt.edu; Fax: +1 412-624-0787; Tel: +1 412-624-8606
First published on 11th November 2008
Abstract
The key steps of a versatile new protocol for the convergent synthesis of 3,4-disubstituted indoles are the addition of an α-lithiated alkylaminofuran to a carbonyl compound, a microwave-accelerated intramolecular Diels–Alder cycloaddition and an in situ double aromatization reaction.
The indole moiety is a ubiquitous structural motif that is found in a wide array of naturally occurring alkaloids and designed therapeutic agents. Interestingly, a relatively uniform fraction of ∼4% of all pharmaceuticals, high-throughput screening samples, as well as natural products, contain an aromatic or partially saturated indole core (Fig. 1).1 Accordingly, the search for efficient protocols for the synthesis of substituted indoles has remained an important research topic for over a century.2 As part of a program aimed at the synthesis of Ergot alkaloids,3,4 we have investigated suitable methodologies for the late-stage introduction of a 4-substituted indole ring system. Padwa and co-workers have developed an intramolecular Diels–Alder furancycloaddition reaction that provides substituted indolines from N-homoallylic 2-aminofurans.5,6 We envisioned that the addition of an α-lithiated alkylaminofuran, 1, to an α,β-unsaturated carbonyl compound, 2, followed by the in situcycloaddition and dehydrative aromatization of 6, would allow an extension of this methodology to the direct preparation of 4-substituted indoles, 7 (Scheme 1).
 | ||
| Fig. 1 Representative synthetic and natural products with 4-substituted indole scaffolds (outlined in bold).1 | ||
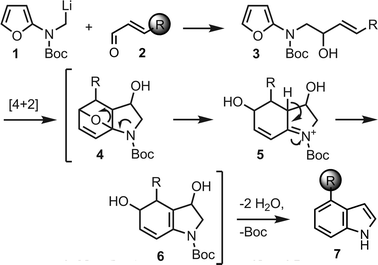 | ||
| Scheme 1 Proposed indole synthesis. | ||
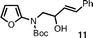
The preparation of lithium reagent 1 was accomplished by transmetalation of stannane10 (Scheme 2), which was readily obtained from known iodide87 and Boc-protected 2-aminofuran98 in the presence of sodium hydride. We next explored conditions for the addition of 10 to α,β-unsaturated carbonyl compounds. Transmetalation with n-butyllithium in THF occurred only sluggishly at −100 °C, and decomposition was found to be the major pathway at 0 °C. However, at −78 °C, transmetalation was complete within 15 min, and upon treatment with an excess of cinnamaldehyde, the expected addition product, 11, was obtained in 66% yield.
 | ||
| Scheme 2 The preparation of furanyl stannane 10, and a typical transmetalation and aldehydeaddition reaction. | ||
After establishing a viable route to homoallylic furanyl amines, we turned our attention to the intramolecular Diels–Alder cascade indole formation process. Initially, alcohol 11 was heated in toluene at reflux. After 48 h, gradual decomposition of the starting material was observed. In contrast, when 11 was heated in o-dichlorobenzene under microwave irradiation for 30 min at 170 °C, complete consumption of the starting material was observed, and 4-phenylindole (12) was isolated in 72% yield, presumably according to the mechanism shown in Scheme 1. Indeed, this process proceeded concomitantly with thermal Boc deprotection.9 In an effort to shorten the reaction time, temperatures were varied, and optimal conditions were found to involve microwave heating of furan derivatives for 20 min at 180 °C. Under these conditions, 12 was obtained in 79% yield from 11 (Table 1, entry 1). The success of the microwave conditions vs. standard heating is likely to be a consequence of the much faster heating process and the higher temperatures in the pressurized reaction vial, which can easily be accomplished with current microwave reactors.
| Entry | Substrate | Time/min | Product | Yield (%) |
|---|---|---|---|---|
| 1 |
|
20 |

|
79 |
| 2 |

|
20 |

|
76 |
| 3 |

|
20 |

|
83 |
| 4 |
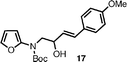
|
20 |

|
69 |
| 5 |

|
20 |

|
77 |
| 6 |

|
20 |

|
81 |
| 7 |

|
20 |
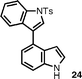
|
71 |
| 8 |
|
20 |

|
61 (7 : 4) |
| 9 |
|
25 |
|
69 |
| 10 |

|
30 |
|
48 |
| 11 |

|
30 |

|
36 |
| 12 |

|
30 |

|
84 |
This annulation strategy is quite tolerant of functional groups. Initially, we varied the 4-aryl substituents from simple electron-rich (entries 2 and 4) to electron-deficient arenes (entry 3), observing a slight increase in yield to 83% in the case of the 4-fluoro aryl substituent. Para- and meta-substituted arenes with ester-functionalized side chains behaved analogously to the methyl group (entries 5 and 6). The use of a tosyl-protected indole ring in substrate 23 provided the corresponding 3,4′-bisindole24 (entry 7).
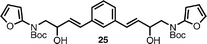
When symmetric bisfuran25 was subjected to the Diels–Alder cascade process, diannulated products 26 and 27 were obtained exclusively in a 7 : 4 ratio, with no monocyclization being observed (entry 8). Interestingly, in this case, the major product still contained a single Bocprotecting group. The formation of a monoprotected derivative of an otherwise symmetrical bisindole could potentially be advantageous for selective functionalizations and desymmetrizations. Alternatively, re-subjecting this compound to thermal reaction conditions (microwave irradiation at 180 °C, 20 min) cleanly removed the residual Boc functionality to afford fully deprotected species 27 in 89% yield after chromatographic purification.
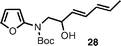
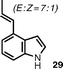
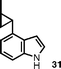
The introduction of alkenyl and alkyl groups in position 4 of the newly formed indole ring was also possible (entries 9–12). Propenyl derivative 29 was isolated in 69% yield in a 7 : 1 ratio of E : Z isomers, while the efficiency of the process was slightly reduced for cyclopropyl compound 31 (48% yield) and isopropyl indole33 (36% yield). In spite of the lower yield observed for 31, the successful use of a cyclopropane-substituted compound in this reaction sequence is noteworthy.
We were also interested in expanding the scope of this reaction to 3,4-disubstituted indoles, derived analogously from additions of lithium reagent 1 to α,β-unsaturated ketones. Specifically, tert-alcohol34 was obtained by the treatment of 1 with 5 equiv. of 2-cyclohexene-1-one. The exposure of 34 to the standard microwave conditions provided cyclohexane-annulated indole35 in 84% yield. Tricycle 35 is representative of the core heterocycle of many Ergot alkaloids.
In conclusion, we have developed a new method for the convergent and rapid preparation of 4-monosubstituted and 3,4-disubstituted indoles featuring the microwave-assisted Diels–Alder cyclization of furans. The cascade process is quite tolerant of functional groups and associated substitution patterns. This strategy is a convenient alternative to the common transition metal-mediated coupling processes for the synthesis of these heterocycles.
This work has been supported by the NIH/NIGMS CMLD program (GM067082), and, in part, by Merck Research Laboratories.
Notes and references
- (a) S. L. Roach, R. I. Higuchi, M. E. Adams, Y. Liu, D. S. Karanewsky, K. B. Marschke, D. E. Mais, J. N. Miner and L. Zhi, Bioorg. Med. Chem. Lett., 2008, 18, 3504 CrossRef CAS; (b) C. S. Li, D. Deschenes, S. Desmarais, J.-P. Falgueyret, J. Y. Gauthier, D. B. Kimmel, S. Leger, F. Masse, M. E. McGrath, D. J. McKay, M. D. Percival, D. Riendeau, S. B. Rodan, M. Therien, V.-L. Truong, G. Wesolowski, R. Zamboni and W. C. Black, Bioorg. Med. Chem. Lett., 2006, 16, 1985 CrossRef CAS; (c) J. Doukas, W. Wrasidlo, G. Noronha, E. Dneprovskaia, R. Fine, S. Weis, J. Hood, A. DeMaria, R. Soll and D. Cheresh, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 19866 CrossRef CAS; (d) N. G. Vinokurova, D. M. Boichenko, B. P. Baskunov, N. F. Zelenkova, I. G. Vepritskaya, M. U. Arinbasarov and T. A. Reshetilova, Appl. Biochem. Microbiol., 2001, 37, 184 Search PubMed; (e) U. Huber, R. E. Moore and G. M. L. Patterson, J. Nat. Prod., 1998, 61, 1304 CrossRef CAS; (f) S. Smith and G. M. Timmis, Nature, 1934, 133, 579 CrossRef CAS.
- (a) R. A. Glennon, J. Med. Chem., 1987, 30, 1 CrossRef CAS; (b) H. M. Hugel and D. Kennaway, Org. Prep. Proced. Int., 1995, 27, 1 CrossRef CAS; (c) M. Lounasmaa and A. Tolvanen, Nat. Prod. Rep., 2000, 17, 175 RSC; (d) M. Somei and F. Yamada, Nat. Prod. Rep., 2004, 21, 278 RSC; (e) T. Kawasaki and K. Higuchi, Nat. Prod. Rep., 2005, 22, 761 RSC; (f) S. E. O’Connor and J. J. Maresh, Nat. Prod. Rep., 2006, 23, 532 RSC.
- P. L. Schiff, Am. J. Pharm. Educ., 2006, 70, 98 Search PubMed.
- For our previous studies on indoline and isoindolinone synthesis using nucleophilic addition and radical annulation strategies, see: (a) P. Wipf and Y. Kim, Tetrahedron Lett., 1992, 33, 5477 CrossRef CAS; (b) J. G. Pierce, D. L. Waller and P. Wipf, J. Organomet. Chem., 2007, 692, 4618 CrossRef CAS; (c) P. Wipf and J. P. Maciejewski, Org. Lett., 2008, 10, 4383 CrossRef CAS; (d) J. G. Pierce, D. Kasi, M. Fushimi, A. Cuzzupe and P. Wipf, J. Org. Chem., 2008, 73, 7807 CrossRef CAS.
- (a) C. O. Kappe, S. S. Murphree and A. Padwa, Tetrahedron, 1997, 53, 14179 CrossRef CAS; (b) A. Padwa, M. A. Brodney and M. Dimitroff, J. Org. Chem., 1998, 63, 5304 CrossRef CAS; (c) A. Padwa, M. A. Brodney, B. Liu, K. Satake and T. Wu, J. Org. Chem., 1999, 64, 3595 CrossRef CAS; (d) A. Padwa, M. A. Brodney, K. Satake and C. S. Straub, J. Org. Chem., 1999, 64, 4617 CrossRef CAS; (e) A. Padwa, M. A. Brodney and S. M. Lynch, J. Org. Chem., 2001, 66, 1716 CrossRef CAS; (f) S. K. Bur, S. M. Lynch and A. Padwa, Org. Lett., 2002, 4, 473 CrossRef CAS; (g) J. D. Ginn and A. Padwa, Org. Lett., 2002, 4, 1515 CrossRef CAS; (h) H. Zhang, J. Boonsombat and A. Padwa, Org. Lett., 2007, 9, 279 CrossRef CAS.
- For other pertinent intramolecular furan Diels–Alder reactions, see: (a) L. L. Klein, J. Org. Chem., 1985, 50, 1770 CrossRef CAS; (b) M. E. Jung and J. Gervay, J. Am. Chem. Soc., 1989, 111, 5469 CrossRef CAS.
- J. Ahman and P. Somfai, Synth. Commun., 1994, 24, 117 CAS.
- A. Padwa, M. A. Brodney and S. M. Lynch, Org. Synth., 2002, 78, 202 CAS.
- (a) H. H. Wasserman, G. D. Berger and K. R. Cho, Tetrahedron Lett., 1982, 23, 465 CrossRef CAS; (b) P. Wipf and M. Furegati, Org. Lett., 2006, 8, 1901 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental procedures and full characterization of all the final products. See DOI: 10.1039/b816989f |
| ‡ Current address: Department of Chemistry and Physics, Southwestern Oklahoma State University, Weatherford, Oklahoma, 73096, USA. |
| § Current address: Cytopia Research PTY Ltd., Richmond, 3121, Australia |
| This journal is © The Royal Society of Chemistry 2009 |