Anti-Markovnikov hydroamination and hydrothiolation of electron-deficient vinylarenes catalyzed by well-defined monomeric copper(I) amido and thiolate complexes†
Colleen
Munro-Leighton
,
Samuel A.
Delp
,
Nikki M.
Alsop
,
Elizabeth D.
Blue
and
T. Brent
Gunnoe
*
Department of Chemistry, North Carolina State University, Raleigh, 27695-8204, USA. E-mail: brent_gunnoe@ncsu.edu; Fax: +1 919-515-8909; Tel: +1 919-513-3704
First published on 24th October 2007
Abstract
Monomeric Cu(I) amido and thiolate complexes that are supported by the N-heterocyclic carbene ligand 1,3-bis(2,6-diisopropylphenyl)imidazol-2-ylidene (IPr) catalyze the hydroamination and hydrothiolation of electron-deficient vinylarenes with reactivity patterns that are consistent with an intermolecular nucleophilic addition of the amido/thiolate ligand of (IPr)Cu(XR) (X = NH or S; R = Ph, CH2Ph) to free vinylarene.
The formation of C–S and C–N bonds via the addition of S–H/N–H bonds of thiols/amines across olefins is a promising atom efficient synthetic method.1 Only a few transition metal catalysts for the regioselective hydroamination of styrenes have been reported.2 Rarely observed “anti-Markovnikov” selectivity has been reported with Rh and Ru systems,2e–h while metal halides in combination with triflate salts or metal triflate systems catalyze “Markovnikov” addition of arylsulfonamides to vinylarenes and other olefins.3 It is likely that many of these metal halide or triflate systems generate protic acid that functions as the active catalyst.4 Similar to hydroamination, examples of transition metal catalyzed hydrothiolation of vinylarenes are relatively rare.5
We have recently prepared a series of monomeric CuI complexes with formally anionic heteroatomic ligands including amido, alkoxo, aryloxo and thiolate fragments.6 Herein, we report that (IPr)Cu(X) {X = NHR or SR; IPr = 1,3-bis(2,6-diisopropylphenyl)imidazol-2-ylidene, Chart 1} systems catalyze regioselective “anti-Markovnikov” hydroamination and hydrothiolation of activated vinylarenes.
![(IPr)Cu(X) [X = NHPh (1), NHCH2Ph (2), SPh (3) or SCH2Ph (4)] catalyst precursors.](/image/article/2008/CC/b715507g/b715507g-c1.gif) | ||
| Chart 1 (IPr)Cu(X) [X = NHPh (1), NHCH2Ph (2), SPh (3) or SCH2Ph (4)] catalyst precursors. | ||
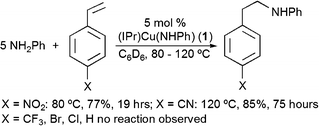
A benzene solution of 5 mol% (based on olefin) (IPr)Cu(NHPh) (1) with aniline and para-nitrostyrene produces the “anti-Markovnikov” product N-phenyl-para-nitrophenethylamine in 77% yield after 21 h at 80 °C (eqn 1). All yields given in eqn 1–5 have been determined by 1H NMR spectroscopy against an internal standard. All organic products have been isolated and fully characterized, and isolated yields are provided in the ESI.† As previously discussed by Hartwig et al., the use of excess amine is required to produce substantial or complete conversion.7Control reactions in the absence of Cucatalyst do not result in hydroamination of vinylarene.
 | (1) |
The reaction of aniline and para-cyanostyrene is also catalyzed by 1, but a higher temperature (120 °C) and longer reaction time are required (eqn 1). Attempts at the catalytic hydroamination with aniline for vinylarenes that possess para-substituents with reduced electron-withdrawing ability do not yield new organic products. For example, heating solutions of 1 (C6D6) and aniline with para-X-styrene {where X = CF3 (σp− = 0.65), Br (σp− = 0.25), Cl (σp− = 0.19), or H (σp− = 0)} to 120 °C results in no change after 48 h (σp− for NO2 = 1.27 and CN = 1.00).
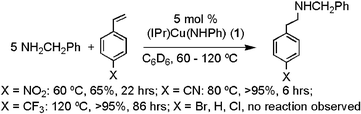
Although we have not been able to isolate the CuI amido complex formed from benzylamine, (IPr)Cu(NHCH2Ph) (2), this system can likely be generated in situ upon reaction of (IPr)Cu(NHPh) and benzylamine. The combination of 1 and benzylamine at 100 °C produces a new (IPr)Cu system and free aniline. Attempted isolation of 2 results in decomposition (see the ESI†). Use of 1 as catalyst precursor with benzylamine extends the range of substituted styrenes that undergo hydroamination to the p-CF3 compound (eqn 2). Thus, the transformations with benzylamine are successful when the para substituent of the vinylarene is nitro, cyano or perfluoromethyl (eqn 2), while the analogous reaction of the p-CF3–styrene with aniline does not undergo reaction after 7 days (120 °C). Apparently, complex 1 is not sufficiently reactive to enter the catalytic cycle for the less activated perfluoromethyl styrene, and we presume that catalysis with the p-CF3–styrene and benzylamine occurs via initial formation of complex 2. For catalysis with benzylamine, yields by 1H NMR are quantitative for X = CN or CF3.
 | (2) |
In addition to control reactions in the absence of Cu amido complexes, which do not give the hydroamination product (see above), we attempted to catalyze these reactions with lithium amido or pyridine as base. Under conditions identical to the Cu-catalyzed reactions shown in eqn 1, aniline/para-nitrostyrene and aniline/para-cyanostyrene do not react in the presence of 5 mol% [Li][NHPh]. Likewise, attempted reaction (again, under conditions analogous to the Cu-catalyzed reactions) of benzylamine with para-cyanostyrene and aniline with para-nitrostyrene in the presence of pyridine do not give the hydroamination product.
Several mechanisms have been elucidated for transition metal catalyzed hydroamination (and related reactions) of olefins. For example, early transition metal imido complexes have been proposed to undergo initial net [2 + 2] cycloaddition of olefins.8Hydroamination of olefins by late transition metal systems have been proposed to occur by olefin coordination and subsequent nucleophilic addition of the amine,1a formation of a π–arene complex and activation toward nucleophilic addition of the amine,2holefin insertion into M–NR2 bonds,1a and formation of η3-allyl complexes (for styrenes) followed by amine addition to the allyl ligand.2a,c
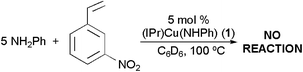
The hydroamination reactions catalyzed by complexes 1 and 2 appear to be dependent upon the electron withdrawing ability of the para-substituent of the vinylarene. For the closely related addition of aniline to acrylonitrile, we have reported evidence supporting a pathway that involves rate-determining nucleophilic addition of the amido ligand of (IPr)Cu(NHPh) to freeacrylonitrile.6c Similarly, we suggest that a related mechanism may be operative for the hydroamination of vinylarenes in which the rate-determining step is intermolecular nucleophilic addition of the heteroatom to free vinylarene (Scheme 1). Although the data are limited, the relative predilection toward hydroamination of vinylarenes as a function of the para-substituent (i.e., reactivity observed only for more electron-withdrawing groups) is consistent with the proposed pathway. In addition, the more nucleophilic benzylamine reacts with the p-NO2 and p-CN vinylarenes at lower temperature than does aniline, and benzylamine reacts with the p-CF3 while aniline does not (eqn 1–2). Furthermore, although catalytic conversion of aniline and p-NO2–styrene is successful, attempted catalysis with aniline and m-NO2–styrene fails to yield hydroamination at temperatures up to 100 °C after 24 h (eqn 3).
![Proposed mechanism for hydroamination and hydrothiolation of vinylarenes [X = NHPh (1), NHCH2Ph (2), SPh (3) or SCH2Ph (4); Y = electron withdrawing group].](/image/article/2008/CC/b715507g/b715507g-s1.gif) | ||
| Scheme 1 Proposed mechanism for hydroamination and hydrothiolation of vinylarenes [X = NHPh (1), NHCH2Ph (2), SPh (3) or SCH2Ph (4); Y = electron withdrawing group]. | ||
| (3) |
As an additional confirmation of the importance of the nucleophile, we attempted hydrothiolation of para-nitrostyrene with (IPr)Cu(SR)/thiols, which are presumably more nucleophilic than the (IPr)Cu(NHR)/amine analogs. Similar to the hydroamination reactions, using (IPr)Cu(SR) {R = Ph (3) or CH2Ph (4)} as catalyst affords the hydrothiolation of the activated p-NO2 compound at room temperature (eqn 4). Under these conditions (i.e., <80 °C), reactions with less electron-withdrawing groups (e.g., X = CN, CF3, Br, Cl or H) do not proceed to products.
 | (4) |
In an effort to demonstrate the potential utility of these Cu catalysts, we have prepared N-methyl-N-benzyl-para-nitrophenethylamine as shown in eqn 5. This compound is a precursor to a class III antiarrhythmic agent and has been prepared in four steps (overall yield 77%) from commercially available reagents in reaction sequences that incorporate HBr, HCl, benzene, ethanol, MeI, acetonitrile, benzaldehyde, potassium borohydride, formaldehyde and formic acid.9 Using (IPr)Cu(NHPh) as catalyst, we have synthesized and isolated the amine in a single step from commercially available reagents (eqn 5). By 1H NMR, the reaction proceeds to ∼70% yield, and the amine has been isolated in ∼50% yield. Although our unoptimized yield is lower than the previous process, the procedure is simpler and generates fewer byproducts and waste.
 | (5) |
In summary, we have reported well-defined monomeric Cu(I) amido and thiolate complexes that catalyze the addition of N–H and S–H bonds of amines and thiols to activated vinylarenes. The anti-Markovnikov selectivity, which has only rarely been achieved with transition metal catalysts, is consistent with a mechanism that involves intermolecular X–C (X = N or S) bond formation and is inconsistent with transformations catalyzed by Brønsted acid impurities. Although non-catalyzed additions of amines to activated vinylarenes in polar solvents have been reported,10 such transformations were not accessible with aniline or alkyl anilines, nor have they been reported for activating groups other than nitro. The Cu-catalyzed reactions likely resemble simple base-catalyzed olefin hydroamination,1f while potentially offering the flexibility of transition metal catalysis [via adjustment of ligand(s) and metal oxidation state] and milder reaction conditions (most base-catalyzed hydroamination reactions of vinylarenes occur at higher temperatures, often >150 °C).1f To our knowledge, reports of transition metal catalyzed “anti-Markovnikov” hydroamination of styrenes are limited to Ru and Rh, which are relatively expensive metals. Even though more active catalysts will be required for general synthetic utility, the development of Cu catalysts that are active for the hydroamination of activated vinyl arenes is a first step toward relatively inexpensive metals.
We acknowledge the NSF (CAREER Award, CHE 0238167) and the Alfred P. Sloan Foundation for financial support.
Notes and references
- (a) T. E. Müller and M. Beller, Chem. Rev., 1998, 98, 675–703 CrossRef; (b) M. Beller, J. Seayad, A. Tillack and H. Jiao, Angew. Chem., Int. Ed., 2004, 43, 3368–3398 CrossRef CAS; (c) S. Hong and T. Marks, Acc. Chem. Res., 2004, 37, 673–686 CrossRef CAS; (d) J. F. Hartwig, Pure Appl. Chem., 2004, 76, 507–516 CrossRef CAS; (e) J.-J. Brunet and D. Neibecker, in Catalytic Hetero-functionalization: From Hydroamination to Hydrozirconation, ed. A. Togni and H. Grützmacher, Wiley-VCH, Weinheim, 2001; pp. 91–132 Search PubMed; (f) J. Seayad, A. Tillack, C. Hartung and M. Beller, Adv. Synth. Catal., 2002, 344, 795–813 CrossRef CAS.
- (a) U. Nettekoven and J. F. Hartwig, J. Am. Chem. Soc., 2002, 124, 1166–1167 CrossRef CAS; (b) M. Utsunomiya and J. F. Hartwig, J. Am. Chem. Soc., 2003, 125, 14286–14287 CrossRef CAS; (c) L. K. Vo and D. A. Singleton, Org. Lett., 2004, 6, 2469–2472 CrossRef CAS; (d) K. Li, P. N. Horton, M. B. Hursthouse and K. K. Hii, J. Organomet. Chem., 2003, 665, 250–257 CrossRef CAS; (e) M. Utsunomiya, R. Kuwano, M. Kawatsura and J. F. Hartwig, J. Am. Chem. Soc., 2003, 125, 5608–5609 CrossRef CAS; (f) M. Beller, H. Trauthwein, M. Eichberger, C. Breindl, J. Herwig, T. E. Müller and O. R. Thiel, Chem.–Eur. J., 1999, 5, 1306–1319 CrossRef CAS; (g) M. Utsunomiya and J. F. Hartwig, J. Am. Chem. Soc., 2004, 126, 2702–2703 CrossRef CAS; (h) J. Takaya and J. F. Hartwig, J. Am. Chem. Soc., 2005, 127, 5756–5757 CrossRef CAS; (i) L. T. Kaspar, B. Fingerhut and L. Ackermann, Angew. Chem., Int. Ed., 2005, 44, 5972–5974 CrossRef.
- (a) J. G. Taylor, N. Whittall and K. K. Hii, Org. Lett., 2006, 8, 3561–3564 CrossRef CAS; (b) J. Zhang, C.-G. Yang and C. He, J. Am. Chem. Soc., 2006, 128, 1798–1799 CrossRef; (c) L. T. Kaspar, B. Fingerhut and L. Ackermann, Angew. Chem., Int. Ed., 2005, 44, 5972–5974 CrossRef; (d) H. Qin, N. Yamagiwa, S. Matsunaga and M. Shibasaki, Chem.–Asian J., 2007, 2, 150–154 CrossRef CAS.
- D. C. Rosenfeld, S. Shekhar, A. Takemiya, M. Utsunomiya and J. F. Hartwig, Org. Lett., 2006, 8, 4179–4182 CrossRef CAS.
- T. Kondo and T.-a. Mitsudo, Chem. Rev., 2000, 100, 3205–3220 CrossRef CAS.
- (a) C. Munro-Leighton, E. D. Blue and T. B. Gunnoe, J. Am. Chem. Soc., 2006, 128, 1446–1447 CrossRef; (b) L. A. Goj, E. D. Blue, S. A. Delp, T. B. Gunnoe, T. R. Cundari, A. W. Pierpont, J. L. Petersen and P. D. Boyle, Inorg. Chem., 2006, 45, 9032–9045 CrossRef CAS; (c) C. Munro-Leighton, S. A. Delp, E. D. Blue and T. B. Gunnoe, Organometallics, 2007, 26, 1483–1493 CrossRef CAS.
- A. M. Johns, N. Sakai, A. Ridder and J. F. Hartwig, J. Am. Chem. Soc., 2006, 128, 9306–9307 CrossRef CAS.
- B. F. Straub and R. G. Bergman, Angew. Chem., Int. Ed., 2001, 40, 4632–4635 CrossRef CAS.
- H. Liu, M. Ji, X. Luo, J. Shen, X. Huang, W. Hu, H. Jiang and K. Chen, J. Med. Chem., 2002, 45, 2953–2969 CrossRef CAS.
- W. J. Dale and G. Buell, J. Org. Chem., 1956, 21, 45–47 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Details of synthetic procedures, characterization of organic compounds and complete tables of catalytic results. See DOI: 10.1039/b715507g |
| This journal is © The Royal Society of Chemistry 2008 |