Metal-specific allosteric activation and deactivation of a diamine†
Hayley J.
Clayton
a,
Lindsay P.
Harding
a,
John P.
Irvine
a,
John C.
Jeffery
b,
T.
Riis-Johannessen
b,
Andrew P.
Laws
a,
Craig R.
Rice
*a and
Martina
Whitehead
a
aDepartment of Chemical and Biological Sciences, University of Huddersfield, Huddersfield, UK HD1 3DH. E-mail: c.r.rice@hud.ac.uk; Fax: (+44)148-447-2182
bSchool of Chemistry, University of Bristol, Cantocks Close, Bristol, UK BS8 1TS
First published on 15th November 2007
Abstract
The reaction of a potentially tetradentate bis(pyridyl–thiazole) ligand with acetone is allosterically activated upon complexation with Cd(II) but deactivated by reaction with Cu(I), demonstrating metal-specific allosteric controlled reactivity.
Since the pioneering work of Rebek in the 1980s, research into artificial allosteric systems has received much attention; correspondingly there are now numerous examples of allosteric receptors and catalysis.1–5 However, the use of allosteric interactions to control reactivity has received less attention. Notable exceptions include the use of metal ions to control the reactions in 3,3′-disubstituted-2,2′-bipyridines.6 For example, Rebek demonstrated that the rate of intramolecular condensation reaction of a 2,2′-bipyridine derivative with amines (Scheme 1) was increased by the coordination of metal ions at the diimine unit.6
 | ||
| Scheme 1 | ||
Thus, upon complexation with NiCl2 the half life for the reaction is 60 min whereas without metal ions no cyclisation is observed. Rebek postulated that, at least in part, the cyclisation was accelerated by the coordination of the bipyridine unit to the Ni(II) centre since this would bring both the electrophile and nucleophile units in close proximity to one another. Other notable systems include the allosteric enhancement of both substitution and elimination reactions.6
Although in these systems an enhancement in the rate of reaction is observed by coordination of metal ions, the actual nature of the metal ion is unimportant, as long as the bipyridine unit is coordinated. In this paper we describe, for the first time, a system in which allosteric control of reactivity is dependent upon the metal ion used.
The potentially tetradentate ligand L1 was prepared from 3′-acetylamino-2,2′-bipyridine7 in seven steps (Scheme 2).
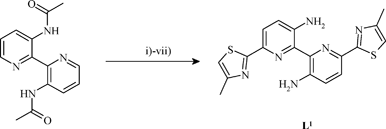 | ||
| Scheme 2 Reagents and yields: (i) m-CPBA (3.0 equiv.), CH2Cl2, 80% yield; (ii) (MeO)2SO2 (excess), 65% yield; (iii) NaCN (3.0 equiv.), NaHCO3, H2O, 60% yield; (iv) H2S, Et3N, DMF, 90% yield; (v) ClCH2COCH3, DMF, 76% yield; (vi) HCl (conc.), 90% yield; (vii) NH3 (conc.), 90% yield. | ||
Reaction of equimolar amounts of ligand L1 with [Cd(ClO4)2]·6H2O in MeCN, followed by slow diffusion of CH3CO2Et, produced a dark yellow crystalline material. Single crystal X-ray diffraction studies revealed the dicadmium(II) double-stranded helicate [Cd2(L1)2]4+ (Fig. 1) in which the ligands partition into two bis-bidentate py–tz (pyridyl–thiazole) binding domains in order to coordinate both Cd(II) ions, with Cd–N bond lengths ranging from 2.311(1)–2.365(1) Å.‡
![Solid-state structure of the [Cd2(L1)2(MeCN)2(ClO4)2]2+ complex cation (thermal ellipsoids are shown at a 50% probability level). All hydrogens (except amine) have been omitted for clarity. The “_2” symbol in the atom labels indicates that these atoms are at equivalent position (1 − x, y, − z).](/image/article/2008/CC/b714256k/b714256k-f1.gif) | ||
Fig. 1 Solid-state structure of the [Cd2(L1)2(MeCN)2(ClO4)2]2+ complex cation (thermal ellipsoids are shown at a 50% probability level). All hydrogens (except amine ) have been omitted for clarity. The “_2” symbol in the atom labels indicates that these atoms are at equivalent position (1 − x, y, ![[/]](https://www.rsc.org/images/entities/char_e0ee.gif) − z). − z). | ||
In the solid state the molecule lies astride a two-fold axis and has crystallographically imposed C2 symmetry. Stabilisation of the metal centres is supplemented by a molecule of acetonitrile and a perchlorate anion, producing two identical distorted octahedral geometries. Due to the ligand's inability to fully stabilise both metal centres this species may be considered an unsaturated double-stranded helicate.
We have previously shown that reaction of Cd(II) with a related pyridyl–thiazole ligand that does not contain amine functional groups on the 3,3′-position of the central bipyridine unit results in the formation of a mononuclear species. In this structure the ligand acts as a planar tetradentate donor coordinating the Cd(II) centre in the equatorial positions.8 However, in order for L1 to bind one metal via all four N-donors, it has to approach planarity and this gives rise to unfavourable steric interactions between the two amine groups on the 3,3′-positions of the central bipyridyl core. Thus, in the solid state, a dinuclear double helicate is preferred for L1 because it minimises unfavourable interactions between these two substituents. The 1H NMR spectrum of crystals dissolved in deuterated acetonitrile reveals that, in solution, the CdL1 complex exists as an equilibrium mixture of dinuclear helicate and mononuclear species: [Cd2(L1)2(S)4]4+ ⇆ 2[Cd(L1)(S)2]2+ (S = deuterated MeCN). The proposed solution structure of the D2 symmetric [Cd2(L1)2(MeCN)4]4+ cation is closely related to that seen in the solid state and it seems likely that the mononuclear species [Cd(L1)(S)2]2+ has a pseudo-octahedral geometry with the ligand acting as a planar tetradentate donor coordinating the Cd(II) centre in the equatorial positions as found for the complex formed between Cd(II) and the unsubstituted pyridyl–thiazole ligand chain. As expected on symmetry grounds, the 1H NMR spectra of both the mononuclear and dinuclear species show three aromatic resonances and additional signals assigned to the NH2 and CH3 moieties (Fig. 2a). Signals ascribed to the helicate on average appear more upfield and we have previously noted that they adopt this position because they are shielded by the aromatic rings of the adjacent helical ligand strand.
![Selected regions of the 1H NMR spectra of CD3CN solutions of (a) [Cd(L1)]2+ and [Cd2(L1)2]4+ and (b) [Cd(L1a)]2+.](/image/article/2008/CC/b714256k/b714256k-f2.gif) | ||
| Fig. 2 Selected regions of the 1H NMR spectra of CD3CN solutions of (a) [Cd(L1)]2+ and [Cd2(L1)2]4+ and (b) [Cd(L1a)]2+. | ||
ESI-MS studies are consistent with the NMR data and show singly charged ions at both m/z 593 and 1285, corresponding to mononuclear {[Cd(L1)](ClO4)}+ and dinuclear double-stranded {[Cd2(L1)2](ClO4)3}+ species. Re-dissolving the crystalline material and immediately obtaining a 1H NMR spectrum (after approx. 2–3 min, in CD3CN) results in a spectrum that contains an identical ratio of mononuclear and double helicate species to that observed in an equilibrated sample. This demonstrates that equilibration is rapid and that there is a fast interconversion between the mononuclear and double helicate species.
Reaction of equimolar amounts of [Cu(MeCN)4]PF6 with L1 produced a dark red coloured solution which, after slow diffusion of Et2O, gave a red crystalline material. ESI-MS studies show two ions at m/z 444 and 1033 corresponding to {[Cu2(L1)2]}2+ and {[Cu2(L1)2]PF6}+. The formation of a copper-containing dinuclear double helicate was confirmed by X-ray crystallography (Fig. 3).§
![Solid-state structure of the [Cu2(L1)2]2+ complex cation (thermal ellipsoids are shown at a 50% probability level). All hydrogens (except amine) have been omitted for clarity.](/image/article/2008/CC/b714256k/b714256k-f3.gif) | ||
| Fig. 3 Solid-state structure of the [Cu2(L1)2]2+ complex cation (thermal ellipsoids are shown at a 50% probability level). All hydrogens (except amine ) have been omitted for clarity. | ||
In the crystal, in a similar manner to the Cd(II) helicate, the ligand partitions into two bidentate binding domains with each Cu(I) centre coordinated by two pyridyl–thiazole units, one from each ligand. The metal centre adopts a 4-coordinate tetrahedral geometry with Cu(I)–N bond lengths ranging from 2.008(3)–2.103(3) Å. The twist of the helicate arises from the partitioning of the two pyridyl–thiazole bidentate domains with NCCN torsion angles of 65.2(5) and 66.9(5)°. The 1H NMR spectrum of the crystalline material in CD3CN solution showed three signals in the aromatic region and one signal for both methyl and amino groups indicating that, in solution, only one complex is formed. The chemical shifts of these signals are consistent with the formation of a dinuclear double helicate in solution and are in good agreement with the signals that have been attributed to a helicate species in the [Cd2(L1)2]2+ system.
Reaction of the mixture of the Cd(II)-containing dinuclear double helicate and mononuclear complexes ([Cd2(L1)2]4+ ⇆ 2[Cd(L1)]2+)) with acetone results in the solution turning from yellow to dark orange over a period of 24 h. 1H NMR studies show a simplified spectrum with only one new set of peaks present in the aromatic region (Fig. 2b). The chemical shifts of the peaks for the new species are more deshielded than those expected for a dinuclear helicate, which indicates that a new mononuclear species is present. This was confirmed by a single crystal diffraction study which revealed a mononuclear Cd(II) complex.¶ The axial positions on the metal are occupied by two monodentate perchlorate anions, whilst the ligand acts as a near planar “equatorial” ligand with a NCCN torsion angle of 3.9(5)° about the central bipyridine core (Fig. 4). The most remarkable feature about the structure is that the amine groups of L1 have reacted with one equivalent of acetone and both are now covalently bonded to the 2-position of a propyl unit. The resulting ligand (L1a) is a derivative of L1 but is now capable of forming only the mononuclear complex [Cd(L1a)]2+.
![Solid-state structure of the [Cd(L1a)(ClO4)2] complex molecule (thermal ellipsoids are shown at a 50% probability level). All hydrogen atoms (except aminal) have been omitted for clarity.](/image/article/2008/CC/b714256k/b714256k-f4.gif) | ||
| Fig. 4 Solid-state structure of the [Cd(L1a)(ClO4)2] complex molecule (thermal ellipsoids are shown at a 50% probability level). All hydrogen atoms (except aminal ) have been omitted for clarity. | ||
Interestingly reaction of [Cd2(L1)2]4+ with acetone in the presence of a catalytic amount of camphorsulfonic acid gives an identical 1H NMR spectrum to that observed when just acetone is added, but the reaction is much faster. The formation of this species is also supported by ESI-MS, which shows an ion at m/z 633 corresponding to {[Cd(L1a)](ClO4)}+. No peaks corresponding to either [Cd(L1)]2+, [Cd2(L1)2]4+ or [Cd2(L1a)2]4+ are observed.
Reaction of [Cu2(L1)2]2+ with acetone in CD3CN either with or without catalytic amounts of acid shows no observable change in the 1H NMR spectrum, indicating that no reaction has occurred and that the dinuclear double helicate is still present. This is further supported by ESI-MS, which showed that even after prolonged reaction time (>10 days), the only ion present corresponded to {[Cu2(L1)2]+(PF6)}+ (m/z 1033).
The rate constants for acid catalysed aminal formation were determined by following the decrease in concentration (as a function of time) of the Cd(II) complex by UV-Vis spectroscopy and this demonstrated clear single phase kinetics with sharp isosbestic points; there was no evidence for the formation and/or decay of intermediate species. All experiments were performed with acetone present in large excess of the mononuclear species, which itself was present in large excess compared to the concentration of the acid catalyst. Under these conditions, the absorbance decreased linearly with time, generating a pseudo-zero-order rate profile, with a rate constant of 6.43 × 103 l mol−1 s−1. However, as mentioned previously, no such reaction is observed with Cu(I).
This difference in reactivity can be attributed to the coordination preferences of the metal ions. It is clear from the 1H NMR studies that in solution the Cd(II) complex forms both the mononuclear and helicate species, whereas with the Cu(I) complex there is no evidence in the 1H NMR spectrum for any species other than the helicate. The formation of this sole species is a consequence of the preference of Cu(I) to adopt tetrahedral coordination geometry.
Although the mechanism in this system is unknown, it is plausible that, upon reaction with acetone, one of the amines forms the corresponding Schiff base . This then undergoes an intramolecular reaction with the other amine resulting in the formation of a 7-membered aza-heterocycle. This latter reaction would require that the ligand change its coordination behaviour and go from a bis-bidentate donor (which forms the dinuclear double helicate) to a tetradentate donor, which results in the mononuclear species. As the Cu(I) helicate does not form this mononuclear species the reduced flexibility of the ligand strand would thus prevent cyclisation. Furthermore the reaction of L1 with acetone is clearly metal-templated because the free ligand does not react with acetone.
This differing reactivity has demonstrated that different metal ions can allosterically activate or deactivate the reactivity of a diamino-containing ligand.
Notes and references
- L. Kovbasyuk and R. Krämer, Chem. Rev., 2004, 104, 3161 CrossRef CAS.
- S. Shinkai, M. Ikeda, A. Sugaskai and M. Takeuchi, Acc. Chem. Res., 2001, 34, 494 CrossRef CAS.
- J. Rebek, Jr., J. E. Trend, R. V. Wattley and S. J. Chachravorti, J. Am. Chem. Soc., 1979, 101, 4333 CrossRef CAS.
- J. Rebek, Jr. and R. V. Wattley, J. Am. Chem. Soc., 1980, 102, 4853 CrossRef.
- J. Rebek, Jr., Acc. Chem. Res., 1984, 17, 258 CrossRef.
- J. Rebek, Jr., J. Am. Chem. Soc., 1985, 107, 7487 CrossRef.
- C. R. Rice, S. Onions, N. Vidal, J. D. Wallis, M. C. Senna, M. Pilkington and H. Stoeckli-Evans, Eur. J. Inorg. Chem., 2002, 1985.
- T. Riis-Johannessen, J. C. Jeffery, A. P. H. Robson, C. R. Rice and L. P. Harding, Inorg. Chim. Acta, 2005, 358, 2781 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental and crystallographic details and UV-Vis spectrum of [Cd(L1)]2+ on acid catalysed reaction with acetone. See DOI: 10.1039/b714256k |
| ‡ X-Ray single crystal diffraction data for [Cd2(L1)2(MeCN)2(ClO4)2](ClO4)2·2MeCN was collected using a Bruker APEX CCD area-detector diffractometer under a stream of cold nitrogen. Crystal data: M = 1547.79, monoclinic, C2/c, a = 22.217(4), b = 11.429(2), c = 23.837(5) Å, β = 112.17(3)°, V = 5605.4(19) Å3, Z = 4, ρc = 1.834 Mg m−3, F(000) = 3104, µ(Mo-Kα) = 1.182 mm−1, T = 100 K. A total of 31695 reflections were measured in the range 1.85 ≤ θ ≤ 27.49° (hkl range indices −28 ≤ h ≤ 28, −14 ≤ k ≤ 14, −30 ≤ l ≤ 30), 6443 independent reflections (Rint = 0.0162). The structure was refined on F2 to Rw = 0.0548, R = 0.0222 (6443 reflections with I > 2σ(I)) and GOF = 1.073 on F2 for 476 refined parameters, largest difference peak and hole 0.796 and −0.560 e Å−3. CCDC 606009. For crystallographic data in CIF format see DOI: 10.1039/b714256k |
| § X-Ray single crystal diffraction data for [Cu2(L1)2](PF6)2·3MeCN was collected using a PROTEUM CCD area-detector diffractometer under a stream of cold nitrogen. Crystal data: M = 1301.16, monoclinic, P21/c, a = 12.1593(2), b = 36.2844(7), c = 12.3673(3) Å, β = 103.137(1), V = 5313.6(2) Å3, Z = 4, ρc = 1.626 Mg m−3, F(000) = 2632, µ(Cu-Kα) = 3.836 mm−1, T = 200 K. A total of 30844 reflections were measured in the range 2.44 ≤ θ ≤ 70.26° (hkl range indices −14 ≤ h ≤ 14, −32 ≤ k ≤ 41, −13 ≤ l ≤ 13), 8480 independent reflections (Rint = 0.0482). The structure was refined on F2 to Rw = 0.1134, R = 0.0579 (8480 reflections with I > 2σ(I)) and GOF = 1.050 on F2 for 713 refined parameters, largest difference peak and hole 0.407 and −0.336 e Å−3. CCDC 660889. For crystallographic data in CIF format see DOI: 10.1039/b714256k |
¶ X-Ray single crystal diffraction data for [Cd(L1a)(ClO4)2] was collected using a PROTEUM CCD area-detector diffractometer under a stream of cold nitrogen. Crystal data: M = 731.85, triclinic, P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) , a = 8.9198(3), b = 11.5523(4), c = 13.7786(5) Å, α = 96.685(2), β = 104.617(2), γ = 105.987(2)°, V = 1294.07(8) Å3, Z = 2, ρc = 1.878 Mg m−3, F(000) = 732, µ(Cu-Kα) = 10.709 mm−1, T = 100 K. A total of 9914 reflections were measured in the range 3.38 ≤ θ ≤ 70.21° (hkl range indices −10 ≤ h ≤ 10, −13 ≤ k ≤ 13, −16 ≤ l ≤ 16), 4397 independent reflections (Rint = 0.0370). The structure was refined on F2 to Rw = 0.0905, R = 0.0445 (4379 reflections with I > 2σ(I)) and GOF = 1.024 on F2 for 441 refined parameters, largest difference peak and hole 1.248 and −0.633 e Å−3. CCDC 606008. For crystallographic data in CIF format see DOI: 10.1039/b714256k , a = 8.9198(3), b = 11.5523(4), c = 13.7786(5) Å, α = 96.685(2), β = 104.617(2), γ = 105.987(2)°, V = 1294.07(8) Å3, Z = 2, ρc = 1.878 Mg m−3, F(000) = 732, µ(Cu-Kα) = 10.709 mm−1, T = 100 K. A total of 9914 reflections were measured in the range 3.38 ≤ θ ≤ 70.21° (hkl range indices −10 ≤ h ≤ 10, −13 ≤ k ≤ 13, −16 ≤ l ≤ 16), 4397 independent reflections (Rint = 0.0370). The structure was refined on F2 to Rw = 0.0905, R = 0.0445 (4379 reflections with I > 2σ(I)) and GOF = 1.024 on F2 for 441 refined parameters, largest difference peak and hole 1.248 and −0.633 e Å−3. CCDC 606008. For crystallographic data in CIF format see DOI: 10.1039/b714256k |
| This journal is © The Royal Society of Chemistry 2008 |