Tagging alcohols with cyclic carbonate: a versatile equivalent of (meth)acrylate for ring-opening polymerization †
Russell C.
Pratt
a,
Fredrik
Nederberg
ab,
Robert M.
Waymouth
b and
James L.
Hedrick
*a
aIBM Almaden Research Center, 650 Harry Road, San Jose, CA, USA 95120. E-mail: hedrick@almaden.ibm.com; Fax: (1)408-927-3310
bDepartment of Chemistry, Stanford University, Stanford, CA, USA 94305
First published on 25th October 2007
Abstract
Cyclic carbonate monomers based on a single biocompatible scaffold allow for incorporation of a wide range of functional groups into macromolecules via ring-opening polymerization .
Acrylic and methacrylic polymers are ubiquitous in modern applications due to the wide variety of properties that can be introduced by variation of the pendant groups. The wide variety of commercially available monomers , coupled with recent advances in controlled radical polymerization techniques, have enabled the generation of new classes of well-defined functional macromolecules. In comparison, the inventory of cyclic ester and carbonate monomers available and suitable for ring-opening polymerization (ROP) is more limited. Methods have been developed for the synthesis of functional lactones1–5 and cyclic carbonates6 but there is as yet no widely adapted method for incorporating arbitrary functional groups into these ROP monomers .
To provide a more versatile and accessible library of ROP monomers , we have exploited recently developed organocatalytic methods for ROP of cyclic carbonate monomers derived from 2,2-bis(methylol)propionic acid (bis-MPA). Bis-MPA has proven a versatile building block for the construction of biocompatible dendrimers.7 The syntheses of carbonate monomers derived from bis-MPA typically employ acetonide protection–deprotection schemes prior to installation of the pendant functional group prior to forming the cyclic carbonate moiety.8 Our approach involves the synthesis of the cyclic carbonate 1 as a versatile synthon for a family of functionalized carbonated monomers . This synthetic scheme parallels the powerful approach of (meth)acrylate derivatization as a means of creating monomers (Scheme 1).
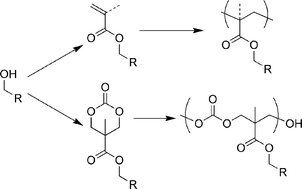 | ||
| Scheme 1 Derivatization of alcohols with (meth)acrylate for radical polymerization (top) compared with cyclic carbonate for ring-opening polymerization (bottom). | ||
The synthesis of 1 can easily be carried out on a 0.1 mol scale (16 g) with typical laboratory apparatus following the procedures outlined in Scheme 2.9 Two procedures for the coupling of 1 to alcohols were used: either direct coupling using DCC, or conversion to the acyl chloride using oxalyl chloride followed by reaction with the alcohol or amine in the presence of base. The latter method has the advantage that the salt byproducts are easily removed. Compared to methods wherein the functional group is attached prior to carbonate formation, the inverse sequence described here more generally requires only a single unique reaction and purification step for new pendant groups. Using these methods, a range of functional groups could be incorporated to generate new ROP monomers offering opportunities for couplings via substitution, cycloaddition, and amide or disulfide linkages, or for introducing strongly hydrophilic or hydrophobic groups (Scheme 3).
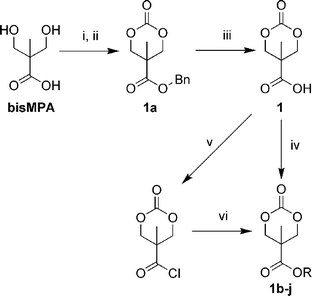 | ||
| Scheme 2 Synthesis of 1-type monomers from bis-MPA. Conditions: (i) BnBr, KOH, DMF, 100 °C, 15 h, 62%. (ii) Triphosgene, pyridine, CH2Cl2, –78 → 0 °C, 95%. (iii) Pd/C (10%), H2 (3 atm), EtOAc, RT, 24 h, 99%. (iv) ROH, DCC, THF, RT, 16 h. (v) (COCl)2, THF, RT, 1 h, 99%. (vi) ROH, NEt3, RT, 3 h. | ||
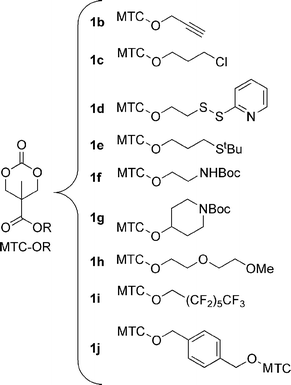 | ||
| Scheme 3 Monomers synthesized via procedures in Scheme 2. | ||
To test the suitability of these bis-MPA-derived monomers for polymerization , we examined the organocatalytic ROP of monomer 1a bearing a simple benzyl group for comparison to the archetypical carbonate monomer , trimethylene carbonate (TMC).10 As summarized in Table 1, both the two-component catalyst consisting of the Lewis acid 1-(3,5-bis(trifluoromethyl)phenyl)-3-cyclohexyl-2-thiourea (TU) with the Lewis base 1,8-diazabicyclo[5.4.0]undec-7-ene (DBU) or alternatively the superbasic catalyst 1,5,7-triazabicyclo[4.4.0]dec-5-ene (TBD) showed high conversions of 1a to polymer in relatively short times. Good control over the molecular weight and polydispersity was achieved with the TU–DBU co-catalyst, while TBD is more active and its use leads to broadening of the polydispersity via transcarbonation of the polymer chains. No scrambling of the pendant benzyl ester into the poly(1a) chains could be observed using 1H-NMR spectroscopy. While excessively bulky substituents (e.g. 2,2-diphenyl) impede the ring-opening of six-membered cyclic carbonates,8a,11 the increased rate of polymerization for 1a when compared to TMC indicates that the methyl and carboxylate substituents are well-suited for polymerization . The location of steric bulk distant from the polymerizing carbonate also avoids interference with the organocatalysts; substituents in the α-position of cyclic ester monomers dramatically reduce the rates of polymerization when organocatalysts are used, making them incompatible with effective derivatization strategies using α-chloro and α-azido groups.
| Monomera | Catalystb | Time/h | Conv.c (%) | M n d /g mol–1 | M w/Mnd |
|---|---|---|---|---|---|
| a Conditions: 1 M monomer in CH2Cl2 with 0.02 M PyBuOH, 20 °C. b TU–DBU: both 0.05 M; TBD: 0.01 M. c By 1H NMR spectroscopy. d By GPC vs. polystyrene standards, uncorrected. | |||||
| 1a | TU–DBU | 0.5 | 93 | 11![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 600 600 |
1.12 |
| TU–DBU | 1 | 94 | 11500 |
1.15 | |
| TU–DBU | 2 | 95 | 12900 |
1.20 | |
| TBD | 5 min | 95 | 13200 |
1.52 | |
| TBD | 1 | 96 | 8000 | 1.76 | |
| TMC | TU–DBU | 1 | 45 | 4400 | 1.03 |
| TU–DBU | 2 | 74 | 7300 | 1.03 | |
| TU–DBU | 3 | 90 | 8600 | 1.03 | |
| TBD | 5 min | 98 | 8900 | 1.08 | |
| TBD | 1 | 98 | 11000 |
1.31 | |
Random copolymerizations of the cyclic carbonates with TMC were conducted using organocatalytic procedures similar to those for the homopolymerizations of TMC and 1a described above (Table 2). Monitoring experiments (1H NMR spectroscopy) reveal that all of the 1b–1i comonomer was incorporated into polymer within 1 h, while the conversion of TMC lagged and did not reach >90% conversions until after 3 h. The relative reactivities match the higher reactivity found for 1avs. TMC in homopolymerization , and suggest that gradient copolymers are formed. The observation that these polymerizations continue on after complete consumption of the faster-reacting monomer contrasts with behavior we have observed for random copolymerizations of lactones, in which the faster reacting monomer reacts exclusively under organocatalytic conditions.12 Block copolymers of different carbonate repeat units can also be constructed by sequential polymerization : for instance, following in situ formation of a PTMC macroinitiator (conditions as per Table 1; [M]0/[I]0 = 37.5, TU–DBU, 3 h; 86% conversion, Mn = 5900, PDI = 1.03), monomer 1a was added to the reaction solution, and after 30 min the chain-extended polymer was obtained (conversion = 92% (TMC), 88% (1a); Mn = 9700, PDI = 1.08).
| Comonomera | Time/h | Conversion TMCb (%) | Conversion comonomerb (%) | M n c /g mol–1 | M w/Mnc |
|---|---|---|---|---|---|
| a Conditions: 1 M monomer in CH2Cl2, 80 : 20 TMC : comonomer (mol : mol), 0.02 M PyBuOH, 0.05 M TU, 0.05 M DBU, 20 °C. b By 1H NMR spectroscopy. c By GPC vs. polystyrene standards, uncorrected. | |||||
| 1b | 1 | 61 | >95 | 4700 | 1.10 |
| 4 | 95 | >95 | 6500 | 1.11 | |
| 1g | 1 | 46 | >95 | 3900 | 1.07 |
| 4 | 91 | >95 | 5700 | 1.07 | |
| 1h | 1 | 49 | >95 | 5300 | 1.09 |
| 4 | 92 | >95 | 7800 | 1.11 | |
| 1i | 1 | 60 | >95 | 6100 | 1.05 |
| 4 | 91 | >95 | 8600 | 1.05 | |
In summary, a general synthetic route to incorporate a broad range of functional groups into cyclic carbonate monomers has been developed. These monomers can be polymerized under mild, one-pot conditions to create random or block copolymers . Further studies are underway to complete procedures for full deprotection and derivatization of polar sidegroups once they are incorporated into polymers .
Notes and references
- T. Mathisen, K. Masus and A.-C. Albertsson, Macromolecules, 1989, 22, 3842 CrossRef CAS; K. Stridsberg and A.-C. Albertsson, J. Polym. Sci., Part A: Polym. Chem., 1999, 37, 3407 CrossRef CAS; R. K. Srivastava and A.-C. Albertsson, J. Polym. Sci., Part A: Polym. Chem., 2005, 43, 4206 CrossRef CAS.
- M. Trollsås, V. Y. Lee, D. Mecerreyes, P. Löwenhielm, M. Möller, R. D. Miller and J. L. Hedrick, Macromolecules, 2000, 33, 4619 CrossRef.
- B. Parrish, J. K. Quansah and T. Emrick, J. Polym. Sci., Part A: Polym. Chem., 2002, 40, 1983 CrossRef CAS; B. Parrish and T. Emrick, Macromolecules, 2004, 37, 5863 CrossRef CAS; B. Parrish, R. B. Breitenkamp and T. Emrick, J. Am. Chem. Soc., 2005, 127, 7404 CrossRef CAS; B. Parrish and T. Emrick, Bioconjugate Chem., 2007, 18, 263 CrossRef CAS.
- R. Riva, S. Schmeits, F. Stoffelbach, C. Jerome, R. Jerome and P. Lecomte, Chem. Commun., 2005, 5334 RSC; P. Lecomte, R. Riva, S. Schmeits, J. Rieger, K. Van Butsele, C. Jerome and R. Jerome, Macromol. Symp., 2006, 240, 157 CrossRef CAS; R. Riva, S. Schmeits, C. Jerome, R. Jerome and P. Lecomte, Macromolecules, 2007, 40, 796 CrossRef CAS; H. Li, R. Riva, R. Jerome and P. Lecomte, Macromolecules, 2007, 40, 824 CrossRef CAS.
- Y. D. Y. L. Getzler, V. Mahadeven, E. B. Lobkovsky and G. W. Coates, J. Am. Chem. Soc., 2002, 124, 1174 CrossRef CAS; V. Mahadevan, Y. D. Y. L. Getzler and G. W. Coates, Angew. Chem., Int. Ed., 2002, 41, 2781 CrossRef CAS; J. A. R. Schmidt, V. Mahadevan, Y. D. Y. L. Getzler and G. W. Coates, Org. Lett., 2004, 6, 373 CrossRef CAS; J. A. R. Schmidt, E. B. Lobkovsky and G. W. Coates, J. Am. Chem. Soc., 2005, 127, 11426 CrossRef CAS; J. W. Kramer, E. B. Lobkovsky and G. W. Coates, Org. Lett., 2006, 8, 3709 CrossRef CAS.
- G. Rokicki, Prog. Polym. Sci., 2000, 25, 259 CrossRef CAS.
- H. Ihre, A. Hult and E. Söderlind, J. Am. Chem. Soc., 1996, 118, 6388 CrossRef CAS; H. Ihre, A. Hult, J. M. J. Fréchet and I. Gitsov, Macromolecules, 1998, 31, 4061 CrossRef CAS; H. Ihre, O. L. Padilla De Jesús and J. M. J. Fréchet, J. Am. Chem. Soc., 2001, 123, 5908 CrossRef CAS; M. Malkoch, E. Malmström and A. Hult, Macromolecules, 2002, 35, 8307 CrossRef CAS; O. L. P. De Jesus, H. R. Ihre, L. Gagne, J. M. J. Fréchet and F. C. Szoka, Bioconjugate Chem., 2002, 13, 453 CrossRef CAS; E. R. Gillies, E. Dy, J. M. J. Fréchet and F. C. Szoka, Mol. Pharmacol., 2005, 2, 129 CrossRef CAS; C. C. Lee, E. R. Gillies, M. E. Fox, S. J. Guillaudeau, J. M. J. Fréchet, E. E. Dy and F. C. Szoka, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 16649 CrossRef CAS.
- (a) K. D. Weilandt, H. Keul and H. Höcker, Macromol. Chem. Phys., 1996, 197, 3851 CrossRef CAS; (b) T. F. Al-Azemi and K. S. Bisht, Macromolecules, 1999, 32, 6536 CrossRef CAS; T. F. Al-Azemi, J. P. Harmon and K. S. Bisht, Biomacromolecules, 2000, 1, 493 CrossRef CAS; T. F. Al-Azemi and K. S. Bisht, Polymer, 2002, 43, 2161 CrossRef CAS; B. D. Mullen, C. N. Tang and R. F. Storey, J. Polym. Sci., Part A: Polym. Chem., 2003, 41, 1978 CrossRef CAS; Z.-L. Liu, Y. Zhou and R.-X. Zhuo, J. Polym. Sci., Part A: Polym. Chem., 2003, 41, 4001 CrossRef CAS; Y. Zhou, R. Zhuo and Z. Liu, Polymer, 2004, 45, 5459 CrossRef CAS; Y. Zhou, R.-X. Zhuo and Z.-L. Liu, Macromol. Rapid Commun., 2005, 26, 1309 CrossRef CAS.
- T. F. Al-Azemi and K. S. Bisht, J. Polym. Sci., Part A: Polym. Chem., 2002, 40, 1267 CrossRef CAS.
- F. Nederberg, B. G. G. Lohmeijer, F. Leibfarth, R. C. Pratt, J. Choi, A. P. Dove, R. M. Waymouth and J. L. Hedrick, Biomacromolecules, 2007, 8, 153 CrossRef CAS.
- T. Hino and T. Endo, Macromolecules, 2003, 36, 5902 CrossRef CAS; H. Keul, R. Bächer and H. Höcker, Makromol. Chem., 1986, 187, 2579 CrossRef CAS; H. R. Kricheldorf, A. Stricker and M. Lossin, J. Polym. Sci., Part A: Polym. Chem., 1999, 37, 2179 CrossRef CAS.
- R. C. Pratt, B. G. G. Lohmeijer, D. A. Long, R. M. Waymouth and J. L. Hedrick, J. Am. Chem. Soc., 2006, 128, 4556 CrossRef CAS; B. G. G. Lohmeijer, R. C. Pratt, F. Leibfarth, J. W. Logan, D. A. Long, A. P. Dove, F. Nederberg, J. Choi, C. Wade, R. M. Waymouth and J. L. Hedrick, Macromolecules, 2006, 39, 8574 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic details and characterization for 1b–1jmonomers . See DOI: 10.1039/b713925j |
| This journal is © The Royal Society of Chemistry 2008 |