Questioning the paradigm of metal complex promoted phosphodiester hydrolysis: [Mo7O24]6− polyoxometalate cluster as an unlikely catalyst for the hydrolysis of a DNA model substrate†
Els
Cartuyvels
,
Gregory
Absillis
and
Tatjana N.
Parac-Vogt
*
Katholieke Universiteit Leuven, Department of Chemistry, Celestijnenlaan 200F, B-3001 Leuven, Belgium. E-mail: Tatjana.Vogt@chem.kuleuven.be; Fax: +32 16 327992; Tel: +32 16 32761
First published on 16th November 2007
Abstract
The first example of a phosphodiester bond cleavage promoted by a highly negatively charged polyoxometalate cluster has been discovered: the hydrolysis of the phosphodiester bond in a DNA model substrate bis(p-nitrophenyl)phosphate (BNPP) is promoted by the heptamolybdate anion [Mo7O24]6− with rates which represent an acceleration of nearly four orders of magnitude compared to the uncatalyzed cleavage.
The mechanism by which metal complexes accelerate the rate of phosphate ester hydrolysis is generally described as an interplay of several factors: (i) Lewis acid activation through the coordination of phosphoryl oxygen(s) to the metal ion, (ii) nucleophile activation of coordinated water or hydroxide ligand, (iii) leaving group activationvia coordination of the leaving group oxygen to the metal, (iv) general base catalysis of solvent water through metal-coordinated hydroxide ligand and (v) acting of metal coordinated water molecules as an intramolecular general acid catalyst.1 As a result, metal complexes that act as artificial phosphoesterases have to meet a number of criteria such as an overall positive charge, free coordination site for the binding of phosphoryl oxygen, the presence of coordinated water or hydroxide, and the presence of functional groups for acid–base catalysis, among others.1,2 In recent years a range of such complexes, containing most notably Co3+, Fe3+, Zn2+, Cu2+, Zr4+, Ln3+, have been shown to exhibit remarkable activity toward phosphodiester cleavage.3 With the exception of the work by Yatsimirsky and his co-workers, in which charge neutral lanthanide complexes have been implicated as hydrolytically active species3h it has been generally assumed that only positively charged metal complexes can act as artificial phosphoesterases. However, in this study we demonstrate, that despite its high negative charge, coordination saturation, and lack of coordinated water or hydroxide, the polyoxomolybdate [Mo7O24]6−cluster efficiently hydrolyzes the phosphodiester bond in commonly used DNA model substrate bis(p-nitrophenyl)phosphate (BNPP).
Our interest in the hydrolytic properties of polyoxomolybdate has been provoked by several reports describing the interaction of [Mo7O24]6− with mono-substituted phosphates, including nucleotides.4 It has been recognized long ago that the presence of molybdate leads to hydrolysis of the labile “high energy” phosphoanhydride bonds in ATP.5 Although the complexation between [Mo7O24]6− and phosphate-containing compounds has been well established, the interactions between [Mo7O24]6− and bis-substituted phosphates, including phosphodiesters, have been virtually unexplored, most likely because they have been presumed to be extremely weak or virtually non-existent.6a
The [Mo7O24]6− is formed by acidification of aqueous molybdate solutions to pH below 6 (eqn (1)). Its structure has been confirmed by numerous studies both in solution and in the solid state.6 The thermodynamic parameters published elsewhere can be used for estimation of [Mo7O24]6− concentration under different reaction conditions.6,7 It has to be noted that the accurate experimental determination of [Mo7O24]6− concentration is still rather difficult, although models for the speciation of molybdate solutions are well-established and based on reliable and reproducible data.‡
| 7[MoO4]2− + 8H+⇌ [Mo7O24]6− + 4H2O | (1) |
The cleavage of phosphodiester model substrate BNPP proceeded smoothly in the presence of an equimolar amount of Na6[Mo7O24] (25 mM, pH 5.5, 50 °C). The release of 2 equivalents of p-nitrophenol (NP) monitored by 1H NMR spectroscopy afforded calculation of the rate constant for the cleavage of BNPP (kobs = 2.3 ± 0.1 × 10−6 s−1 at 50 °C). This represents an acceleration of almost four orders of magnitude compared to the uncatalysed cleavage of BNPP (kobs = 3 × 10−10 s−1).8 The absence of any paramagnetic species during the course of reaction excludes the possibility of oxidative cleavage and the reduction of Mo(VI) to Mo(V). The monophasic kinetic profile suggests that the cleavage of the first phosphoester bond in BNPP is much slower than the cleavage of the phosphoester bond in the first product of hydrolysis (p-nitrophenyl)phosphate (NPP), and is the rate determining step. Indeed, the separate experiments involving NPP and Na6[Mo7O24] showed that the cleavage of the phosphoester bond in NPP proceeded nearly 40 times faster than the cleavage of the phosphodiester bond in BNPP.
The effect of the pH on the cleavage reaction was examined in order to correlate the rate–pH profile with the species distribution diagram.§ As seen in Fig. 1, the pH dependence of the kobs exhibits a bell-shaped profile, with the fastest cleavage observed at pH = 5.3. While depending on the conditions, slightly different speciation models can be found in the literature, however, there is very little doubt that at concentrations of molybdate greater than 1 mM [Mo7O24]6− is the only polyoxoanion that exists at pH = 5.3.6 Indeed, 95Mo NMR spectroscopy, which recently has been proved as an excellent and accurate tool for the examination of equilibria involving molybdate,9 showed the presence of a peak at δ = 34 ppm, confirming that at pH 5.3 [Mo7O24]6− is the only polymolybdate form present in solution. Comparison of the rate profile with the concentration profile of molybdate species, whose existence has been previously well established in solution, shows striking overlap of the kobs profile with the concentration of [Mo7O24]6−.6,7,9 Although within the whole range of examined pH other species, such as MoO42− and [Mo8O26]4− as well as various protonated forms of them, are present in solution, they seem to be catalytically much less active. Since at pH 8, where MoO42− is the only Mo(VI) species found in solution, BNPP hydrolysis hardly occurred it is reasonable to assume that this monomeric complex does not cause hydrolysis. Similarly, slow hydrolysis at the lower pH values implies low catalytic activity of [Mo8O26]4− which is the most abundant polyoxoanion in the pH range from 2 to 4.6,7,9
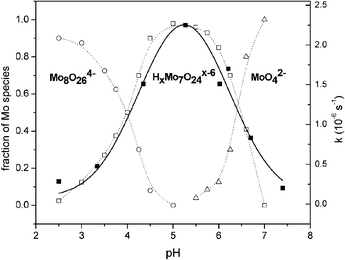 | ||
| Fig. 1 pH dependence of kobs (■, solid line) for the cleavage of BNPP (25 mM) in the presence of Na2MoO4 (175 mM starting conc.). Fractions of Mo species have been added for comparison (points taken from ref. 9). | ||
The significant increase of the rate of BNPP hydrolysis in the presence of heptamolybdate at pH = 5.3 implies that interaction between [Mo7O24]6− and BNPP must take place in the solution. The hydrolysis reaction followed by 31P NMR spectroscopy revealed that upon addition of Na6[Mo7O24] to BNPP the 31P NMR signal of BNPP shifted only slightly upfield by 0.15 ppm. During the course of hydrolysis the concentration of the initial complex decreased, and appearance of NPP, which is the first product of hydrolytic reaction was observed. Interestingly, 31P NMR spectra did not show evidence for the presence of inorganic phosphate, which is expected as the final product of the hydrolytic reaction. Instead, the final species present in solution had the same spectroscopic features as a separately prepared pentamolybdodiphosphate ion [P2Mo5O23]6−, which typically forms in mildly acidic solutions containing molybdate and phosphate ions.10 The hydrolytic activity of [P2Mo5O23]6− toward BNPP was also tested, and no observable cleavage of the phosphoester bond occurred even after prolonged reaction times. Variable temperature 31P NMR spectra of a mixture containing BNPP and [Mo7O24]6− recorded immediately upon mixing, revealed broadening of the BNPP 31P resonance upon an increase of temperature, suggesting a dynamic exchange process between free and bound BNPP at higher temperatures (Fig. 2).
![Half-width (ν1/2) of the BNPP 31P NMR resonance in the presence of Na6[Mo7O24] as a function of temperature.](/image/article/2008/CC/b714860g/b714860g-f2.gif) | ||
| Fig. 2 Half-width (ν1/2) of the BNPP 31P NMR resonance in the presence of Na6[Mo7O24] as a function of temperature. | ||
The binding constant between BNPP and the [Mo7O24]6− was determined by carrying out kinetic experiments using a fixed amount of BNPP and increasing amounts of [Mo7O24]6− at pH 5.3 and 50 °C (Fig. 3).
![Influence of [Mo7O24]6− concentration on kobs for cleavage of BNPP (20 mM, pH = 5.3, T = 50 °C).](/image/article/2008/CC/b714860g/b714860g-f3.gif) | ||
| Fig. 3 Influence of [Mo7O24]6− concentration on kobs for cleavage of BNPP (20 mM, pH = 5.3, T = 50 °C). | ||
By taking into account the fact that the release of the first equivalent of NP from BNPP is the rate limiting step (eqn (2)), the values for the rate constant for cleavage of the phosphodiester bond k2 (3.33 × 10−6 s−1), and the association constant for the BNPP–[Mo7O24] complex K1 (28.7 M−1), were obtained by a computer generated least-squares fit of eqn (3) to kobs.
 | (2) |
 | (3) |
At first glance the interaction between [Mo7O24]6− and BNPP seems counterintuitive, since highly negatively charged metal clusters are not expected to bind or interact with the negatively charged anions. However, the anion–anion interactions in polyoxometalate chemistry are not without precedent, given that interaction between negatively charged polyoxometalate anions have been previously detected both in solid state and in solution.11 The intriguing question is then by which mechanism does the cleavage of the phosphoester bond occur?
Although the bell-shaped pattern of the pH profile is usually indicative of either a deprotonation step of two acidic functions in the metal complex which have opposite effect, or an equilibrium involving metal bound water and metal bound hydroxide that acts as an active nucleophile,1,2 both mechanisms are highly improbable since [Mo7O24]6− does not contain any acidic functions or coordinated water ligands.6 In addition, the polyoxometalate anions are known to be very weak conjugate bases with low negative charge densities.6
We propose that the origin of hydrolytic activity of [Mo7O24]6− lies in its high internal lability and a known intramolecular exchange which results in partial detachment of one MoO4 tetrahedron.9,12 This detachment may allow attachment of the structurally related phosphodiester tetrahedron into the polyoxometalate structure, in a similar manner as in the well-known process that occurs at higher temperatures in which tetrahedral molybdate adds to heptamolybdate to yield an octamolybdate structure.¶,12a Broadening of 31P NMR resonances of BNPP in the presence of [Mo7O24]6− upon increase of temperature is consistent with the interaction between BNPP and [Mo7O24]6− taking place at elevated temperatures.|| The incorporation of the phosphodiester group into the polyxomolybdate skeleton, and sharing of oxygen atoms with the Mo(VI) centre, may lead to bond strain and cause polarization of the P–O ester bond and its activation toward external attack by water. Our hypothesis that the hydrolytic activity of [Mo7O24]6− may be due to its internal flexibility is strengthened by the fact that the analogous and isostructural [W7O24]6− polyoxometalate cluster, which is much more inert and does not undergo fast intramolecular exchange,9 is virtually hydrolytically inactive towards hydrolysis of BNPP.
In conclusion, we report the first example of a DNA model phosphodiester bond cleavage promoted by a highly negatively charged polyoxometalate cluster. The kinetic studies strongly suggest that [Mo7O24]6− is the hydrolytically active complex and that cleavage occurs by a mechanism which is different from that of all currently known hydrolytically active metal complexes. Studies involving other types of polyoxometalate complexes such as artificial phosphoesterases are currently under way. Such studies are of special interest considering numerous reports that express a rapidly growing interest in the biological and medicinal application of polyoxometalates.13
Notes and references
- N. H. Williams, B. Takasaki, M. Wall and J. Chin, Acc. Chem. Res., 1999, 32, 485 CrossRef CAS.
- (a) E. L. Hegg and J. N. Burstyn, Coord. Chem. Rev., 1998, 173, 133 CrossRef CAS; (b) J. R. Morrow and O. Iranzo, Curr. Opin. Chem. Biol., 2004, 8, 192 CrossRef CAS.
- (a) F. Mancin, P. Scrimin, P. Tecilla and U. Tonellato, Chem. Commun., 2005, 2540 RSC and references therein; (b) R. Ott and R. Krämer, Angew. Chem., Int. Ed., 2000, 39, 3255 CrossRef CAS; (c) A. M. Fanning, S. E. Plush and T. Gunnlaugsson, Chem. Commun., 2006, 3791 RSC; (d) G. Feng, D. Natale, R. Prabaharan, J. C. Mareque-Rivas and N. H. Williams, Angew. Chem., Int. Ed., 2006, 45, 7056 CrossRef CAS; (e) H.-J. Schenider, J. Rammo and R. Hettich, Angew. Chem., Int. Ed. Engl., 1993, 32, 1716 CrossRef; (f) A. O'Donoghue, S. Y. Pyun, M. Y. Yang, J. R. Morrow and J. P. Richard, J. Am. Chem. Soc., 2006, 128, 1615 CrossRef CAS; (g) M. Komiyama, S. Kina, K. Matsumura, J. Sumaoka, S. Tobey, V. M. Lynch and E. Anslyn, J. Am. Chem. Soc., 2002, 24, 13731 CrossRef; (h) F. Aguilar-Perez, P. Gomez-Tagle, E. Collado-Fregoso and A. K. Yatsimirsky, Inorg. Chem., 2006, 45, 9502 CrossRef CAS.
- (a) D. E. Katsoulis, A. N. Lambriandou and M. T. Pope, Inorg. Chim. Acta, 1980, 46, 55; (b) C. F. G. C. Geraldes and M. M. C. A. Castro, J. Inorg. Biochem., 1986, 28, 319 CrossRef CAS; (c) P. Piperaki, N. Katsaros and D. Katakis, Inorg. Chim. Acta, 1982, 67, 37 CrossRef CAS; (d) L. M. R. Hill, G. N. George, A. K. Duhm-Klair and C. G. Young, J. Inorg. Biochem., 2002, 88, 274 CrossRef CAS; (e) W. Kwak, M. T. Pope and T. F. Scully, J. Am. Chem. Soc., 1975, 97, 5735 CrossRef CAS.
- (a) H. Weil-Malherbe and R. H. Green, Biochem. J., 1951, 49, 286 CAS; (b) E. Ishikawa and T. Yamase, J. Inorg. Biochem., 2006, 100, 344 CrossRef CAS.
- (a) M. T. Pope, in Heteropoly and Isopoly Oxometalates, Springer Berlin, 1983, pp. 60–117 Search PubMed; (b) J. J. Cruywagen, Adv. Inorg. Chem., 2000, 49, 121 and references therein.
- J. J. Cruywagen, A. G. Draaijer, J. B. B. Heyns and E. A. Rohwer, Inorg. Chim. Acta, 2002, 331, 322 CrossRef CAS.
- J. Chin, M. Banzszczyk, V. Jubian and X. Zou, J. Am. Chem. Soc., 1989, 111, 186 CrossRef CAS.
- R. I. Maksimovskaya and G. M. Maksimov, Inorg. Chem., 2007, 46, 3688 CrossRef CAS.
- (a) R. Strandberg, Acta Chem. Scand., 1973, 27, 1004 CrossRef CAS; (b) L. Pettersson, I. Andersson and L.-O. Öhman, Inorg. Chem., 1986, 25, 4726 CrossRef CAS.
- (a) C. Rocchiccioli-Deltcheff, M. Fournier and R. Franck, Inorg. Chem., 1983, 22, 207 CrossRef CAS; (b) M. Fournier, R. Thouvenot and C. Rocchiccioli-Deltcheff, J. Chem. Soc., Faraday Trans., 1991, 87, 349 RSC.
- (a) O. W. Horwath and P. Kelly, J. Chem. Soc., Chem. Commun., 1988, 1236 RSC; (b) O. W. Horwath and P. Kelly, J. Chem. Soc., Dalton Trans., 1990, 81 RSC.
- For an overview of the biological activity of polyoxomolybdates see: (a) J. T. Rhule, C. L. Hill and D. A. Judd, Chem. Rev., 1998, 98, 327 CrossRef CAS; (b) T. Yamase, J. Mater. Chem., 2005, 15, 4733 Search PubMed; (c) B. Hasenknopf, Front. Biosci., 2005, 10, 275 CrossRef CAS; (d) H. U. V. Gerth, A. Rompel, B. Krebs, J. Boos and C. Lanvers-Kaminsky, Anti-Cancer Drugs, 2005, 16, 101 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: 95Mo NMR spectrum of the sample. See DOI: 10.1039/b714860g |
| ‡ The ionic strength of the solution was not further adjusted, because it was noticed that high salt concentration impedes hydrolysis. Since thermodynamic parameters for the exact reaction conditions used in hydrolysis experiments are not available, an approximation had to be made by applying the thermodynamic parameters that were obtained in 0.6 M NaClO4 solution. |
| § The pH experiments were performed in the absence of any buffer. The pH of the solutions was measured before and after the hydrolysis reaction and the difference in pH was always less than 0.1 pH unit. The hydrolysis of BNPP in the absence of metal complex was measured at each pH and the final values of the kobs have been adjusted for the background cleavage. |
| ¶ Addition of equimolar amounts of phenyl phosphonate or methyl phosphonic acid inhibited BNPP hydrolysis (kobs = 1.1 × 10−6 and 9.1 × 10−7 s−1, respectively) suggesting competition for binding to [Mo7O24]6−. |
| || At 37 °C only 30% of BNPP was cleaved after 70 h, and at room temperature the BNPP hydrolysis was extremely slow implying very weak interactions with [Mo7O24]6−. |
| This journal is © The Royal Society of Chemistry 2008 |