Activation and reduction of diethyl ether by low valent uranium: formation of the trimetallic, mixed valence uranium oxo species [U(CpRR′)(μ-I)2]3(μ3-O) (CpRR′ = C5Me5, C5Me4H, C5H4SiMe3)†
Christopher P.
Larch
,
F. Geoffrey N.
Cloke
* and
Peter B.
Hitchcock
Department of Chemistry and Biochemistry, School of Life Sciences, University of Sussex, Falmer, Brighton, UK BN1 9QJ. E-mail: f.g.cloke@sussex.ac.uk; Fax: +44 1273 677 196; Tel: +44 1273 678735
First published on 13th November 2007
Abstract
The reaction of UI3 and KCpRR′ (CpRR′ = pentamethylcyclopentadienyl, trimethylsilylcyclopentadienyl or tetramethylcyclopentadienyl) in diethyl ether results in the two-electron reduction of the solvent to form trimetallic, mixed valence uranium oxo species.
There is considerable current interest in the binding of small molecules to U(III) centres, studies of which have revealed a number of unprecedented bonding modes or activation routes: for example, end-on coordination of N2 to an f-element,1 end-on coordination of CO2,2 and reductive coordination3 or reductive cyclooligomerisation of CO.4,5 In this context, we have previously reported the synthesis of the mixed sandwich U(III) complex [U{C8H4(SiiPr3)2}Cp*] and its ability to reversibly reduce dinitrogen to form [U{C8H4(SiiPr3)2}(Cp*)]2(μ-η2:η2-N2).6 The synthesis of the former involved the metathesis reaction between UI3 and KCp* in diethyl ether to form a green material which we assumed to be “(UCp*I2)n”, followed by subsequent addition of K2[C8H4(SiiPr3-1,4)2], which proceeds in modest (40%) overall yield. In this communication we show that, in fact, the reaction between a range of substituted cyclopentadienyl potassium reagents and UI3 in diethyl ether results in the activation and reduction of the solvent to afford trimetallic, mixed valence cyclopentadienyl–uranium oxo species.
When UI3 is stirred with an equimolar amount of KCpRR′ (where CpRR′ is pentamethylcyclopentadienyl, trimethylsilylcyclopentadienyl, or tetramethylcyclopentadienyl) in diethyl ether for 16 h a green solution with white precipitate (KI) was observed in each case. Filtration followed by cooling to −50 °C resulted in the formation of green needles of the trinuclear compounds 1, 2 and 3 (Scheme 1).‡ Analytical purities were confirmed by combustion analyses, and mass spectrometry confirmed that 1, 2, and 3 are trinuclear, with molecular ion peaks at m/z 1897, 1902 and 1856, respectively.
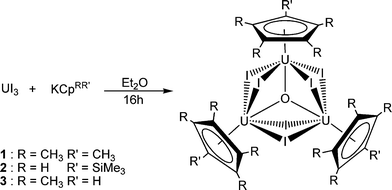 | ||
| Scheme 1 | ||
Crystals of 1 suitable for X-ray diffraction analysis were grown from diethyl ether at −50 °C; the molecular structure is shown in Fig. 1, together with selected bond lengths and angles.§ Compound 1 crystallises in the monoclinic space groupP21/c, with two independent molecules in the asymmetric unit. The structure reveals a trimeric, oxo-centred species in which each uranium centre displays distorted pseudo octahedral geometry. The relative coordinative unsaturation of the uranium centres in 1 is reflected in the shorter U–Cp*(centroid) bond lengths (2.45(2) Å, 2.44(2) Å, and 2.46(2) Å) than that found in the mononuclear UIII complex [UCp*I2(THF)3] (2.80 Å).7 The U–O bond distances are of equal length within esd's (2.234(12) Å, 2.228(12) Å and 2.192(13) Å). Formally 1 contains two U(IV) centres and one U(III) centre, therefore the equivalence of the U–O bond lengths suggests a delocalised mixed valence structure for the trimer. There are no uranium(III) alkoxide, oxide or oxo bond lengths reported in the literature, and limited examples of uranium(IV) oxo bond lengths available for comparison; [{UCp′3}2(μ-O)] (U(IV))8 exhibits a U–O bond length of 2.105(2) Å, which is shorter than all observed U–O distances in 1. The longer uranium–oxygen distance in 1 is attributable to the sharing of the oxo unit between three uranium centres.
 | ||
| Fig. 1 Molecular structure of 1, thermal ellipsoids at 50% probability and hydrogen atoms omitted for clarity; only one of the two independent molecules is shown; only the U and I atoms were refined anisotropically. Selected bond lengths (Å) and angles (°): U(1)–O(1) 2.234(12), U(2)–O(1) 2.228(12), U(3)–O(1) 2.192(13), U(1)–M(1) 2.450(2), U(2)–M(2) 2.440(2), U(3)–M(3) 2.460(2), U(1)–I(1) 3.214(15), U(2)–I(1) 3.179(15); M(1)–U(1)–O(1) 177(1), M(1)–U(1)–I(1) 108(1). M(1), M(2) and M(3) are the centroids of the cyclopentadienyl rings bound to U(1), U(2) and U(3), respectively. | ||
Previously reported examples of related trimetallic uranium complexes are [Cp*3U3(μ3-I)(μ3S)(μ2-I)3(I)3]9 and [U3(μ3-S)(μ3-StBu)(μ2-StBu)3(StBu)6],10 which display similar structural motifs, with a central bridging group 16 atom held between three uranium centres. However both are rigorously U(IV), hence the bridging U–I and U–Cp* bond lengths therein are slightly longer than those in 1, in which the average oxidation state of uranium is 3⅔.
Single crystals of 2 suitable for X-ray crystallographic analysis were grown from diethyl ether at −50 °C and the molecular structure is shown in Fig. 2, together with selected bond lengths and angles.‡ Compound 2 crystallises in the monoclinic space groupP21/c and displays very similar gross structural features to 1.§ The trimethylsilyl groups are orthogonal to each other; crystal packing forces are proposed to be responsible for this alignment since the distance between each silyl group is far too great for intramolecular steric repulsions to be of any effect. The notable difference between 1 and 2 in the solid state is the uranium–oxygen bond lengths. In 1, the U–O bond lengths are all equal within esd's (2.234(12) Å, 2.228(12) Å, 2.192(13) Å), however in 2 only two of the U–O bond lengths are equal (2.168(7) Å and 2.156(7) Å) and the other is significantly longer (2.311(7) Å). Thus, in the solid state the mixed valence nature of 2 appears to be more localised than in 1, which may be a consequence of the higher electron donating ability of Cp*vs. CpTMS.
 | ||
| Fig. 2 Molecular structure of 2, thermal ellipsoids at 50% probability and hydrogen atoms omitted for clarity. Selected bond lengths (Å) and angles (°): U(1)–O(1) 2.168(7), U(2)–O(1) 2.156(7), U(3)–O(1) 2.311(7), U(1)–M(1) 2.430(1), U(2)–M(2) 2.430(1), U(3)–M(3) 2.440(1), U(1)–I(1) 3.142(7) U(2)–I(1) 3.138(8); M(1)–U(1)–O(1) 175.1(2), M(1)–U(1)–I(1) 101.2(2). M(1), M(2) and M(3) are the centroids of the cyclopentadienyl rings bound to U(1), U(2) and U(3), respectively. | ||
The 1H NMR of 1 revealed a broad singlet at δ 20.91 ppm for the Cp* methyl groups. Compound 2 exhibited three broad singlets in the 1H NMR spectrum in the expected 2 : 2 : 9 ratio, consistent with the two pairs of ring protons (δ 3.23 and 1.06 ppm) and the SiMe3 group (δ−3.07 ppm) respectively. Thus the asymmetry present in the solid state structure of 2 appears not to be present in solution, although the NMR line widths may be too broad to resolve it.
Magnetic data for 1 were recorded on a powder sample of 29.6 mg using a SQUID magnetometer between 2 and 300 K in a magnetic field of 0.2 T. The susceptibility values were corrected for the sample diamagnetic contribution using Pascal's constants. The magnetic moment of 1 was temperature dependent, varying from 2.28 μB at 5 K to 2.78 μB at 300 K. Plots of molar susceptibility (χm) and effective magnetic moment (μeff = √(8χmT)μB) vs. T are shown in Fig. 3. Compound 1 exhibits antiferromagnetism at low temperatures, but otherwise obeys the Curie law. The optimised value for the Curie temperature is θ = −0.288 K, therefore overall 1 is effectively a weak antiferromagnet. The experimentally determined effective magnetic moment of 1 at room temperature is considerably lower than the values reported for mononuclear uranium(III) complexes by Stewart and Andersen11 and other researchers in this field;12 it is also much lower than the theoretical values for 5f2 and 5f3 free-ion systems (3.58 μB and 3.69 μB respectively). In fact, μeff for 1 is comparable to that reported (2.80 μB) for the U(IV) complex [UtBuNONCp*Cl] where tBuNON = [tBuN(Si(CH3)2)]2O.13 This lowering of the magnetic moment is ascribed to antiferromagnetic coupling between the three uranium centres, via the central oxo unit.
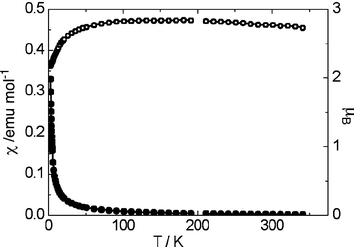 | ||
| Fig. 3 Magnetic data for 1, per mole of uranium; upper curve (open circles) = μeff and lower curve (solid circles) = χm. | ||
The formation of 1, 2 and 3 was quite reproducible, and carried out with strict exclusion of oxygen and water, the obvious potential sources of the oxo unit. The source of the latter was confirmed as the ether solvent by GC-MS analysis of the reaction mixture, which showed the presence of butane. On the basis of the latter, the formation of 1, 2, and 3 presumably involves a radical mechanism. Activation of diethyl ether is rare—during attempts to prepare the highly reducing TmCp*2 in diethyl ether, Evans and co-workers found the unexpected formation of trimetallic Cp*2Tm(μ-OEt)TmCp*2(μ-O)TmCp*2, the product of ether cleavage.14 The formation of 1 also accounts for the modest yields (40%) obtained in the synthesis of [U{C8H4(SiiPr3)2}Cp*], since some of the K2[C8H4(SiiPr3-1,4)2] reagent clearly has to function as a reducing agent to convert the two U(IV) centres in 1 to U(III).
We thank Professor Andrew Harrison (Edinburgh) for the magnetic studies, and EPSRC for financial support.
Notes and references
- W. J. Evans, S. A. Kozimor and J. W. Ziller, J. Am. Chem. Soc., 2003, 125, 14264 CrossRef CAS.
- I. Castro-Rodriguez, H. Nakai, L. N. Zakharov, A. L. Rheingold and K. Meyer, Science, 2004, 305, 1757 CrossRef CAS.
- I. Castro-Rodriguez and K. Meyer, J. Am. Chem. Soc., 2005, 127, 11242 CrossRef CAS.
- O. T. Summerscales, F. G. N. Cloke, P. B. Hitchcock, J. C. Green and N. Hazari, Science, 2006, 311, 829 CrossRef.
- O. T. Summerscales, F. G. N. Cloke, P. B. Hitchcock, J. C. Green and N. Hazari, J. Am. Chem. Soc., 2006, 128, 9602 CrossRef CAS.
- F. G. N. Cloke and P. B. Hitchcock, J. Am. Chem. Soc., 2002, 124, 9352 CrossRef CAS.
- L. R. Avens, C. J. Burns, R. J. Butcher, D. L. Clark, J. C. Gordon, A. R. Schake, B. L. Scott, J. G. Watkin and B. D. Zwick, Organometallics, 2000, 19, 451 CrossRef CAS.
- J.-C. Berthet, J.-F. L. Marechal, M. Nierlich, M. Lance, J. Vigner and M. Ephritikhine, J. Organomet. Chem., 1991, 408, 335 CrossRef CAS.
- D. L. Clark, J. C. Gordon, J. C. Huffman, J. G. Watkin and B. D. Zwick, New J. Chem., 1995, 19, 495 Search PubMed.
- P. C. Leverd, T. Arliguie, M. Ephritikhine, M. Nierlich, M. Lance and J. Vigner, New J. Chem., 1993, 17, 769 Search PubMed.
- J. L. Stewart and R. A. Andersen, Polyhedron, 1998, 17, 953 CrossRef CAS.
- H. Nakai, X. Hu, L. N. Zakharov, A. L. Rheingold and K. Meyer, Inorg. Chem., 2004, 43, 855 CrossRef CAS.
- K. C. Jantunen, R. J. Batchelor and D. B. Leznoff, Organometallics, 2004, 23, 2186 CrossRef CAS.
- W. J. Evans, N. T. Allen and J. W. Ziller, J. Am. Chem. Soc., 2001, 123, 7927 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Crystallographic details (data collection parameters, complete bond lengths and angles) for 1 and 2, experimental procedures and full characterisation data for all new compounds. See DOI: 10.1039/b714211k |
| ‡ Syntheses and characterisation data1: UI3 (0.619 g, 1 mmol) and KCp* (0.174 g, 1 mmol) were placed in an ampoule equipped with a greaseless stopcock and Et2O (40 ml) was added. The reaction was left stirring for 16 h, which resulted in a dark green solution with a creamy white precipitate. The solution was filtered from insolubles (KI) through pre-dried Celite on a glass frit. The solvent was reduced and the solution slowly cooled to −50 °C which after 48 h yielded small green needles. These were washed with cold Et2O and collected (yield 0.48 g, 76%).Anal. Calc. for C30H45I6OU3: C, 18.99; H: 2.39. Found C, 18.92; H: 2.48MS (EI) m/z : 1897 [24%, M+], 1725 [M+− Cp*], 1143 [M+− (UCp*I3)].1H NMR (C6D6) δ : 20.49 (s, 45H, CH3–Cp*).2: Prepared in a similar manner to 1, with sodium trimethylsilylcyclopentadienyl. (0.51 g, 81% yield)Anal. Calc. for C24H27I6Si3U3O: C, 15.15; H, 2.07. Found C, 15.02; H, 1.96.MS (EI) m/z : 1902 [17%, M+], 1147 [M+− (UCp′I3)].1H NMR (C6D6) δ : 3.23 (s, 6H, CH ‘ring’), 1.06 (s, 6H, CH ‘ring’), −3.07 (br s, 27H, SiMe3).3: Prepared in a similar manner to 1, with potassium tetramethylcyclopentadienyl (0.36 g, 58% yield).Anal. Calc. for C27H36I6OU3: C, 17.48; H, 2.12. Found C, 17.51; H, 2.15.MS (EI) m/z : 1856 [24%, M+]. 1H NMR (C6D6) δ : 20.69 (br s, 18H, CH3), 17.84 (br s, 18H, CH3). The ring C–H proton could not be unambiguously assigned. |
| § Crystal data for 1. C30H45I6OU3, Mr = 1897.15, monoclinic, a = 24.5979(5), b = 21.5504(4), c = 16.6282(3) Å, β = 109.891(1)°, U = 8288.7(3) Å3, T = 173(2) K, space groupP21/c, Z = 8, λ = 0.71073 Å, μ = 16.19 mm−1. 55482 reflections collected, 13374 independent reflections. Rint = 0.092. Final R values [I > 2σ(I)]: R1 = 0.048, wR2 = 0.113. Crystal data for 2. C24H39I6OSi3U3, Mr = 1903.31, monoclinic, a = 16.7070(3), b = 8.3748(1), c = 31.3762(4) Å, β = 102.745(1)°, U = 4281.82(11) Å3, T = 173(2) K, space groupP21/c, Z = 4, λ = 0.71073 Å, μ = 15.75 mm−1. 61341 reflections collected, 8349 independent reflections. Rint = 0.125. Final R values [I > 2σ(I)]: R1 = 0.045, wR2 = 0.110. CCDC 661103 and 661104. For crystallographic data in CIF or other electronic format see DOI: 10.1039/b714211k |
| This journal is © The Royal Society of Chemistry 2008 |