Intermolecular radical addition reactions of α-iodo cycloalkenones and a synthetic study of the formal synthesis of enantiopure fawcettimine†
Kuan-Miao
Liu
*a,
Chi-Min
Chau
*a and
Chin-Kang
Sha
b
aSchool of Applied Chemistry, Chung Shan Medical University, Taichung 402, Taiwan, ROC. E-mail: lkm@csmu.edu.tw; cmchau@csmu.edu.tw
bDepartment of Chemistry, National Tsing Hua University, Hsinchu 300, Taiwan, ROC
First published on 21st November 2007
Abstract
The generation of α-carbonyl vinyl radicals from α-iodo cycloalkenones, the scope of their participation in intermolecular addition reactions with electron-withdrawing olefins are studied and a synthetic study of the formal synthesis of enantiopure fawcettimine using this reaction is described.
Inter- and intramolecular radical reactions have been some of the most important and effective synthetic methods in organic synthesis, and they have frequently been used as key steps in the construction of natural products.1 Synthetic applications of vinyl radicals are often limited to intramolecular reactions; this may be due to the fact that vinyl radicals are more reactive than alkyl radicals and can easily be trapped by hydrogen, electron-deficient double or triple bonds in the same molecule, or radical mediators, such as Bu3SnH, in a reaction. In our previous studies, we have demonstrated that the intramolecular cyclization of an α-carbonyl radical could be used as an efficient step in the total synthesis of several natural products, including (±)-modhephene, (−)-dendrobine, (−)-5-oxosilphiperfor-6-exe, (+)-paniculatine and (−)-bakkenolide III, and in the formal synthesis of (−)-pinguisenol and (−)-α-pinguisene.2
To the best of our knowledge, no reports so far have described the use of an α-carbonyl vinyl radical for intermolecular carbon–carbon bond formation. Herein, we report the intermolecular addition reactions of α-carbonyl vinyl radicals generated from α-iodo cycloalkenones with electron-withdrawing olefins (schematically shown in Scheme 1). This reaction has important synthetic value in the preparation of chiral α-substituted cyclic enones that are difficult to obtain by other methods. In order to demonstrate the versatility of this reaction, we also report a synthetic study of the formal synthesis of enantiopure fawcettimine by an intramolecular radical cyclization .
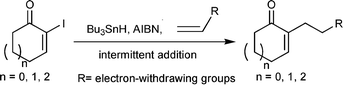 | ||
| Scheme 1 Intermolecular addition reactions of α-carbonyl vinyl radicals with electron-withdrawing olefins . | ||
Our experiment began by treating a refluxing benzene solution of 2-iodocyclohex-2-enone (2) and acrylonitrile (1 equiv.) with a benzene solution of Bu3SnH (1.2 equiv.) and AIBN (0.12 equiv.); however, the expected amount of adduct was not obtained. We believe that one of the reasons leading to the failure of this reaction was the low concentration of acrylonitrile. To solve this problem, we examined the dependence of the amount of acrylonitrile on the yield of the product by increasing its amount from 3 equiv. up to 10 equiv. The desired adduct 2a was then successfully obtained in yields of 13 and 39%, respectively. The yield of the adduct did not increase any further, even when the amount of acrylonitrile was increased above 10 equiv. In addition to the concentration of acrylonitrile, the concentration of the resulting α-carbonyl vinyl radical may also be an important factor in the success of this intermolecular radical addition reaction . If the concentration of the α-carbonyl vinyl radical is very high, the self-coupling reaction would be significant. Based on this consideration, we attempted to maintain a low concentration of generated radical at any given instant by using the slow addition method. We found that the yield of the product increased slightly; however, the reaction became more complicated, and some of the enone was recovered. Therefore, we used the intermittent addition method, in which the reaction was performed by adding a benzene solution (0.5 M with respect to Bu3SnH) of AIBN (0.12 equiv.) and Bu3SnH (1.2 equiv.) to a refluxing benzene solution (0.2 M with respect to α-iodocyclohexenone) of 2-iodocyclohex-2-enone and acrylonitrile (10 equiv.) eight times at 40 min intervals, after which 2a was obtained in 60% yield. These were the most optimized reaction conditions for the intermolecular α-carbonyl vinyl radical addition reaction with olefins , and were applied in many other cases later. Several other sets of modified conditions were also examined; however, none were better than the present set.
In this study, four α-iodocycloalkenones, including 2-iodocyclopent-2-enone (1), 2-iodocyclohex-2-enone (2), 2-iodocyclohept-2-enone (3) and even (R)-2-iodo-5-methylcyclohex-2-enone (5) (in the study of the formal synthesis of fawcettimine), were employed as the radical donors and various electron-withdrawing olefins were used as radical acceptors. The final results are summarized in Table 1. In addition, vinyl acetate, which is an electron-rich olefin , was employed in this reaction; however, none of the desired adduct was obtained. Based on these experimental results, we suggest that α-carbonyl vinyl radicals are ideal electron-rich radical donors under radical reaction conditions. To demonstrate the synthetic utility of the intermolecular radical addition reaction , it was treated as an important step in the formal synthesis of enantiopure fawcettimine.
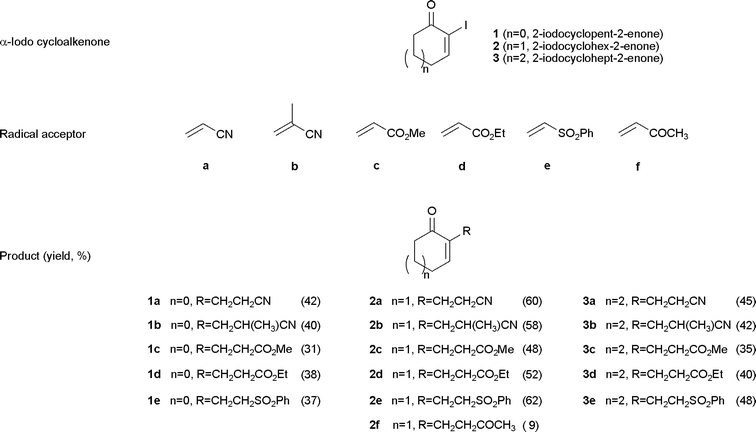
Fawcettimine was first isolated from extracts of the alkaloids of Lycopodium fawcetti collected in the Blue Mountain range of Jamaica by the Burnell group in 1959. Because of their potent acetylcholine esterase inhibition activity,3 coupled with an extremely complex structure,4 various different synthetic methodologies leading to this Lycopodium alkaloid were proposed and studied by many groups.5 Unfortunately, the total or formal synthesis was a really difficult challenge. Until 1979, the first racemic total synthesis of fawcettimine was accomplished by Inubushi et al. using a Diels–Alder reaction as the key step.5a To date, there are only three total synthesis reports (Inubushi et al. in 1979,5a and Heathcock et al. in 19865c and 19895d) and one publication of the core skeleton synthesis (Mehta in 19915e) of fawcettimine; however, none of them deals an enantiopure synthesis.
Herein, we propose a unique methodology for the formal synthesis of enantiopure fawcettimine by using chiral enone 46 as the starting material, and inter- and intramolecular radical reaction sequences as the key steps. The synthetic scheme for obtaining compound 11, whose structure is almost the same as that of the key intermediate in Heathcock et al.'s synthesis of fawcettimine, is illustrated in Scheme 2. We suggest that compound 11 could be converted into fawcettiminevia a procedure similar to Heathcock et al.'s process developed in 1989.5d
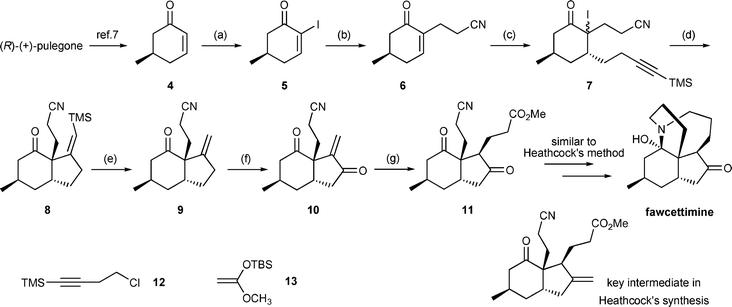 | ||
| Scheme 2 Reagents and conditions: (a) I2, pyridine, CH2Cl2, 95%; (b) acrylonitrile, AIBN, Bu3SnH, PhH, 70%; (c) 12, Mg, CuI, TMSCl, HMPA, THF; NaI, m-CPBA, THF, 78% (2 steps); (d) Bu3SnH, AIBN, PhH, slow addition, 60 °C, 64%; (e) TFA, CH2Cl2, 78%; (f) SeO2, tBuOOH, CH2Cl2; Jones’ reagent, 52%; (g) 13, LiClO4, Et2O; AcOH, THF, H2O, 40%. | ||
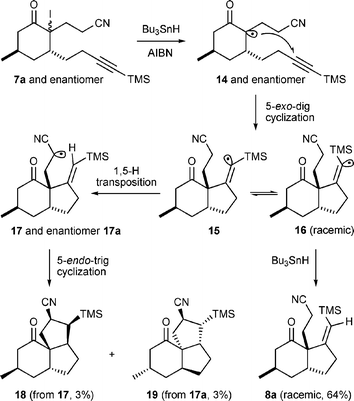
Optically pure cyano-enone 6 could be obtained in 70% yield from chiral α-iodoenone 5via a intermolecular radical addition reaction with acrylonitrile. The CuI-mediated conjugate addition of 4-(trimethylsilyl)-3-butynylmagnesium chloride was followed by the trapping of the resulting enolate with chlorotrimethylsilane (TMSCl) to obtain the trimethylsilyl enol ether . Without purification, the resulting enol ether was then treated with a mixture of NaI and m-CPBA to afford the unstable iodo ketone 7 in 78% yield. The intramolecular radical cyclization of 7 was then carried out by using a benzene solution of AIBN and Bu3SnH, which was introduced using a syringe pump at reflux, and the desired cyclized product 8 was obtained in 64% yield. It is noteworthy that in the model study of fawcettimine, wherein the racemic 5-methylcyclohex-2-enone was used as the starting material, trace amounts of compounds 18 and 19 were separately isolated during the radical cyclization reaction and their structures ascertained by single crystal X-ray analysis.‡ This result may be rationalized by the 1,5-hydrogen transposition of vinyl radical 15, formed by the 5-exo-dig cyclization of radical 14, followed by the intramolecular 5-endo-trig radical cyclization of the resulting radical 17. The proposed mechanism is illustrated in Scheme 3. Based on the single crystal X-ray analyses, we suggested that the relative stereochemistry of compounds 18 and 19 could be the indirect evidence that allows the relative stereochemistry of optically pure compound 11 to be assigned.
The exposure of compound 9 to SeO2, followed by Jones’ oxidation, gave the allylic oxidation product, enone 10. The lithium perchlorate-mediated conjugate addition of freshly prepared ketene silyl acetal , 13, to enone 10 afforded compound 11, which was almost identical to the key intermediate in Heathcock's synthesis of fawcettimine, except for the carbonyl functionality on the five-membered ring.
In conclusion, a useful intermolecular radical addition reaction of α-iodo cycloalkenone has been described. The formal synthesis of enantiopure fawcettimine has been accomplished stereoselectively, during which inter- and intramolecular radical reactions were employed as key steps to facilitate the construction of the core skeleton of the product. Further applications using this strategy are currently under development in our laboratory.
Notes and references
- (a) B. Giese, in Radicals in Organic Synthesis: Formation of Carbon–Carbon Bonds, ed. J. E. Baldwin, Pergamon Press, Oxford, 1986, ch. 3–4, pp. 36–209 Search PubMed; (b) B. Giese, Angew. Chem., Int. Ed. Engl., 1989, 28, 969 CrossRef; (c) D. P. Curran, W. Shen, J. Zhang and T. A. Heffner, J. Am. Chem. Soc., 1990, 112, 6738 CrossRef CAS; (d) N. A. Porter, D. M. Scott, I. J. Rosenstein, B. Giese, A. Veit and H. G. Zeitz, J. Am. Chem. Soc., 1991, 113, 1791 CrossRef CAS; (e) D. P. Curran, in Comprehensive Organic Synthesis, ed. B. M. Trost and I. Fleming, Pergamon Press, Oxford, 1991, vol. 4, ch. 4.2, pp. 779–831 Search PubMed.
- (a) C. K. Sha, T. S. Jean and D. C. Wang, Tetrahedron Lett., 1990, 31, 3745 CrossRef CAS; (b) C. K. Sha, K. C. Santhosh and S. H. Lih, J. Org. Chem., 1998, 63, 2699 CrossRef CAS; (c) C. K. Sha, R. T. Chiu, C. F. Yang, N. T. Yao, W. H. Tseng, F. L. Liao and S. L. Wang, J. Am. Chem. Soc., 1997, 119, 4130 CrossRef CAS; (d) C. K. Sha, F. K. Lee and C. J. Chang, J. Am. Chem. Soc., 1999, 121, 9875 CrossRef CAS; (e) C. K. Sha, H. W. Liao, P. C. Cheng and S. C. Yen, J. Org. Chem., 2003, 68, 8704 CrossRef CAS; (f) C. H. Jiang, A. Bhattacharyya and C. K. Sha, Org. Lett., 2007, 9, 3241 CrossRef CAS.
- (a) J. Kobayashi and H. Morita, in The Alkaloid, ed. G. A. Cordell, Academic Press, New York, 2005, vol. 61, pp. 1–57 Search PubMed; (b) X. Ma and D. R. Gang, Nat. Prod. Rep., 2004, 21, 752 RSC; (c) X. C. He, S. Feng, Z. F. Wang, Y. Shi, S. Zheng, Y. Xia, H. Jiang, X. C. Tang and D. Bai, Bioorg. Med. Chem., 2007, 15, 1394 CrossRef CAS.
- (a) K. Nishio, T. Fujiwara, K. Tomiza, H. Ishii, Y. Inubushi and T. Harayama, Tetrahedron Lett., 1961, 861; (b) W. A. Ayer, B. Altenkirk and Y. Fukazawa, Tetrahedron, 1974, 30, 4213 CrossRef CAS; (c) Y. Inubushi, T. Harayama, K. Yamaguchi and H. Ishii, Chem. Pharm. Bull., 1981, 29, 3418 CAS.
- (a) T. Harayama, M. Takatani and Y. Inubushi, Tetrahedron Lett., 1979, 4307 CrossRef CAS; (b) T. A. Blumenkopf and C. H. Heathcock, J. Am. Chem. Soc., 1983, 105, 2354 CrossRef CAS; (c) C. H. Heathcock, K. M. Smith and T. A. Blumenkopf, J. Am. Chem. Soc., 1986, 108, 5022 CrossRef CAS; (d) C. H. Heathcock, T. A. Blumenkopf and K. M. Smith, J. Org. Chem., 1989, 54, 1548 CrossRef CAS; (e) G. Mehta, S. Reddy Sreenivasa, R. Radhakrishnan, M. V. Manjula and M. A. Viswamitra, Tetrahedron Lett., 1991, 32, 6219 CrossRef CAS.
- D. Caine, K. Procter and R. A. Cassell, J. Org. Chem., 1984, 49, 2647 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Detailed experimental procedures, 1H and 13C NMR spectroscopic data, and analytical data of all the compounds in Table 1, and Scheme 1, Scheme 2 and Scheme 3. See DOI: 10.1039/b714078a |
| ‡ Crystal data: For 18: C17H27NOSi, M = 289.49, T = 296(2) K, monoclinic, space group P21/c, a = 7.5791(14), b = 12.075(2), c = 19.243(4) Å, β = 91.904(3)°, V = 1760.1(6) Å3, Z = 4, F(000) = 632, reflections collected: 10942, independent reflections: 4155 (Rint = 0.0240). Final R indices [I > 2σ(I)]: R1 = 0.0499, wR2 = 0.1341; R indices (all data): R1 = 0.0780, wR2 = 0.1503. For 19: C17H27NOSi, M = 289.49, T= 296(2) K, monoclinic, space group P21/c, a = 11.6687(8), b = 12.3423(9), c = 12.2850(9) Å, β = 95.921(2)°, V = 1759.8(2) Å3, Z = 4, F(000) = 632, reflections collected: 10218, independent reflections: 3920 (Rint = 0.0201). Final R indices [I > 2σ(I)]: R1 = 0.0463, wR2 = 0.1348; R indices (all data): R1 = 0.0577, wR2 = 0.1435. CCDC 663530 and 663530. For crystallographic data in CIF or other electronic format see DOI: 10.1039/b714078a |
| This journal is © The Royal Society of Chemistry 2008 |